A Rare Cause of Chronic Hypokalemia with Metabolic Alkalosis: Case Report and Differential Diagnosis

 , and
, and
Abstract
1. Introduction
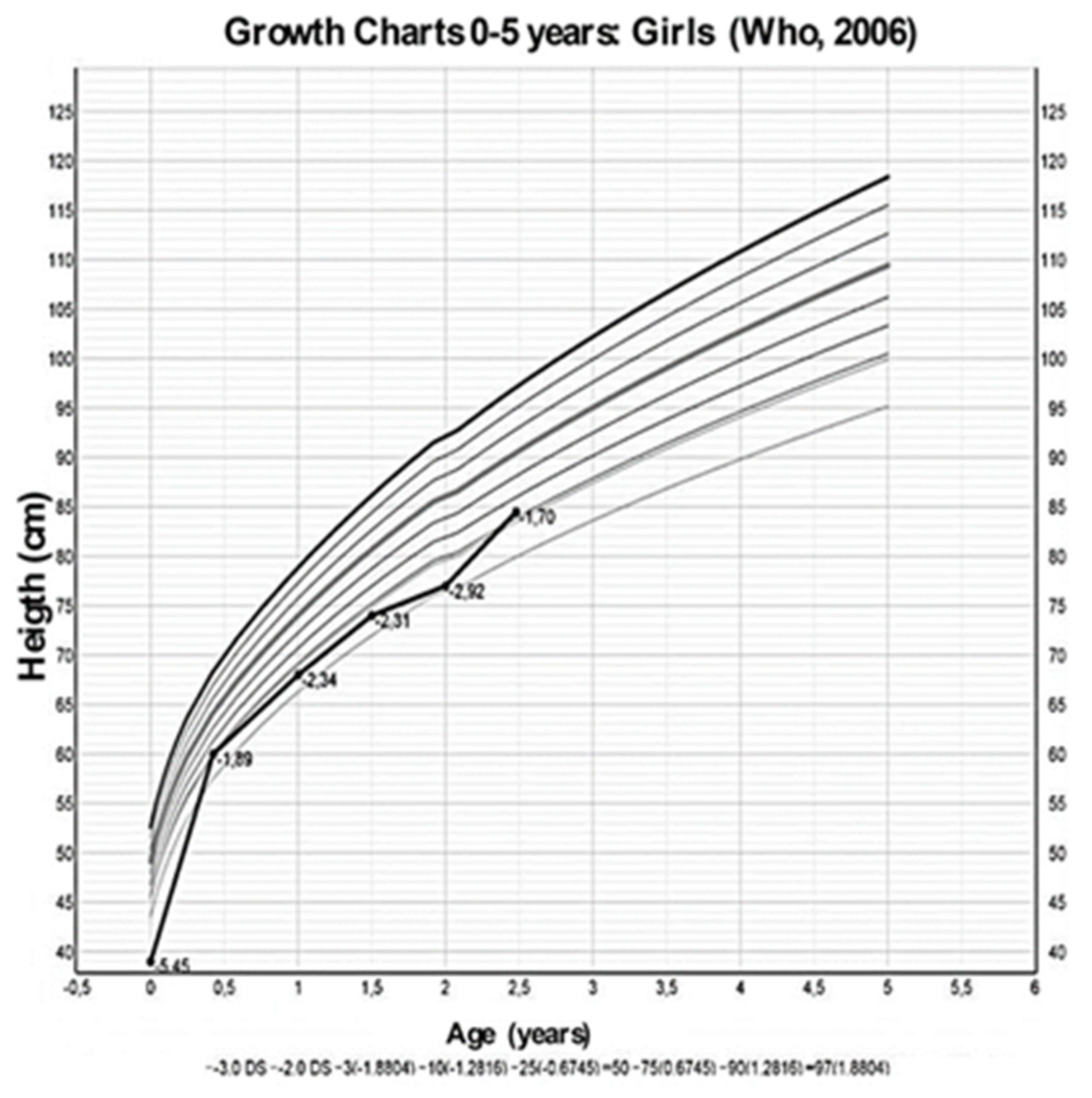
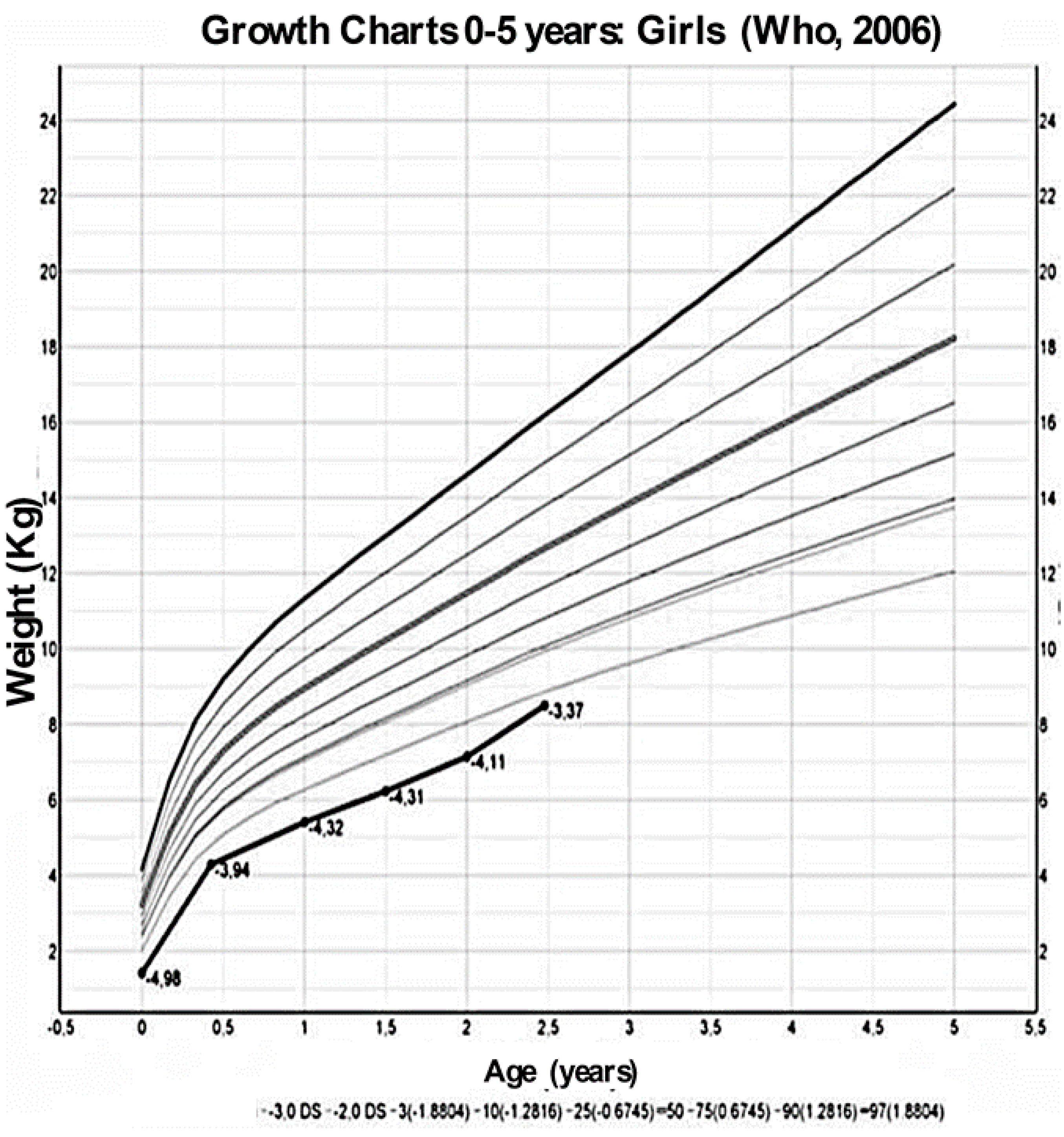
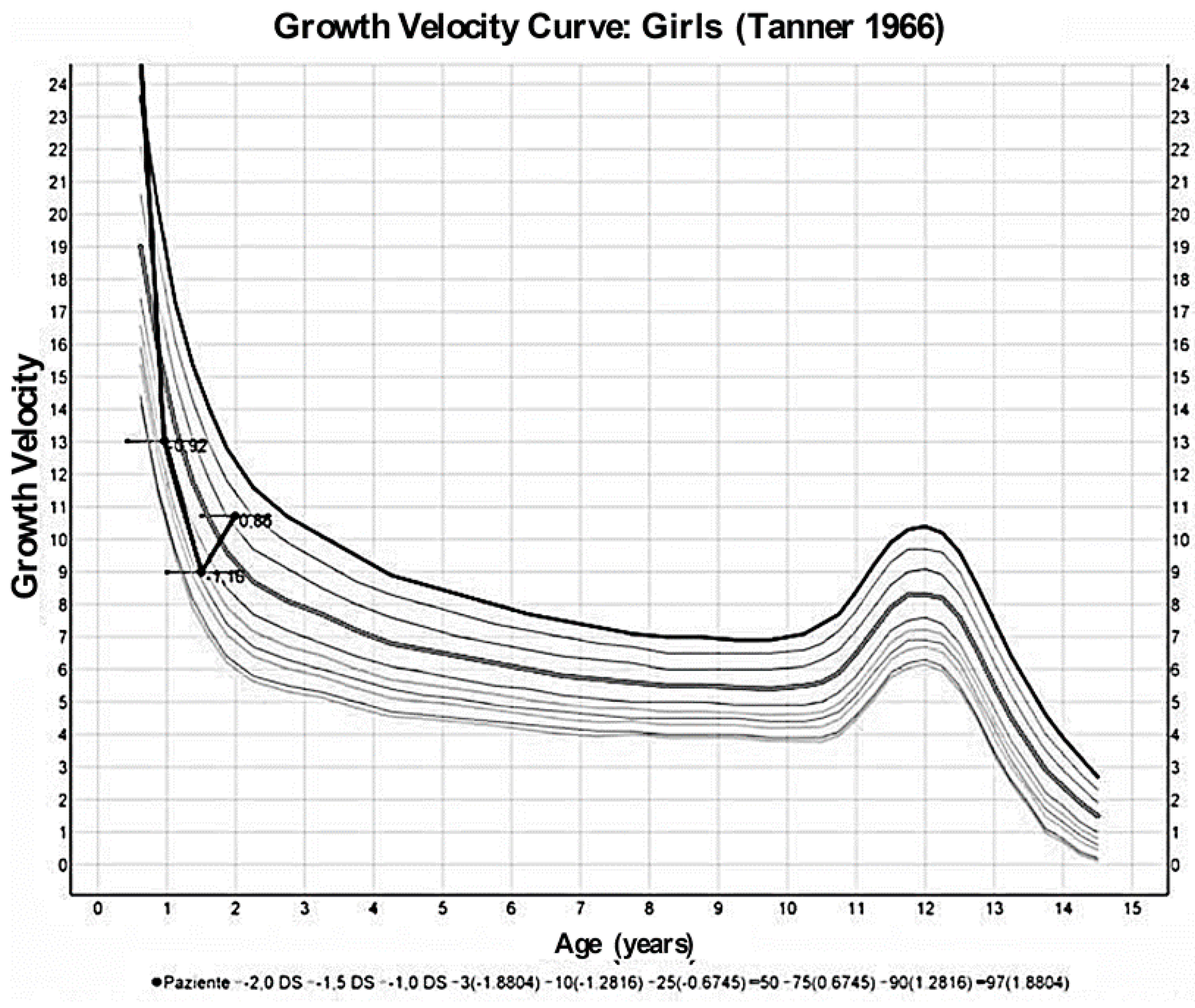
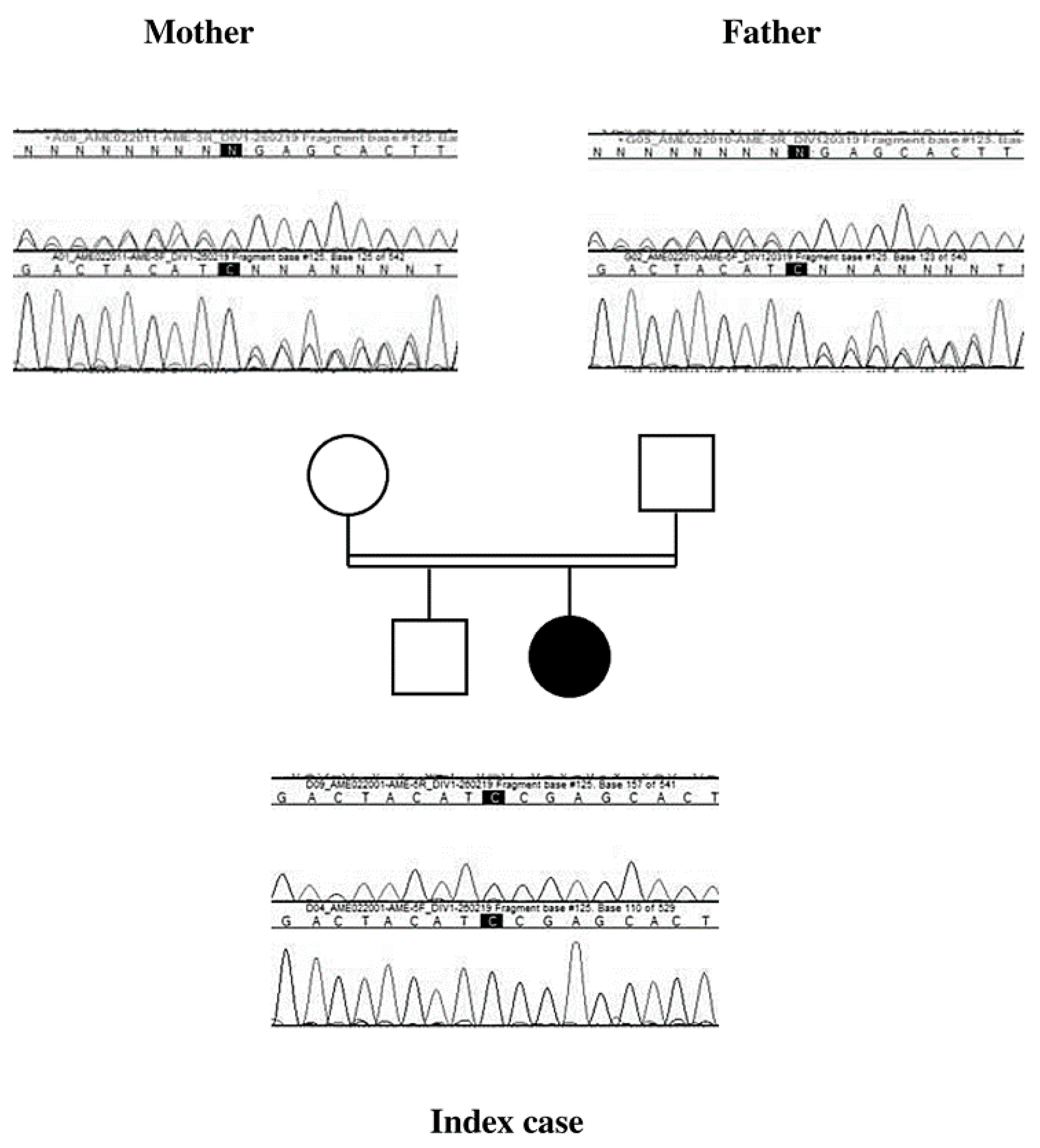
2. Case Presentation
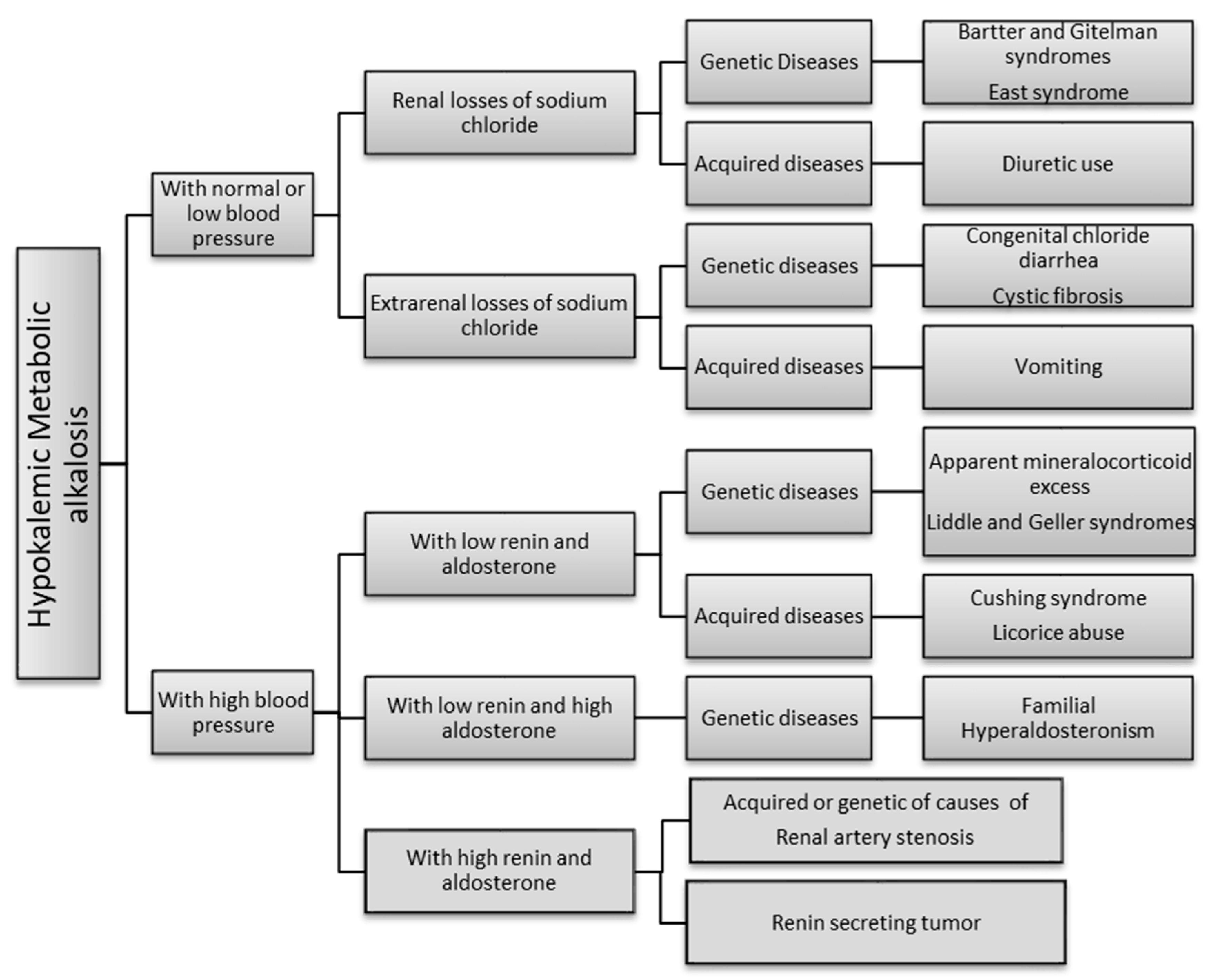
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zieg, J.; Gonsorcikova, L.; Landau, D. Current views on the diagnosis and management of hypokalaemia in children. Acta Paediatr. 2016, 105, 762–772. [Google Scholar] [CrossRef]
- Gennari, F.J.; Hussain-Khan, S.; Segal, A. An unusual case of metabolic alkalosis: A window into the pathophysiology and diagnosis of this common acid-base disturbance. Am. J. Kidney Dis. 2010, 55, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Kleta, R.; Bockenhauer, D. Salt-Losing Tubulopathies in Children: What’s New, What’s Controversial? J. Am. Soc. Nephrol. 2018, 29, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, G.D.; Mohaupt, M.G.; Bianchetti, M.G. Monogenic forms of hypertension. Eur. J. Pediatr. 2012, 171, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Raina, R.; Krishnappa, V.; Das, A.; Amin, H.; Radhakrishnan, Y.; Nair, N.R.; Kusumi, K. Overview of Monogenic or Mendelian Forms of Hypertension. Front. Pediatr. 2019, 7, 263. [Google Scholar] [CrossRef]
- Parsa, A.A.; New, M.I. Low-renin hypertension of childhood. Endocrinol. Metab. Clin. N. Am. 2011, 40, 369–377. [Google Scholar] [CrossRef]
- Morineau, G.; Sulmont, V.; Salomon, R.; Fiquet-Kempf, B.; Jeunemaître, X.; Nicod, J.; Ferrari, P. Apparent mineralocorticoid excess: Report of six new cases and extensive personal experience. J. Am. Soc. Nephrol. 2006, 17, 3176–3184. [Google Scholar] [CrossRef]
- Najafi, M.; Kordi-Tamandani, D.M.; Behjati, F.; Sadeghi-Bojd, S.; Bakey, Z.; Karimiani, E.G.; Schüle, I.; Azarfar, A.; Schmidts, M. Mimicry and well known genetic friends: Molecular diagnosis in an Iranian cohort of suspected Bartter syndrome and proposition of an algorithm for clinical differential diagnosis. Orphanet J. Rare Dis. 2019, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Yau, M.; Haider, S.; Khattab, A.; Ling, C.; Mathew, M.; Zaidi, S.; Bloch, M.; Patel, M.; Ewert, S.; Abdullah, W.; et al. Clinical, genetic, and structural basis of apparent mineralocorticoid excess due to 11β-hydroxysteroid dehydrogenase type 2 deficiency. Proc. Natl. Acad. Sci. USA 2017, 114, E11248–E11256. [Google Scholar] [CrossRef]
- Dave-Sharma, S.; Wilson, R.C.; Harbison, M.D.; Newfield, R.; Azar, M.R.; Krozowski, Z.S.; Funder, J.W.; Shackleton, C.H.; Bradlow, H.L.; Wei, J.Q.; et al. Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. J. Clin. Endocrin. Metab. 1998, 83, 2244–2254. [Google Scholar] [CrossRef]
- Wilson, R.C.; Krozowski, Z.S.; Li, K.; Obeyesekere, V.R.; Razzaghy-Azar, M.; Harbison, M.D.; Wei, J.Q.; Shackleton, C.H.; Funder, J.W.; New, M.I. A mutation in the HSD11B2 gene in a family with apparent mineralocorticoid excess. J. Clin. Endocrin. Metab. 1995, 80, 2263–2266. [Google Scholar]
- Condon, J.; Gosden, C.; Gardener, D.; Nickson, P.; Hewison, M.; Howie, A.J.; Stewart, P.M. Expression of type 2 11beta-hydroxysteroid dehydrogenase and corticosteroid hormone receptors in early human fetal life. J. Clin. Endocrin. Metab. 1998, 83, 4490–4497. [Google Scholar]
- WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr Suppl. 2006, 450, 76–85. [Google Scholar]
- Tanner, J.M.; Whitehouse, R.H. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch. Dis. Child. 1976, 51, 170–179. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Ehret, G.B.; Caulfield, M.J. Genes for blood pressure: An opportunity to understand hypertension. Eur. Heart J. 2013, 34, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Van’T Hoff, W.; Dattani, M.; Lehnhardt, A.; Subtirelu, M.; Hildebrandt, F.; Bichet, D.G. Secondary nephrogenic diabetes insipidus as a complication of inherited renal diseases. Nephron Physiol. 2010, 116, 23. [Google Scholar] [CrossRef] [PubMed]
- Knops, N.B.; Monnens, L.A.; Lenders, J.W.; Levtchenko, E.N. Apparent mineralocorticoid excess: Time of manifestation and complications despite treatment. Pediatrics 2011, 127, e1610–e1614. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, T.; Al-Shaikh, A. Apparent mineralocorticoid excess syndrome: Report of one family with three affected children. J. Pediatr. Endocrinol. Metab. 2012, 25, 1083–1088. [Google Scholar] [CrossRef]
- Kitanaka, S.; Tanae, A.; Hibi, I. Apparent mineralocorticoid excess due to 11 beta-hydroxysteroid dehydrogenase deficiency: A possible cause of intrauterine growth retardation. Clin. Endocrinol. (Oxf.) 1996, 44, 353–359. [Google Scholar] [CrossRef]
- Moudgil, A.; Rodich, G. Nephrocalcinosis and renal cysts associated with apparent mineralocorticoid excess syndrome. Pediatr. Nephrol. 2000, 15, 60–62. [Google Scholar] [CrossRef]
- Adamidis, A.; Cantas-Orsdemir, S.; Tsirka, A.; Abbott, M.A.; Visintainer, P.; Tonyushkina, K. Apparent Mineralocorticoid Excess in the Pediatric Population: Report of a Novel Pathogenic Variant of the 11β-HSD2 Gene and Systematic Review of the Literature. Pediatr. Endocrinol. Rev. 2019, 16, 335–358. [Google Scholar]
- Palermo, M.; Delitala, G.; Mantero, F.; Stewart, P.M.; Shackleton, C.H. Congenital deficiency of 11beta-hydroxysteroid dehydrogenase (apparent mineralocorticoid excess syndrome): Diagnostic value of urinary free cortisol and cortisone. J. Endocrinol. Investig. 2001, 24, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Parfait, B.; Vidaud, D.; Vidaud, M. Mécanismes et consequences des mutations. Med. Sci. 2005, 21, 969–980. [Google Scholar] [CrossRef]
- Conti, E.; Izaurralde, E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005, 17, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.R.; Bailey, M.A.; Soares, D.C.; Dunbar, D.R.; Mullins, J.J. In silico structure-function analysis of pathological variation in the HSD11B2 gene sequence. Physiol. Genom. 2010, 42, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.P.; Ganjam, V.; Chen, Y.J.; Liu, Y.; Clark, S.A.; Gomez-Sanchez, C.E. The 11beta hydroxysteroid dehydrogenase 2 exists as an inactive dimer. Steroids 2001, 66, 845–848. [Google Scholar] [CrossRef]
- Stewart, P.M.; Krozowski, Z.S.; Gupta, A.; Milford, D.V.; Howie, A.J.; Sheppard, M.C.; Whorwood, C.B. Hypertension in the syndrome of apparent mineralocorticoid excess due tomutation of the 11 beta-hydroxysteroid dehydrogenase type 2 gene. Lancet 1996, 347, 88–91. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FE | 4 dd | 7 mo | 9 mo | 12 mo | 15 mo | 18 mo | 30 mo | 36 mo | |
|---|---|---|---|---|---|---|---|---|---|
| Creatinine | 0.2 | 0.3 | 0.26 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | mg/dL |
| Na | 137 | 138 | 140 | 139 | 140 | 139 | 139 | 139 | mmol/L |
| K | 3.3 | 3.3 | 3.1 | 3.3 | 3.3 | 3.6 | 4.2 | 4.6 | mmol/L |
| Cl | 101 | 102 | 101 | 102 | 101 | 101 | 101 | 100 | mmol/L |
| pH | 7.51 | 7.51 | 7.51 | 7.47 | 7.45 | 7.37 | 7.37 | 7.37 | |
| HCO3− | 27 | 27 | 31.1 | 33.5 | 31 | 24 | 28.3 | 28.3 | mmol/L |
| Renine | − | 2.1 | 1.8 | 1.7 | 1.8 | − | 3.6 | − | pg/mL |
| Aldosterone | − | 3.6 | 3.6 | 3.6 | 3.6 | − | 2.6 | − | microU/mL |
| CaU/CrU | − | <0.2 | <0.2 | 0.5 | 0.8 | 0.5 | 0.5 | 0.5 | mg/mg |
| FENa | − | − | 0.7 | − | − | 0.22 | 0.26 | - | % |
| uK/uCr | − | − | 17 | − | − | 31 | 27 | − | mmol/mmol |
| OSM U | − | − | 130 | − | 132 | 236 | 350 | mOsm/L | |
| Renal ultrasound | − | Nephrocalcinosis | − | − | − | − | − | Nephrocalcinosis | |
| Cardiac ultrasound | − | Normal | Mild ventricular septal hypertrophy (IVSD 6.3 mm) | − | Ventricular hypertrophy (IVSD 7–8 mm) | − | − | Normal | |
| Therapies | − | None | None | None | Carvedilol Amlodipine | Carvedilol Amlodipine | Spironolactone Amiloride Amlodipine | Spironolactone Amiloride Amlodipine |
| Bartter | Gitelman | Liddle | AME | Geller | Congenital Adrenal Hyperplasia | Familial Hyperaldosteronism | |
|---|---|---|---|---|---|---|---|
| Potassium | Low | Low | Low | Low | Low | Low | Normal or Low |
| Sodium Bicarbonate | High | High | High | High | High | High | High |
| Renin | High | High | Low | Low | Low | Low | Low |
| Aldosterone | High | High | Low | Low | Low | Low | High |
| Hypertension | No | No | Yes | Yes | Yes | Yes | Yes |
| Cortisol/ACTH | − | − | − | N/N | − | Low/High | |
| Age of onset | Infancy | Childhood/Adulthood | Childhood/Adulthood | Every age | Adulthood | Infancy | Childhood/Adulthood |
| Transmission | AD-AR | AR | AD | AR | AD | AD | AD |
| Alteration | NaKCl cotransporter 2 Renal outer medullary K channel Cl channel Barttin Ca sensing receptor | NaCl cotransporter Cl channel Kb | Epithelial Na channel gain of function | Defect in11-beta-hydroxysteroid dehydrogenase type 2 | Gain of function mutation of mineralocorticoid receptor | Steroid synthesis defect with gain of function of mineralocorticoid receptor | Increased levels of 18-oxocortisol and 18-hydroxycortisol |
| Gene | SLC12A1 KCNJ1 CLCNKB BSND CASR | SLC12A3 CLCNKB | SCNN1B SCNN1G | HSD11B2 | NR3C2 | CYP21A2 | CYP11B1/CYP11B2 CLCN2 KCNJ5 CACNA1H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertulli, C.; Hureaux, M.; De Mutiis, C.; Pasini, A.; Bockenhauer, D.; Vargas-Poussou, R.; La Scola, C. A Rare Cause of Chronic Hypokalemia with Metabolic Alkalosis: Case Report and Differential Diagnosis. Children 2020, 7, 212. https://doi.org/10.3390/children7110212
Bertulli C, Hureaux M, De Mutiis C, Pasini A, Bockenhauer D, Vargas-Poussou R, La Scola C. A Rare Cause of Chronic Hypokalemia with Metabolic Alkalosis: Case Report and Differential Diagnosis. Children. 2020; 7(11):212. https://doi.org/10.3390/children7110212
Chicago/Turabian StyleBertulli, Cristina, Marguerite Hureaux, Chiara De Mutiis, Andrea Pasini, Detlef Bockenhauer, Rosa Vargas-Poussou, and Claudio La Scola. 2020. "A Rare Cause of Chronic Hypokalemia with Metabolic Alkalosis: Case Report and Differential Diagnosis" Children 7, no. 11: 212. https://doi.org/10.3390/children7110212
APA StyleBertulli, C., Hureaux, M., De Mutiis, C., Pasini, A., Bockenhauer, D., Vargas-Poussou, R., & La Scola, C. (2020). A Rare Cause of Chronic Hypokalemia with Metabolic Alkalosis: Case Report and Differential Diagnosis. Children, 7(11), 212. https://doi.org/10.3390/children7110212