Persistent Pulmonary Hypertension in the Newborn

Abstract
:1. Introduction
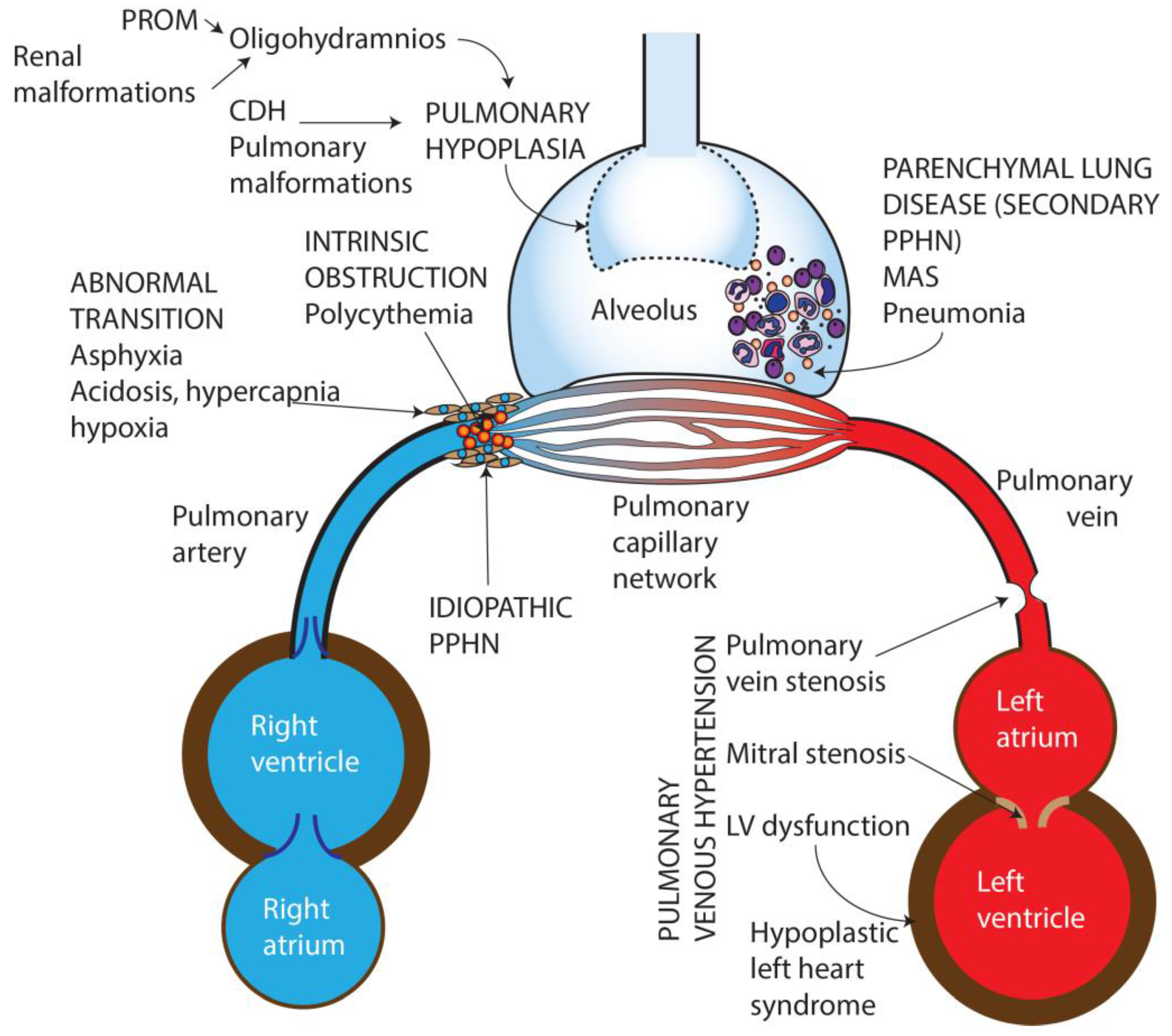
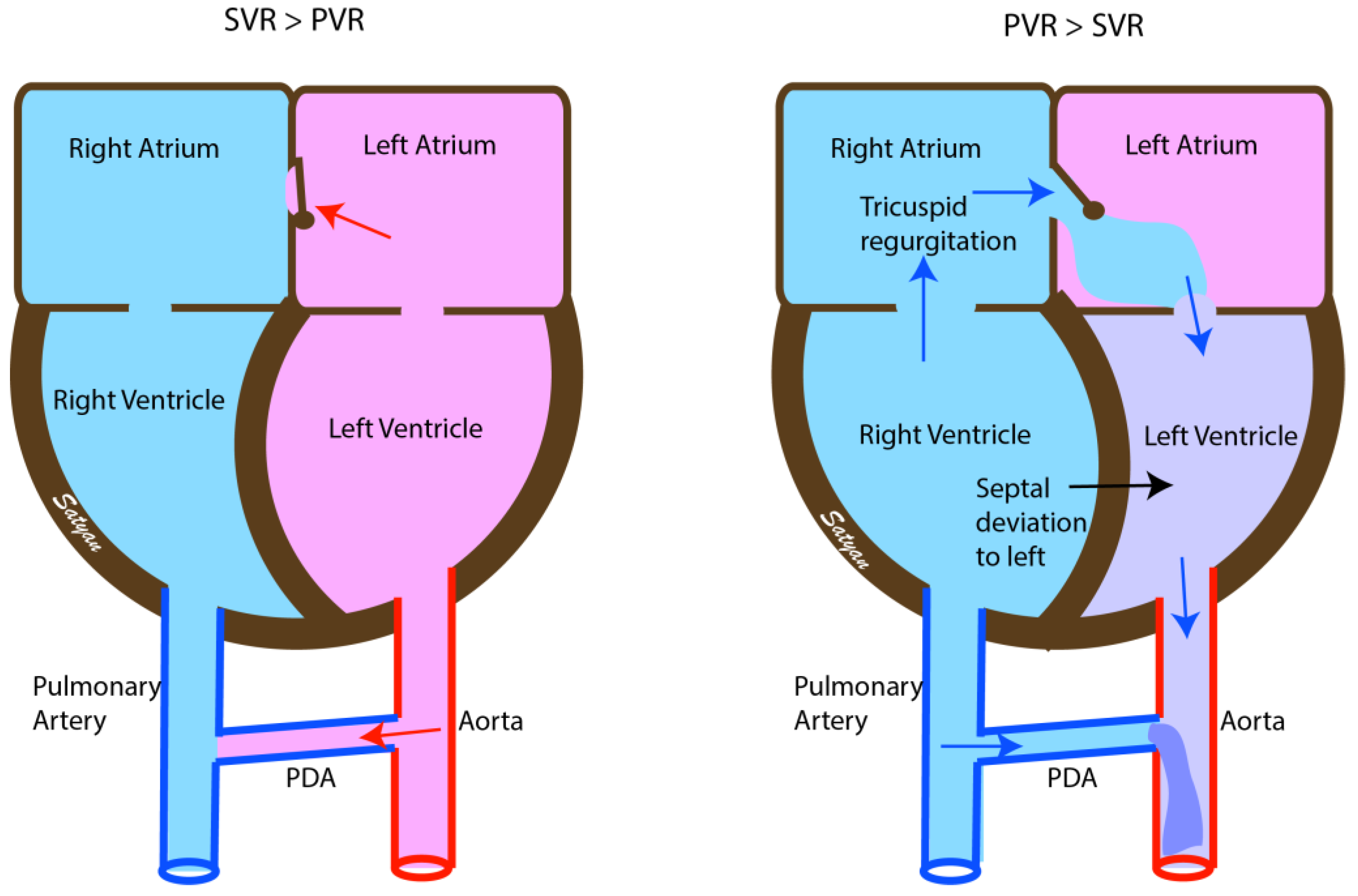
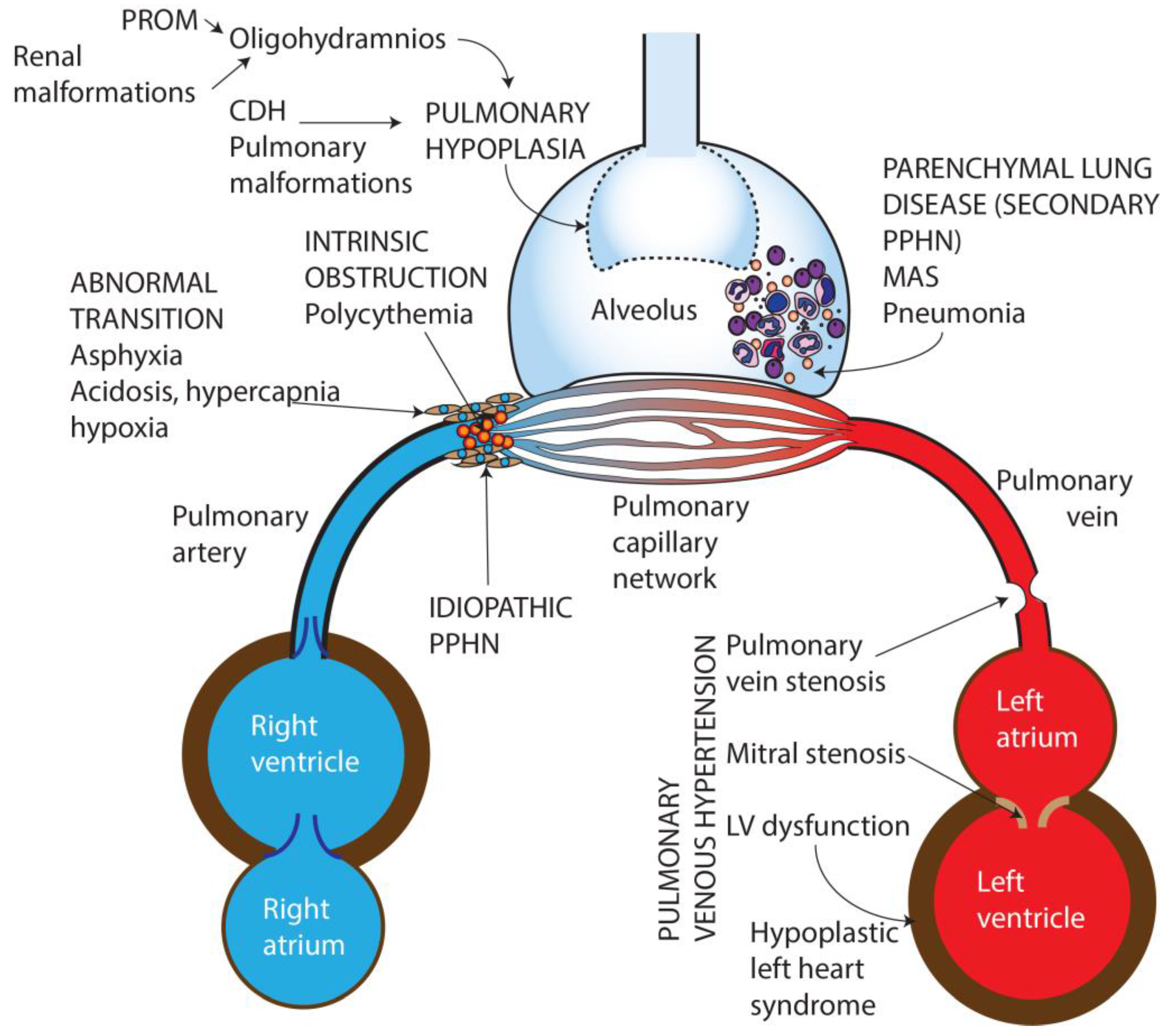
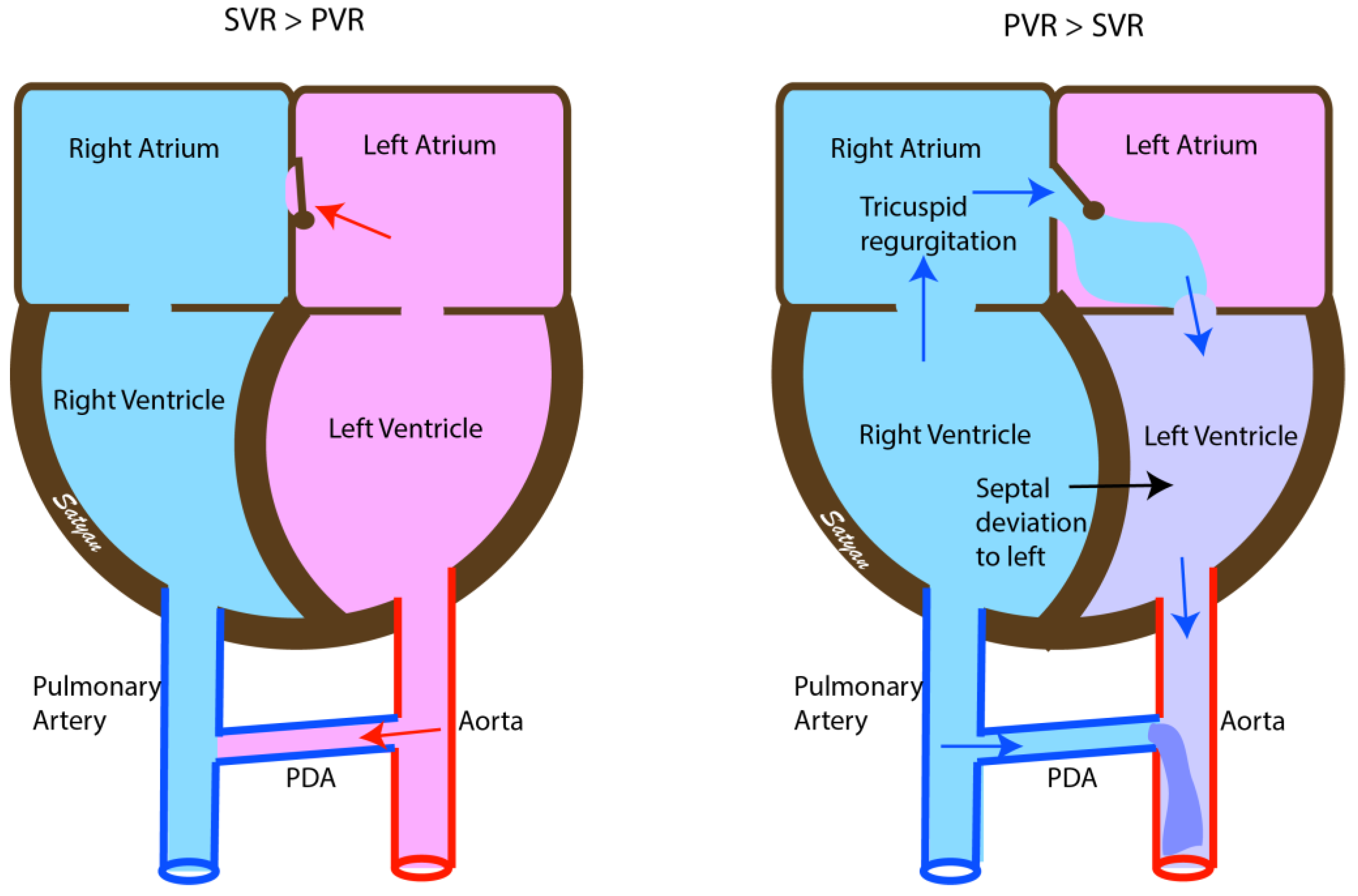
2. Pathophysiology of Persistent Pulmonary Hypertension
3. Etiology of Persistent Pulmonary Hypertension of the Newborn
- Idiopathic—No lung disease is present and Qp is decreased due to abnormal vascular remodeling leading to pulmonary vasoconstriction.
- Abnormal transition at birth—perinatal asphyxia, RDS, and transient tachypnea of newborn, (TTN) resulting in impaired pulmonary vasodilation at birth.
- Parenchymal disorders (also known as “secondary PPHN”)—such as due to meconium aspiration syndrome (MAS) and pneumonia.
- Abnormal lung development—pulmonary hypoplasia due to oligohydramnios secondary to renal dysfunction/anomalies or prolonged rupture of membranes, or congenital diaphragmatic hernia (CDH) and other pulmonary malformations.
- Intravascular obstruction due to hyperviscosity—polycythemia.
- Pulmonary hypertension (PH) in preterm infants in the initial phase of RDS [10].
- Pulmonary venous hypertension [11].
3.1. Idiopathic PPHN
3.2. Abnormal Pulmonary Transition
3.3. Parenchymal Lung Disease
3.4. Pulmonary Hypoplasia
3.5. Prematurity
3.6. Pulmonary Venous Hypertension
4. Clinical Presentation
4.1. Echocardiography
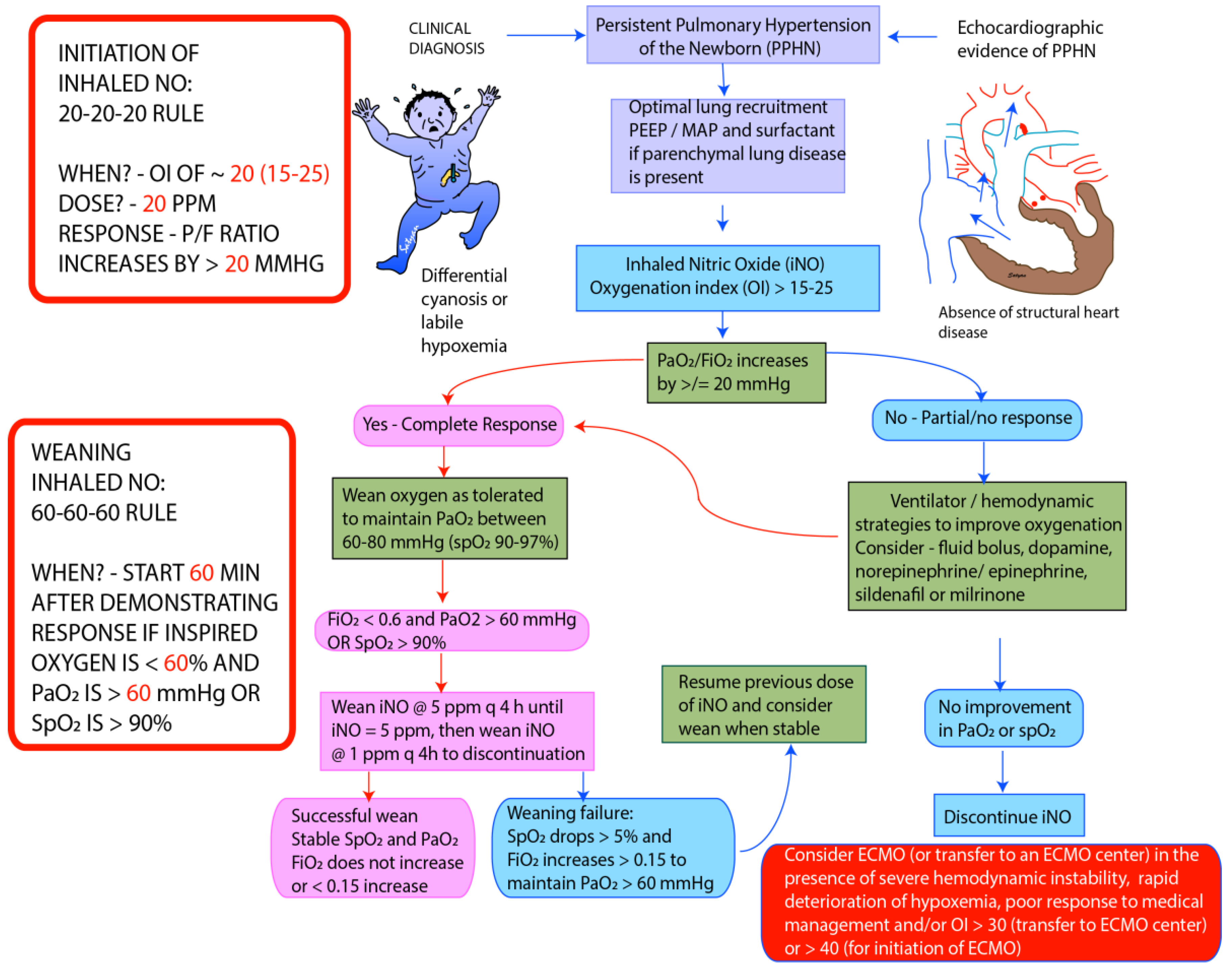
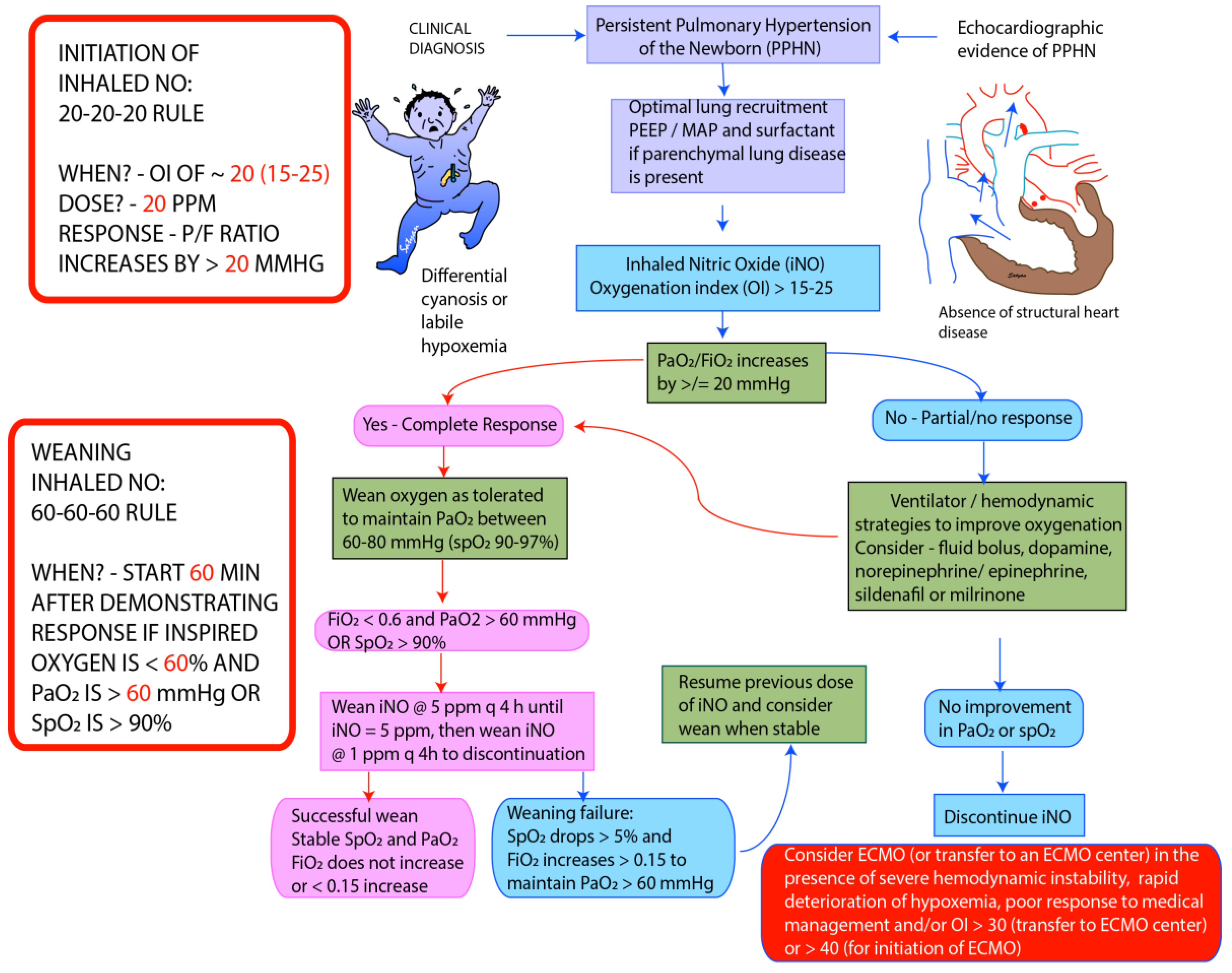
5. Assessment of Severity of HRF and Monitoring Response to Therapy
- Oxygenation index (OI) = FiO2 × mean airway pressure × 100/PaO2Severity of HRF based on OI:
- ○
- Mild ≤15
- ○
- Moderate 15 to ≤25
- ○
- Severe 25 to ≤40 and
- ○
- Very severe >40
- Alveolar—arterial oxygen gradient (A-a gradient or A-aDO2). This estimates the partial pressure gradient of oxygen from the alveolus to the aorta.A-aDO2 = [FiO2 × (Barometric pressure − water vapor pressure) − PaCO2/R)] − PaO2.
- ○
- Where R is the respiratory quotient (R = 1, in an infant receiving exclusive intravenous dextrose and approximately 0.8 when on mixed diet).
- ○
- The normal A-aDO2 is 4–20 mm of Hg. A-aDO2 can be above 600 mm Hg in very severe cases of HRF. An online calculator is available at http://perinatology.com/calculators/A-a%20gradient.htm.
- P/F ratio is the ratio of the partial pressure of oxygen (in mm Hg) in the arterial blood to the fractional inspired oxygen concentration. P/F ratio = PaO2/ FiO2Severity assessment based on P/F ratio:
- ○
- Mild >200 to ≤300.
- ○
- Moderate >100 to ≤200 and
- ○
- Severe ≤100 mm Hg.
- Note: For calculation of OI and P/F ratios, it is preferable to use preductal blood gases [25]. Preductal PaO2 accurately predicts oxygen delivery to vital organs such as the brain and heart and is not altered by right to left shunting at the PDA. However, many patients with PPHN have umbilical arterial access (postductal blood gases) resulting in lower PaO2 and higher OI and lower P/F ratio compared to preductal evaluation.
- Oxygen saturation index (OSI)—All of the above indices require arterial blood gas for evaluation and hence the need for arterial access. OSI is a noninvasive index of gas exchange and is calculated as follows:
- ○
- OSI = Mean airway pressure × FiO2 × 100/Preductal SpO2.
- ○
- OSI has been shown to correlate well in infants with HRF, OI ≈ 2 × OSI [26].
6. Management
6.1. Supportive Therapies
6.2. Lung Recruitment
6.3. Oxygenation
6.4. Surfactant Replacement Therapy
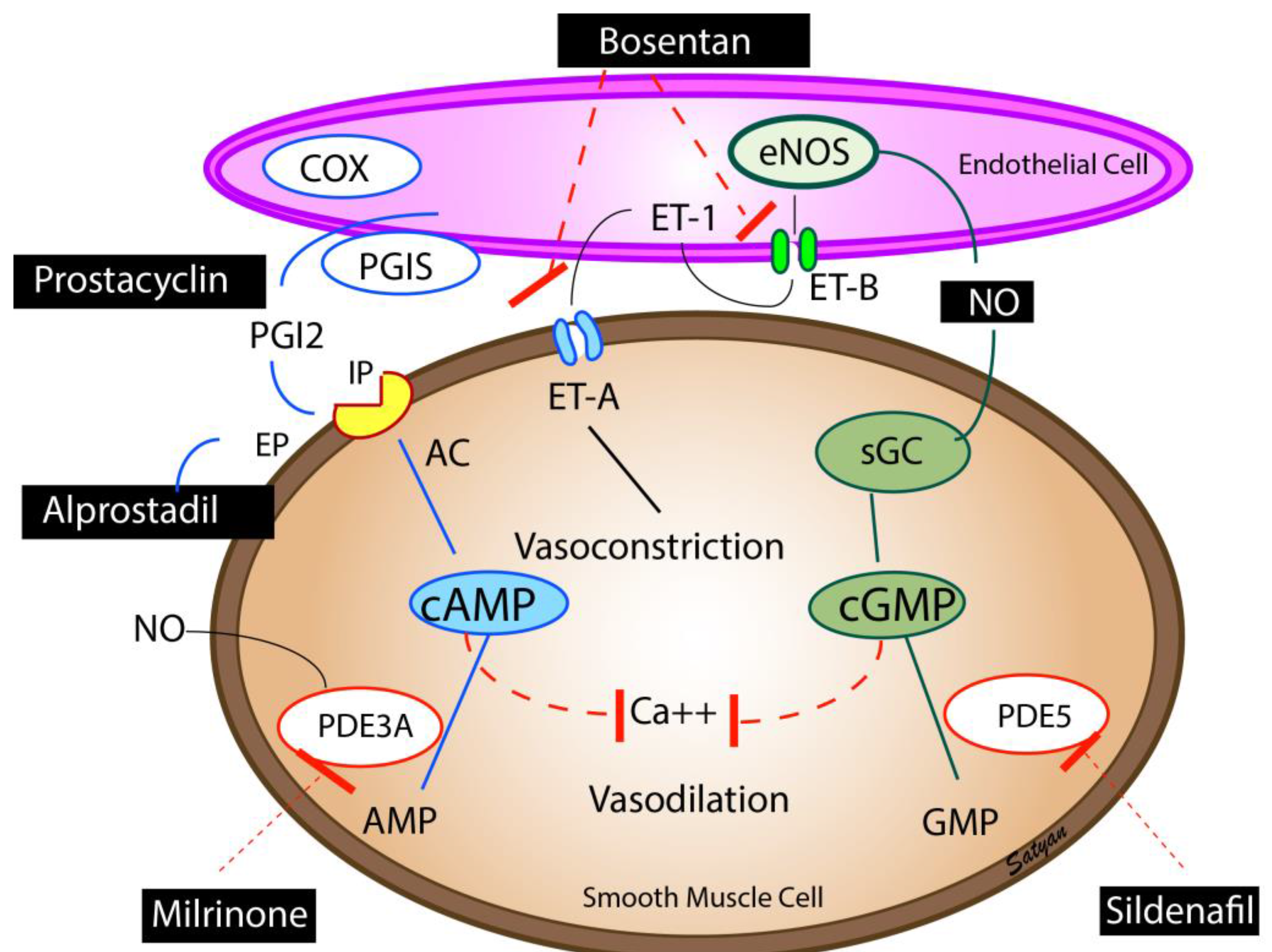
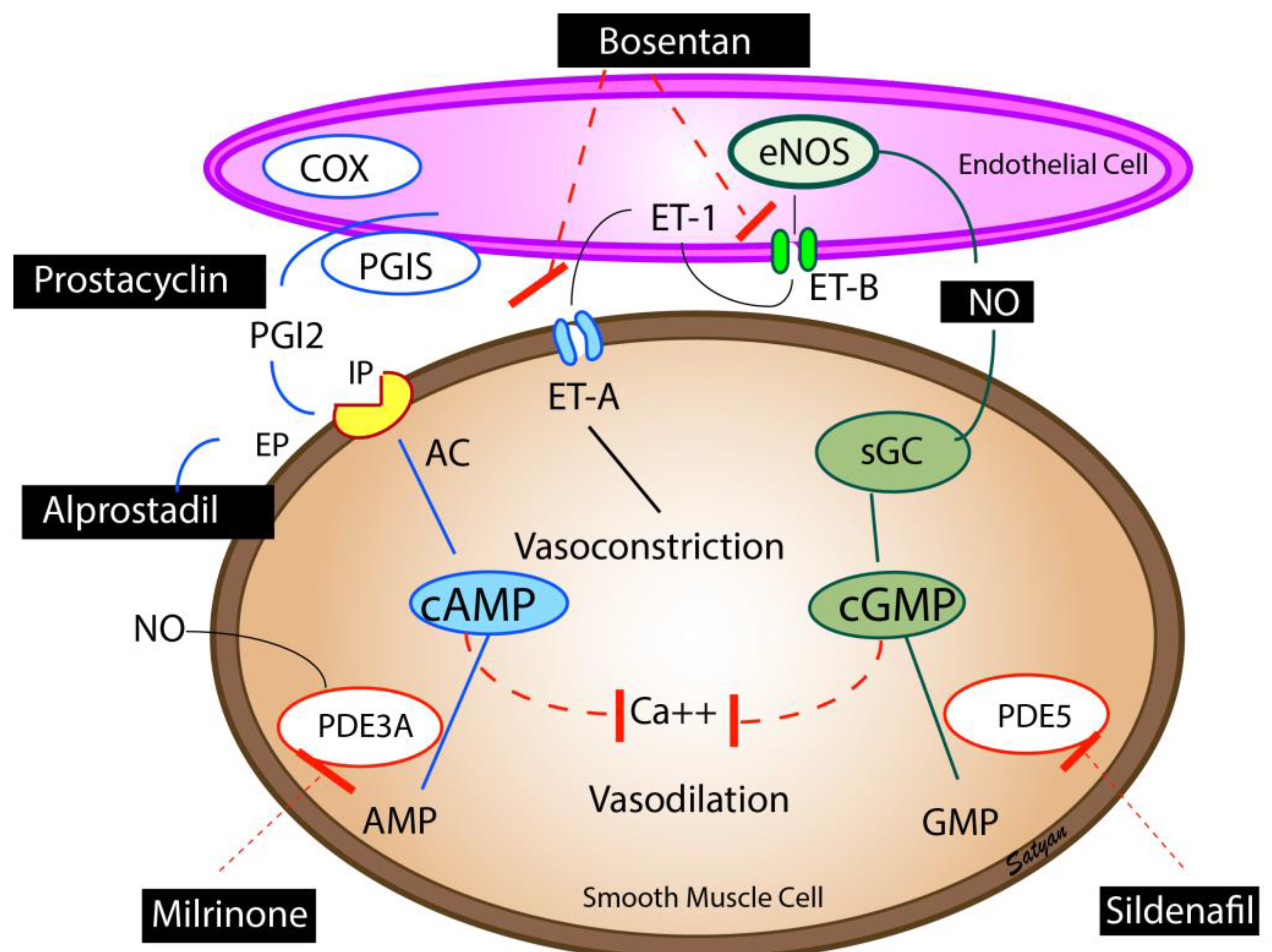
6.5. Inhaled Nitric Oxide (iNO)
6.6. Prostaglandins
6.7. Phosphodiesterase Inhibitors
6.8. Inotropes
- (1)
- In the presence of systemic hypotension without cardiac dysfunction, the agents of choice are dopamine, norepinephrine and vasopressin (pressor support).
- (2)
- When systemic hypotension is associated with cardiac dysfunction, epinephrine or a combination of dopamine/vasopressin and milrinone are the agents of choice.
- (3)
- In the presence of stable systemic blood pressure and cardiac dysfunction, milrinone is the agent of choice.
- (4)
- Studies in the lamb model of PPHN have shown increase in pulmonary arterial pressure and decreased pulmonary blood flow following initiation of dopamine. In control lambs with normal pulmonary vasculature, dopamine increases the systemic blood pressure with relatively small increase in pulmonary arterial pressure. In contrast, lambs with remodeled pulmonary vasculature and PPHN induced by antenatal ductal ligation, dopamine, especially at doses >10 µg/kg/min, results in a greater increase in pulmonary arterial pressure [9].
- (5)
- The use of dobutamine is often associated with a fall in systemic blood pressure resulting in exacerbation of right to left shunting and systemic desaturation. Dobutamine causes increase in myocardial oxygen requirement which may worsen myocardial dysfunction in PPHN.
6.9. Sedation/Paralysis
6.10. Nutrition
6.11. Acid–Base Balance
6.12. Extracorporeal Membrane Oxygenation (ECMO)
7. Follow-Up
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Kiserud, T. Physiology of the fetal circulation. Semin. Fetal Neonat. Med. 2005, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Steinhorn, R.H. Pulmonary vascular biology during neonatal transition. Clin. Perinatol. 1999, 26, 601–619. [Google Scholar] [PubMed]
- Nair, J.; Lakshminrusimha, S. Update on pphn: Mechanisms and treatment. Semin. Perinatol. 2014, 38, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Walsh-Sukys, M.C.; Tyson, J.E.; Wright, L.L.; Bauer, C.R.; Korones, S.B.; Stevenson, D.K.; Verter, J.; Stoll, B.J.; Lemons, J.A.; Papile, L.A.; et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: Practice variation and outcomes. Pediatrics 2000, 105, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Steurer, M.A.; Jelliffe-Pawlowski, L.L.; Baer, R.J.; Partridge, J.C.; Rogers, E.E.; Keller, R.L. Persistent pulmonary hypertension of the newborn in late preterm and term infants in California. Pediatrics 2017, 139, e20161165. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Saugstad, O.D. The fetal circulation, pathophysiology of hypoxemic respiratory failure and pulmonary hypertension in neonates, and the role of oxygen therapy. J. Perinatol. 2016, 36 (Suppl. 2), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Prsa, M.; Sun, L.; van Amerom, J.; Yoo, S.J.; Grosse-Wortmann, L.; Jaeggi, E.; Macgowan, C.; Seed, M. Reference ranges of blood flow in the major vessels of the normal human fetal circulation at term by phase-contrast magnetic resonance imaging. Circ. Cardiovasc. Imaging 2014, 7, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Rasanen, J.; Wood, D.C.; Weiner, S.; Ludomirski, A.; Huhta, J.C. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation 1996, 94, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S. The pulmonary circulation in neonatal respiratory failure. Clin. Perinatol. 2012, 39, 655–683. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, P.; Kozielski, R.; Kumar, V.H.; Rawat, M.; Manja, V.; Ma, C.; Lakshminrusimha, S. Early use of inhaled nitric oxide in preterm infants: Is there a rationale for selective approach? Am. J. Perinatol. 2016, 34, 428–440. [Google Scholar] [PubMed]
- Swier, N.L.; Richards, B.; Cua, C.L.; Lynch, S.K.; Yin, H.; Nelin, L.D.; Smith, C.V.; Backes, C.H. Pulmonary vein stenosis in neonates with severe bronchopulmonary dysplasia. Am. J. Perinatol. 2016, 33, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Alano, M.A.; Ngougmna, E.; Ostrea, E.M., Jr.; Konduri, G.G. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics 2001, 107, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.D.; Hernandez-Diaz, S.; Van Marter, L.J.; Werler, M.M.; Louik, C.; Jones, K.L.; Mitchell, A.A. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N. Engl. J. Med. 2006, 354, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Van Marter, L.J.; Hernandez-Diaz, S.; Werler, M.M.; Louik, C.; Mitchell, A.A. Nonsteroidal antiinflammatory drugs in late pregnancy and persistent pulmonary hypertension of the newborn. Pediatrics 2013, 131, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Pearson, D.L.; Dawling, S.; Walsh, W.F.; Haines, J.L.; Christman, B.W.; Bazyk, A.; Scott, N.; Summar, M.L. Neonatal pulmonary hypertension--urea-cycle intermediates, nitric oxide production, and carbamoyl-phosphate synthetase function. N. Engl. J. Med. 2001, 344, 1832–1838. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, P.K.; Rawat, M.; Madappa, R.; Rothstein, D.H.; Lakshminrusimha, S. Congenital diaphragmatic hernia–A review. Matern. Health Neonatol. Perinatol. 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, J.P.; Ivy, D.D.; Abman, S.H. Pulmonary vasodilator therapy in congenital diaphragmatic hernia: Acute, late, and chronic pulmonary hypertension. Semin. Perinatol. 2005, 29, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.H.; Hutchison, A.A.; Lakshminrusimha, S.; Morin, F.C., 3rd; Wynn, R.J.; Ryan, R.M. Characteristics of pulmonary hypertension in preterm neonates. J. Perinatol. 2007, 27, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Chock, V.Y.; Van Meurs, K.P.; Hintz, S.R.; Ehrenkranz, R.A.; Lemons, J.A.; Kendrick, D.E.; Stevenson, D.K.; Network, N.N.R. Inhaled nitric oxide for preterm premature rupture of membranes, oligohydramnios, and pulmonary hypoplasia. Am. J. Perinatol. 2009, 26, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, R.G.; Tyson, R.W.; Ivy, D.D.; Abman, S.H.; Kinsella, J.P. Congenital pulmonary venous stenosis presenting as persistent pulmonary hypertension of the newborn. Pediatr. Pulmonol. 1999, 28, 301–306. [Google Scholar] [CrossRef]
- Lakshminrusimha, S.; Wynn, R.J.; Youssfi, M.; Pabalan, M.J.; Bommaraju, M.; Kirmani, K.; Carrion, V. Use of ct angiography in the diagnosis of total anomalous venous return. J. Perinatol. 2009, 29, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.S.; Havalad, V.; Aponte-Patel, L.; Ravindranath, T.M.; October, T.W.; Starc, T.J.; Smerling, A.J. Nitric oxide-associated pulmonary edema in children with pulmonary venous hypertension. Pediatr. Cardiol. 2012, 34, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Konduri, G.G.; Steinhorn, R.H. Considerations in the management of hypoxemic respiratory failure and persistent pulmonary hypertension in term and late preterm neonates. J. Perinatol. 2016, 36 (Suppl. 2), S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Giesinger, R.E.; McNamara, P.J. Hemodynamic instability in the critically ill neonate: An approach to cardiovascular support based on disease pathophysiology. Semin. Perinatol. 2016, 40, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Gien, J.; Kinsella, J.P. Differences in preductal and postductal arterial blood gas measurements in infants with severe congenital diaphragmatic hernia. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101, F314–F318. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Chandrasekharan, P.K.; Williams, A.; Gugino, S.; Koenigsknecht, C.; Swartz, D.; Ma, C.X.; Mathew, B.; Nair, J.; Lakshminrusimha, S. Oxygen saturation index and severity of hypoxic respiratory failure. Neonatology 2015, 107, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, A.M.; Yuan, S. Response of the pulmonary vasculature to hypoxia and h+ ion concentration changes. J. Clin. Invest. 1966, 45, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Russell, J.A.; Steinhorn, R.H.; Swartz, D.D.; Ryan, R.M.; Gugino, S.F.; Wynn, K.A.; Kumar, V.H.; Mathew, B.; Kirmani, K.; et al. Pulmonary hemodynamics in neonatal lambs resuscitated with 21%, 50%, and 100% oxygen. Pediatr. Res. 2007, 62, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Swartz, D.D.; Gugino, S.F.; Ma, C.X.; Wynn, K.A.; Ryan, R.M.; Russell, J.A.; Steinhorn, R.H. Oxygen concentration and pulmonary hemodynamics in newborn lambs with pulmonary hypertension. Pediatr. Res. 2009, 66, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Morin, F.C., 3rd; Steinhorn, R.H.; Gugino, S.F.; Ryan, R.M.; Kumar, V.H.; Russell, J.A. Ovine bronchial-derived relaxing factor: Changes with development and hyperoxic ventilation. J. Appl. Physiol. 2006, 101, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Russell, J.A.; Steinhorn, R.H.; Ryan, R.M.; Gugino, S.F.; Morin, F.C., 3rd; Swartz, D.D.; Kumar, V.H. Pulmonary arterial contractility in neonatal lambs increases with 100% oxygen resuscitation. Pediatr. Res. 2006, 59, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Russell, J.A.; Wedgwood, S.; Gugino, S.F.; Kazzaz, J.A.; Davis, J.M.; Steinhorn, R.H. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Steinhorn, R.H.; Wedgwood, S.; Savorgnan, F.; Nair, J.; Mathew, B.; Gugino, S.F.; Russell, J.A.; Swartz, D.D. Pulmonary hemodynamics and vascular reactivity in asphyxiated term lambs resuscitated with 21 and 100% oxygen. J. Appl. Physiol. 2011, 111, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Konduri, G.G.; Sokol, G.M.; Van Meurs, K.P.; Singer, J.; Ambalavanan, N.; Lee, T.; Solimano, A. Impact of early surfactant and inhaled nitric oxide therapies on outcomes in term/late preterm neonates with moderate hypoxic respiratory failure. J. Perinatol. 2013, 33, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Konduri, G.G. New approaches for persistent pulmonary hypertension of newborn. Clin. Perinatol. 2004, 31, 591–611. [Google Scholar] [CrossRef] [PubMed]
- Thelitz, S.; Bekker, J.M.; Ovadia, B.; Stuart, R.B.; Johengen, M.J.; Black, S.M.; Fineman, J.R. Inhaled nitric oxide decreases pulmonary soluble guanylate cyclase protein levels in 1-month-old lambs. J. Thorac. Cardiovasc. Surg. 2004, 127, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Berkelhamer, S.K.; Lakshminrusimha, S. Persistent pulmonary hypertension of the newborn. Matern. Health Neonatol. Perinatol. 2015, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Mathew, B.; Leach, C.L. Pharmacologic strategies in neonatal pulmonary hypertension other than nitric oxide. Semin. Perinatol. 2016, 40, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.K.; Porta, N.F.; Goodman, D.M.; Carroll, C.L.; Steinhorn, R.H. Inhaled prostacyclin for term infants with persistent pulmonary hypertension refractory to inhaled nitric oxide. J. Pediatr. 2002, 141, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Wardle, A.J.; Wardle, R.; Luyt, K.; Tulloh, R. The utility of sildenafil in pulmonary hypertension: A focus on bronchopulmonary dysplasia. Arch. Dis. Child. 2013, 98, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lee, K.J.; Perez, M.; Schriewer, J.M.; Wedgwood, S.; Lakshminrusimha, S.; Smith, C.L.; Steinhorn, R.H.; Schumacker, P.T. Brief hyperoxia increases mitochondrial oxidation and increases phosphodiesterase 5 activity in fetal pulmonary artery smooth muscle cells. Antioxid. Redox Signal. 2012, 17, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Steinhorn, R.H.; Kinsella, J.P.; Pierce, C.; Butrous, G.; Dilleen, M.; Oakes, M.; Wessel, D.L. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J. Pediatr. 2009, 155, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Keszler, M. Persistent pulmonary hypertension of the newborn. Neoreviews 2015, 16, e680–e692. [Google Scholar] [CrossRef] [PubMed]
- Hendricks-Munoz, K.D.; Walton, J.P. Hearing loss in infants with persistent fetal circulation. Pediatrics 1988, 81, 650–656. [Google Scholar] [PubMed]
- Aschner, J.L.; Gien, J.; Ambalavanan, N.; Kinsella, J.P.; Konduri, G.G.; Lakshminrusimha, S.; Saugstad, O.D.; Steinhorn, R.H. Challenges, priorities and novel therapies for hypoxemic respiratory failure and pulmonary hypertension in the neonate. J. Perinatol. 2016, 36, S32–S36. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lung Disease without PPHN | Cyanotic Congenital Heart Disease | PPHN | |
|---|---|---|---|
| History | Fetal distress, PROM, chorioamnionitis | Antenatal diagnosis | Often negative other than in secondary PPHN |
| Respiratory distress | Present | Usually absent | Often present |
| Oxygen saturation on pulse oximetry | Improves with supplemental oxygen | Fixed low saturations Minimal response to supplemental oxygen | Labile saturations. Differential cyanosis |
| Hyperoxia test * | PaO2 often > 150 mm Hg | PaO2 often < 100 mm Hg | PaO2 often > 100 mm Hg |
| PaCO2 | Elevated | Normal/low | Often elevated (except in idiopathic PPHN) |
| Hyperoxia-Hyperventilation * | PaO2 > 150 mm Hg | PaO2 often < 100 mm Hg | PaO2 improves with hyperventilation |
| Chest X ray | Abnormal | Abnormalities of cardiac silhouette and pulmonary vascularity | Decreased vascularity in idiopathic PPHN |
| Echocardiogram | Normal | Structural cardiac abnormalities | Structurally normal heart (see text for characteristic echo findings of PPHN) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, B.; Lakshminrusimha, S. Persistent Pulmonary Hypertension in the Newborn. Children 2017, 4, 63. https://doi.org/10.3390/children4080063
Mathew B, Lakshminrusimha S. Persistent Pulmonary Hypertension in the Newborn. Children. 2017; 4(8):63. https://doi.org/10.3390/children4080063
Chicago/Turabian StyleMathew, Bobby, and Satyan Lakshminrusimha. 2017. "Persistent Pulmonary Hypertension in the Newborn" Children 4, no. 8: 63. https://doi.org/10.3390/children4080063
APA StyleMathew, B., & Lakshminrusimha, S. (2017). Persistent Pulmonary Hypertension in the Newborn. Children, 4(8), 63. https://doi.org/10.3390/children4080063