
Pediatric-Onset Multiple Sclerosis (POMS) and Epilepsy: Exploring Etiological Complexity—Outcomes from a Single-Center Experience


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Concerning Seizures and Epilepsy
3.2. Concerning MS
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MS | Multiple Sclerosis |
| ILAE | International League Against Epilepsy |
| EDSS | Expanded Disability Status Scale |
| BBB | Blood–Brain Barrier |
| MRI | Magnetic Resonance Imaging |
| EEG | Electroencephalography |
| JASP | Jeffreys’s Amazing Statistics Program |
| AEDs | Anti-Epileptic Drugs |
| IGEs | Idiopathic Generalized Epilepsies |
| ICS | Isolated Clinical Syndrome |
| DMTs | Disease-Modifying Therapies |
| POMS | Pediatric-Onset Multiple Sclerosis |
| HLA | Human Leukocyte Antigen |
| GWAS | Genome-Wide Association Studies |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| ARR | Annualized Relapse Rate |
| HET | High-Efficacy Therapies |
Appendix A
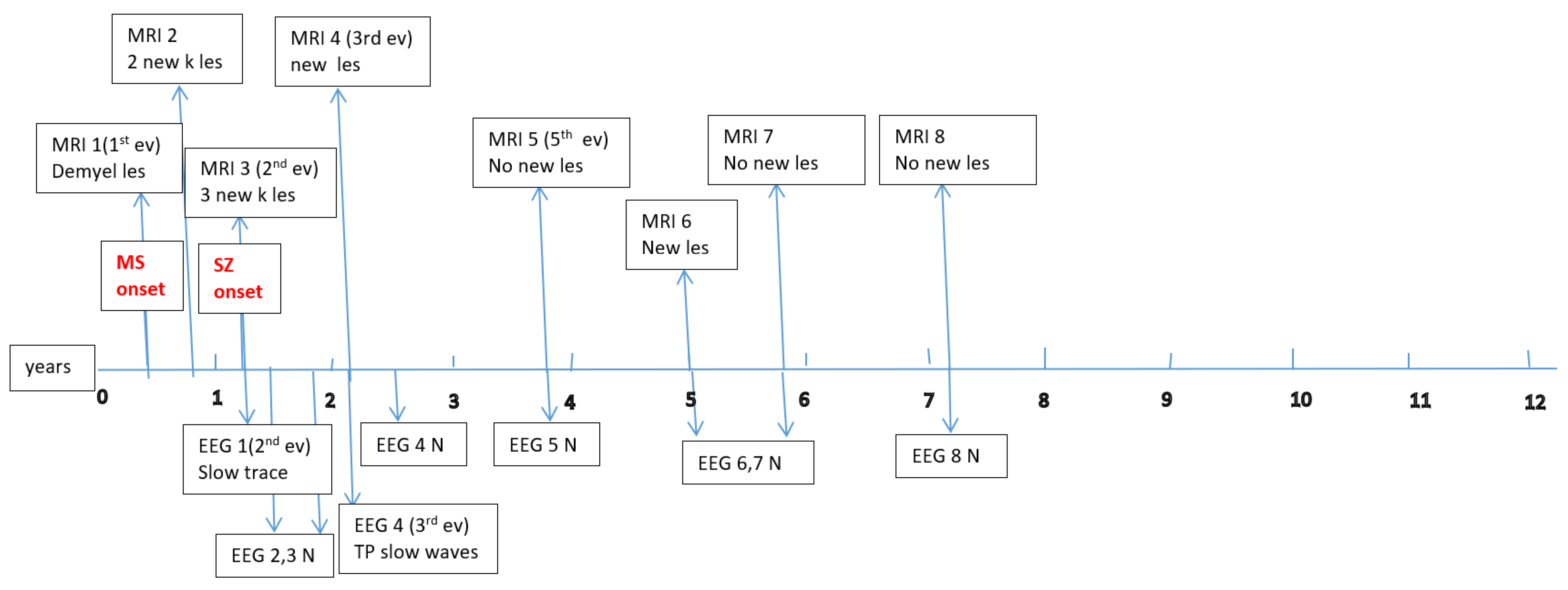
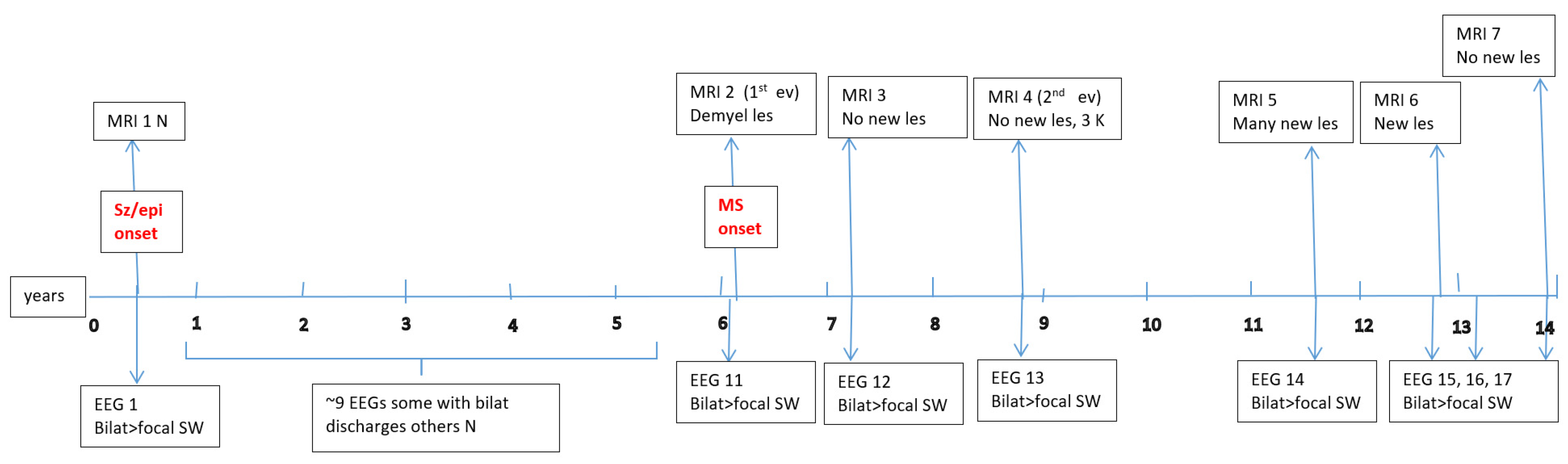




Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | MS with Epilepsy Group (6 Patients) | MS Without Epilepsy Group (114 Patients) | p Value (Where Appropriate) |
| Gender (F:M) | 3 F, 3 M | 76 F, 38 M | |
| Age at MS onset | 13.5 [5.25] | 15 [3] | 0.63 |
| Age at MS diagnosis | 14 [5] | 15 [2] | 0.55 |
| Duration MS dg–DMT onset | 1 [0.75] | 1 [0] | 0.057 |
| Duration MS monitoring during study | 3.5 [1] | 3 [1] | 0.034 |
| ARR before treatment (1 y *) | 1.5 [1] (6) | 1 [1] (114) | 0.68 |
| ARR before treatment (2 y *) | 1 [0.5] (2) | 0.5 [0.5] (41) | 0.77 |
| ARR before treatment (3 y *) | 0.83 [0.5] (2) | 0.66 [0.66] (24) | 0.79 |
| ARR after treatment initiation (1 y *) | 0 [0.75] (6) | 0 [0] (114) | 0.5 |
| ARR after treatment initiation (2 y *) | 0 [0.5] (5) | 0.25 [0.5] (58) | 0.82 |
| ARR after treatment initiation (3 y *) | 0.91 [0.41] (2) | 0.33 [0.83] (30) | 0.17 |
| Actual/last EDSS score | 1.25 [1.25] | 0 [0] | 0.003 # |
References
- Rayatpour, A.; Farhangi, S.; Verdaguer, E.; Olloquequi, J.; Ureña, J.; Auladell, C.; Javan, M. The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies. Pharmaceuticals 2021, 14, 1031. [Google Scholar] [CrossRef]
- Pozzilli, V.; Haggiag, S.; Di Filippo, M.; Capone, F.; Di Lazzaro, V.; Tortorella, C.; Gasperini, C.; Prosperini, L. Incidence and determinants of seizures in multiple sclerosis: A meta-analysis of randomised clinical trials. J. Neurol. Neurosurg. Psychiatry 2024, 95, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Durmus, H.; Kurtuncu, M.; Tuzun, E.; Pehlivan, M.; Akman-Demir, G.; Yapıcı, Z.; Eraksoy, M. Comparative clinical characteristics of early- and adult-onset multiple sclerosis patients with seizures. Acta Neurol. Belg. 2013, 113, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Koch, M.; Uyttenboogaart, M.; Polman, S.; De Keyser, J. Seizures in multiple sclerosis. Epilepsia 2008, 49, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Fisher, K.S.; Cuascut, F.X.; Rivera, V.M.; Hutton, G.J. Current Advances in Pediatric Onset Multiple Sclerosis. Biomedicines 2020, 8, 71. [Google Scholar] [CrossRef]
- Aaberg, K.M.; Gunnes, N.; Bakken, I.J.; Lund Søraas, C.; Berntsen, A.; Magnus, P.; Lossius, M.I.; Stoltenberg, C.; Chin, R.; Surén, P. Incidence and Prevalence of Childhood Epilepsy: A Nationwide Cohort Study. Pediatrics 2017, 139, e20163908. [Google Scholar] [CrossRef]
- Camfield, P.; Camfield, C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord. 2015, 17, 117–123. [Google Scholar] [CrossRef]
- Krupp, L.B.; Vieira, M.C.; Toledano, H.; Peneva, D.; Druyts, E.; Wu, P.; Boulos, F.C. A Review of Available Treatments, Clinical Evidence, and Guidelines for Diagnosis and Treatment of Pediatric Multiple Sclerosis in the United States. J. Child. Neurol. 2019, 34, 612–620. [Google Scholar] [CrossRef]
- Chitnis, T.; Aaen, G.; Belman, A.; Benson, L.; Gorman, M.; Goyal, M.S.; Graves, J.S.; Harris, Y.; Krupp, L.; Lotze, T.; et al. US Network of Paediatric Multiple Sclerosis Centers. Improved relapse recovery in paediatric compared to adult multiple sclerosis. Brain 2020, 143, 2733–2741. [Google Scholar] [CrossRef]
- Yan, K.; Balijepalli, C.; Desai, K.; Gullapalli, L.; Druyts, E. Epidemiology of pediatric multiple sclerosis: A systematic literature review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 44, 102260. [Google Scholar] [CrossRef] [PubMed]
- Sandesjö, F.; Tremlett, H.; Fink, K.; Marrie, R.A.; Zhu, F.; Wickström, R.; McKay, K.A. Incidence rate and prevalence of pediatric-onset multiple sclerosis in Sweden: A population-based register study. Eur. J. Neurol. 2024, 31, e16253. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Reider, N.; Cohen, J.; Trojano, M.; Sorensen, P.S.; Cutter, G.; Reingold, S.; Stuve, O. A systematic review of the incidence and prevalence of sleep disorders and seizure disorders in multiple sclerosis. Mult. Scler. 2015, 21, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.; Zelano, J. Epilepsy in multiple sclerosis: A nationwide population-based register study. Neurology 2017, 89, 2462–2468. [Google Scholar] [CrossRef]
- Grothe, M.; Ellenberger, D.; von Podewils, F.; Stahmann, A.; Rommer, P.S.; Zettl, U.K. Epilepsy as a predictor of disease progression in multiple sclerosis. Mult. Scler. 2022, 28, 942–949. [Google Scholar] [CrossRef]
- Grothe, M.; Ellenberger, D.; Rommer, P.S.; Stahmann, A.; Zettl, U.K. Epileptic seizures at multiple sclerosis onset and their role in disease progression. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231192826. [Google Scholar] [CrossRef]
- Chabas, D.; Strober, J.; Waubant, E. Pediatric multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2008, 8, 434–441. [Google Scholar] [CrossRef]
- Kavčič, A.; Hofmann, W.E. Seizures in Multiple Sclerosis are, above all, a Matter of Brain Viability. SciBase Neurol. 2024, 2, 1011. [Google Scholar] [CrossRef]
- Martínez-Lapiscina, E.H.; Ayuso, T.; Lacruz, F.; Gurtubay, I.G.; Soriano, G.; Otano, M.; Bujanda, M.; Bacaicoa, M.C. Corticojuxtacortical involvement increases risk of epileptic seizures in multiple sclerosis. Acta Neurol. Scand. 2013, 128, 24–31. [Google Scholar] [CrossRef]
- Calabrese, M.; Grossi, P.; Favaretto, A.; Romualdi, C.; Atzori, M.; Rinaldi, F.; Perini, P.; Saladini, M.; Gallo, P. Cortical pathology in multiple sclerosis patients with epilepsy: A 3 year longitudinal study. J. Neurol. Neurosurg. Psychiatry 2011, 83, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Uribe-San-Martín, R.; Ciampi-Díaz, E.; Suarez-Hernández, F.; Vásquez-Torres, M.; Godoy-Fernández, J.; Cárcamo-Rodríguez, C. Prevalence of epilepsy in a cohort of patients with multiple sclerosis. Seizure 2014, 23, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.J.; Barkhof, F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008, 7, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Progressive multiple sclerosis. Semin. Immunopathol. 2009, 31, 455–465. [Google Scholar] [CrossRef]
- Kuntz, S.; Wu, A.S.; Matheson, E.; Vyas, I.; Vyas, M.V. Association between multiple sclerosis and epilepsy: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2023, 69, 104421. [Google Scholar] [CrossRef] [PubMed]
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the Significance of Blood-Brain Barrier Disruption in Multiple Scle-rosis Pathology and Treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef]
- Mahamud, Z.; Burman, J.; Zelano, J. Risk of epilepsy after a single seizure in multiple sclerosis. Eur. J. Neurol. 2018, 25, 854–860. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, D.W. Focal Cortical Dysplasia and Epilepsy Surgery. J. Epilepsy Res. 2013, 3, 43–47. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Hirsch, E.; French, J.; Scheffer, I.E.; Bogacz, A.; Alsaadi, T.; Sperling, M.R.; Abdulla, F.; Zuberi, S.M.; Trinka, E.; Specchio, N.; et al. ILAE definition of the Idiopathic Generalized Epilepsy Syndromes: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1475–1499. [Google Scholar] [CrossRef]
- Riney, K.; Bogacz, A.; Somerville, E.; Hirsch, E.; Nabbout, R.; Scheffer, I.E.; Zuberi, S.M.; Alsaadi, T.; Jain, S.; French, J.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1443–1474. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Nabbout, R.; Scheffer, I.E.; Alsaadi, T.; Bogacz, A.; French, J.A.; Hirsch, E.; Jain, S.; Kaneko, S.; Riney, K.; et al. Methodology for classification and definition of epileptic syndromes with list of syndromes: Report of the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mannan, O.A.; Manchoon, C.; Rossor, T.; Southin, J.C.; Tur, C.; Brownlee, W.; Byrne, S.; Chitre, M.; Coles, A.; Forsyth, R.; et al. Use of Disease-Modifying Therapies in Pediatric Relapsing-Remitting Multiple Sclerosis in the United Kingdom. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1008. [Google Scholar] [CrossRef]
- Margoni, M.; Rinaldi, F.; Perini, P.; Gallo, P. Therapy of Pediatric-Onset Multiple Sclerosis: State of the Art, Challenges, and Opportunities. Front Neurol. 2021, 12, 676095. [Google Scholar] [CrossRef]
- Zuo, H.; Peng, L.; Li, W.; Wang, Y.; Du, X.; Zou, X.; Dong, Z.; Yi, L.; Yin, H.; Quan, F.; et al. Assessment of bidirectional relationships between multiple sclerosis and epilepsy: A two-sample Mendelian randomization study. Mult. Scler. Relat. Disord. 2024, 81, 105148. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.J.; Rodriguez, M. Seizures in patients with multiple sclerosis: Epidemiology, pathophysiology and management. CNS Drugs 2009, 23, 805–815. [Google Scholar] [CrossRef]
- Neuß, F.; Von Podewils, F.; Wang, Z.I.; Süße, M.; Zettl, U.K.; Grothe, M. Epileptic seizures in multiple sclerosis: Prevalence, competing causes and diagnostic accuracy. J. Neurol. 2021, 268, 1721–1727. [Google Scholar] [CrossRef]
- You, Y.; Zhao, Y.; Bai, H.; Liu, Z.; Meng, F.; Zhang, H.; Xu, R. Glatiramer acetate, an anti-demyelination drug, reduced rats’ epileptic seizures induced by pentylenetetrazol via protection of myelin sheath. Eur. J. Pharm. Sci. 2013, 49, 366–370. [Google Scholar] [CrossRef]
- Yazdi, A.; Baharvand, H.; Javan, M. Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015, 311, 34–44. [Google Scholar] [CrossRef]
- Saridas, F.; Mesut, G.; Ozpar, R.; Koc, E.R.; Hakyemez, B.; Bican Demir, A.; Turan, O.F. Coexistence of epilepsy or seizure and multiple sclerosis; review of the literature with a single center experience. Mult. Scler. Relat. Disord. 2024, 92, 105948. [Google Scholar] [CrossRef] [PubMed]
- Messina, S. Small GTPase RAS in multiple sclerosis—Exploring the role of RAS GTPase in the etiology of multiple sclerosis. Small GTPases 2020, 11, 312–319. [Google Scholar] [CrossRef]
- Goris, A.; Vandebergh, M.; McCauley, J.L.; Saarela, J.; Cotsapas, C. Genetics of multiple sclerosis: Lessons from polygenicity. Lancet Neurol. 2022, 21, 830–842. [Google Scholar] [CrossRef]
- Cotsapas, C.; Mitrovic, M.; Hafler, D. Multiple sclerosis. Handb. Clin. Neurol. 2018, 148, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Meijer, M.; Agirre, E.; Kabbe, M.; van Tuijn, C.A.; Heskol, A.; Zheng, C.; Mendanha Falcão, A.; Bartosovic, M.; Kirby, L.; Calini, D.; et al. Epigenomic priming of immune genes implicates oligodendroglia in multiple sclerosis susceptibility. Neuron 2022, 110, 1193–1210.e13. [Google Scholar] [CrossRef] [PubMed]
| Patient No. | Age at sz Onset | Time in Relation to MS Onset | Time Between sz Onset- and MS Onset | Time Between MS Onset and sz Onset | Seizure Aspects | Seizure Duration | Seizure Frequency | Epilepsy Diagnosis | AED Treatment | Seizure Control | Personal and Family History |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 9 y 7 mo | after | NA | 9 mo | Left focal motor SE, +LOC, at the end | 3–4 h | Only 1 SE, awake | NO, acute, probable autoimmune | LEV 6 mo | YES, ~7 y, no AED at present | Neg |
| P2 | 4 y 6 mo | before | 5 y 8 mo | NA | Type 1: Focal se generalized (head + eyes right deviation, oral automatism, gen hypertonia+/−clonic mov Type 2: myoclonic–atonic Type 3: absence of sz; rare GTC or focal sec gen | Type 1 <1′ Type 2 1–2” Type 3 5–10” absence of sz; 1–5′ GTC | Type 1 3/day, daily, awake and asleep Type 2 5–10/day, daily, awake Type 3 3–10/day, 2–3 times/week, awake; GTC/focal 2–3/year | YES, non-syndromic, probable genetic-autoimmune | Type 1 VPA, then * ACTH Type 2 VPA+CNZ, * ACTH Type 3 +CBZ, +LEV, –CBZ | Type 1 Yes, after ACTH Type 2 Yes, after ACTH Type 3 NO, 2–4 sz/day VPA+CNZ+LEV at present | 6 FS, between 2 and 4 y; mild cognitive decline; + some non-epileptic (reactive) events |
| P3 | 10 y 1 mo | before | 6 y 5 mo | NA | Type 1: Generalized Type 2: Focal+/− sec gen(tinnitus/diplopia, headache, left mouth dev, L limb dystonic posture, +/−gen hypertonia and clonic mov, headache +/− vomiting after sz) | Type 1 1–2′ Type 2 1–2′ | Type 1 1/5–6 mo, awake Type 21/1–2 mo, most of them awake | YES, non-syndromic, probable genetic–autoimmune | Type 1: VPA Type 2 + LEV + CBZ, + LTG, -CBZ | NO, sz freq stable in the last 3 y ** LEV+VPA+ LTG+CBZ at present | 1 parent with epilepsy (adolescence); 2 FS before 2 y |
| P4 | 13 y 1 mo | before | 1 y 8 mo | NA | GTCsz | 1–2′ | 2 GTC sz, 2y apart, from awake | YES, GIEs (JME vs. GTCA) | LEV | YES, 1 y, LEV at present | Neg |
| P5 | 6 y 5 mo | before | 4 y 9 mo | NA | Right focal motor sz (speech impairment, hypersalivation, R facio-brachial clonic mov, NO LOC) | <1′ | 2 focal sz, 2 mo apart, before sleep | YES, SeLECTS | VPA 2.5 y | YES,~12 y, no AED at present | 1 parent with MS (adult onset) |
| P6 | 15 y 3 mo | after | NA | 2 wks | Focal +/− sec gen sz 1st: nausea, dizziness, R head dev, RUL hypertonia + clonic mov, no LOC, 5 min aphasia 2nd: nausea, L head dev, gen hypertonia and clonic mov, left hemibody transient paresis | <1′, 1 asleep, 1 awake | 2 sz, 4 days apart | YES, non-syndromic, probable auto-immune | LEV 2y | YES, ~3 y, no AED at present | Neg |
| Chi-Squared Test | Value | df | p |
|---|---|---|---|
| Χ2 | 7.536 | 1 | 0.006 |
| Χ2 continuity correction | 5.003 | 1 | 0.025 |
| N | 120 |
| Log Odds Ratio | ||||
|---|---|---|---|---|
| 95% Confidence Interval | ||||
| Log Odds Ratio | Upper | p | ||
| Odds ratio | 2.124 | 0.364 | 3.884 | |
| Fisher’s exact test | 2.099 | 0.088 | 4.560 | 0.020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dică, A.D.; Craiu, D.; Iliescu, C.; Găină, M.-A.; Sandu, C.; Pomeran, C.; Bârcă, D.; Butoianu, N.; Burloiu, C.; Minciu, I.; et al. Pediatric-Onset Multiple Sclerosis (POMS) and Epilepsy: Exploring Etiological Complexity—Outcomes from a Single-Center Experience. Children 2025, 12, 631. https://doi.org/10.3390/children12050631
Dică AD, Craiu D, Iliescu C, Găină M-A, Sandu C, Pomeran C, Bârcă D, Butoianu N, Burloiu C, Minciu I, et al. Pediatric-Onset Multiple Sclerosis (POMS) and Epilepsy: Exploring Etiological Complexity—Outcomes from a Single-Center Experience. Children. 2025; 12(5):631. https://doi.org/10.3390/children12050631
Chicago/Turabian StyleDică, Alice Denisa, Dana Craiu, Catrinel Iliescu, Marcel-Alexandru Găină, Carmen Sandu, Cristina Pomeran, Diana Bârcă, Niculina Butoianu, Carmen Burloiu, Ioana Minciu, and et al. 2025. "Pediatric-Onset Multiple Sclerosis (POMS) and Epilepsy: Exploring Etiological Complexity—Outcomes from a Single-Center Experience" Children 12, no. 5: 631. https://doi.org/10.3390/children12050631
APA StyleDică, A. D., Craiu, D., Iliescu, C., Găină, M.-A., Sandu, C., Pomeran, C., Bârcă, D., Butoianu, N., Burloiu, C., Minciu, I., Găină, A.-M., Șurlică, D., Moțoescu, C., Tarța-Arsene, O., Cazacu, C., Badea, A., Niculae, A. Ș., & Ion, D. A. (2025). Pediatric-Onset Multiple Sclerosis (POMS) and Epilepsy: Exploring Etiological Complexity—Outcomes from a Single-Center Experience. Children, 12(5), 631. https://doi.org/10.3390/children12050631