
The Role of miRNA Expression in Congenital Heart Disease: Insights into the Mechanisms and Biomarker Potential

 ,
,  , , and
, , and 
Abstract
1. Introduction
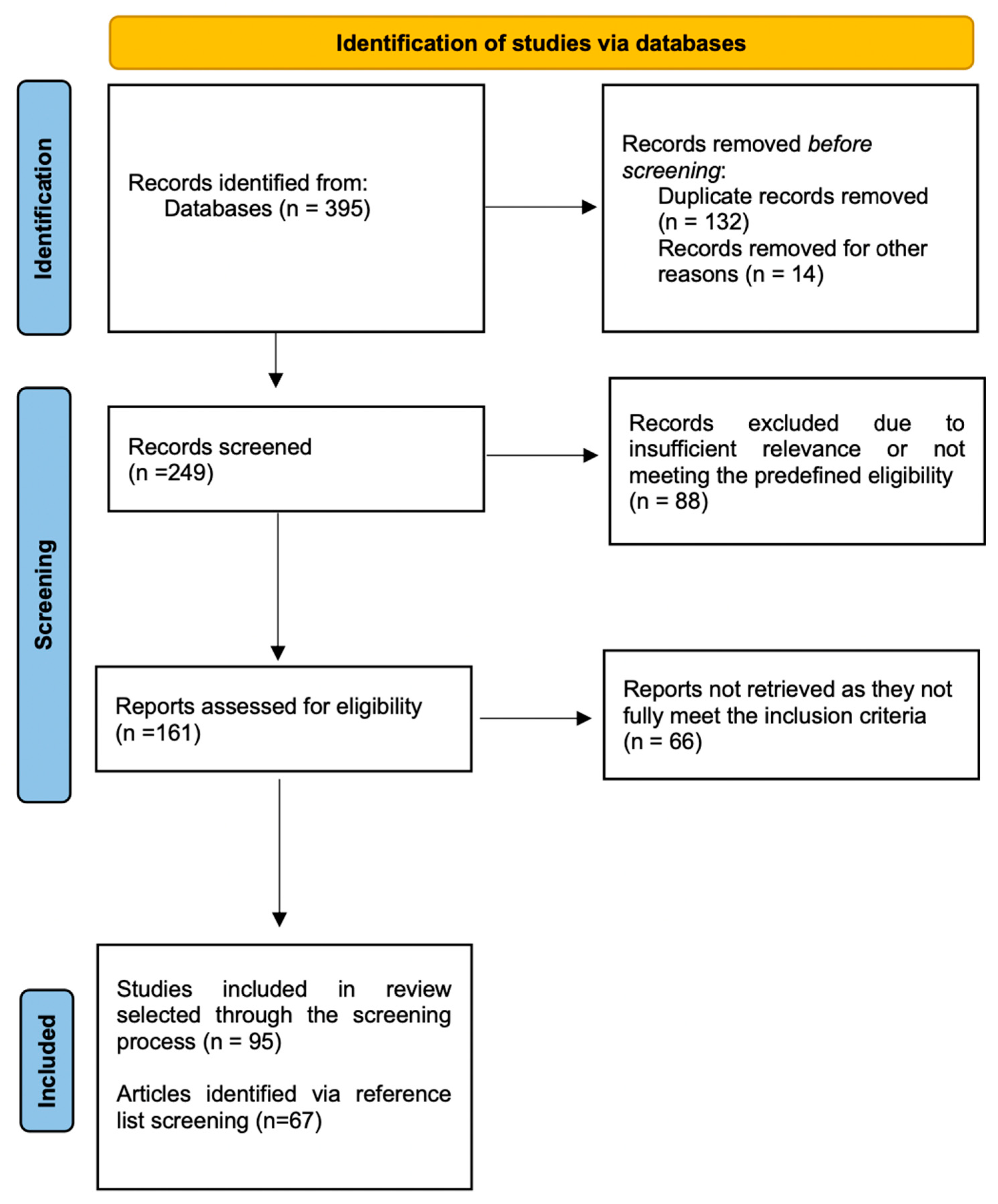
2. Methods
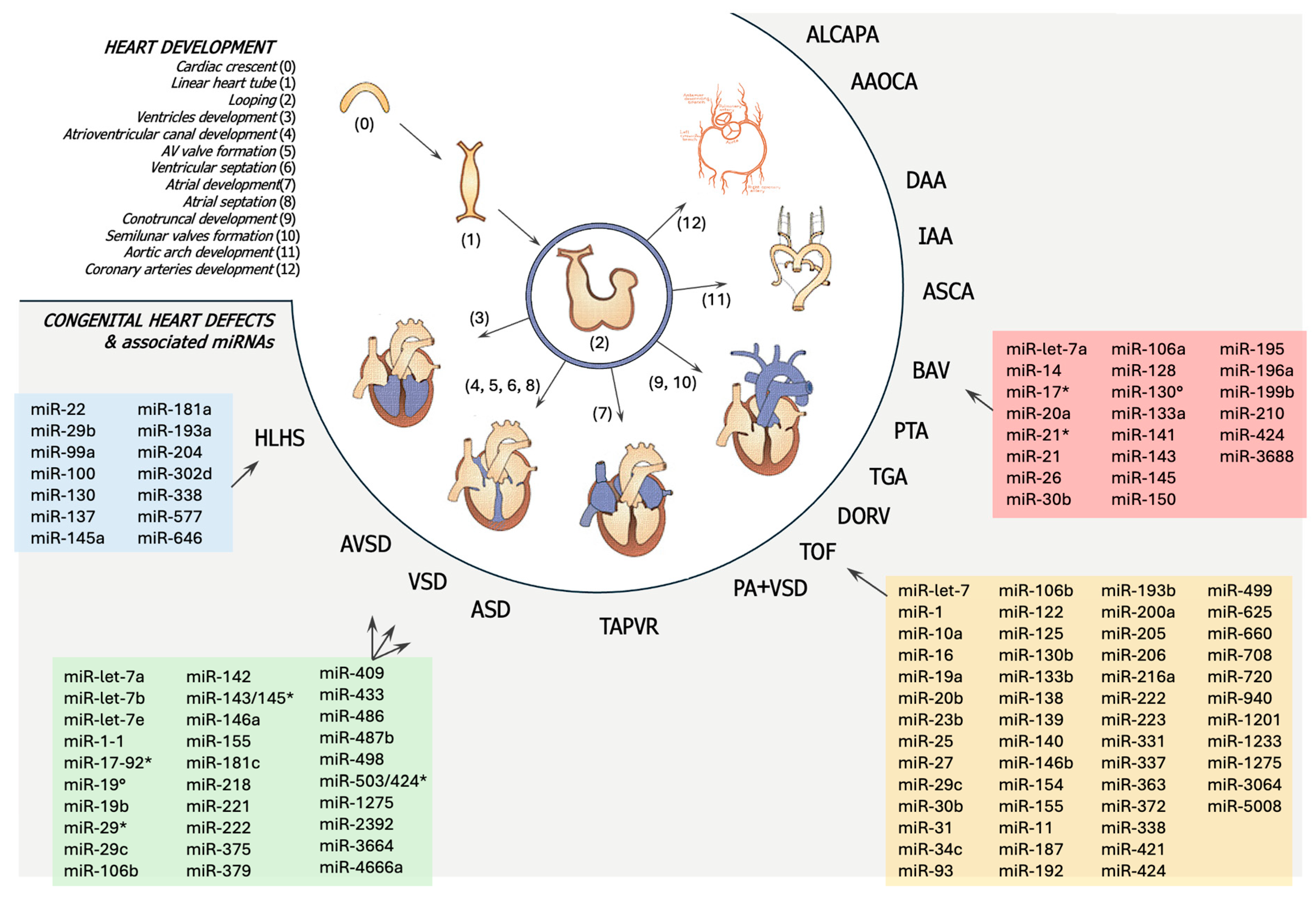
3. Heart Development and Congenital Heart Diseases
3.1. Heart Development
- (1)
- Early cardiogenesis (days 8–18 of embryonic development): This developmental phase starts with the establishment of the cardiac fields and the crescent-like cardiac primordium during gastrulation, and concludes with the emergence of two endocardial tubes encased by myocardial cells.
- (2)
- Morphogenetic phase (weeks 4–8 of embryonic development): This starts with the formation of the linear heart tube originating from the first heart field (FHF) and concludes with the assembly of all primordial components into the fully developed four-chambered heart, primarily derived from the second heart field (SHF).
- (3)
- Septation and remodeling of the heart chamber (~day 30 of embryonic development): During this phase, differential growth and remodeling occur, forming the valves and septa, thereby defining the distinct identities of the atrial and ventricular cavities.
- (4)
- Maturation and histodifferentiation (weeks 16–18 of embryonic development): this phase includes the maturation of the ventricular and atrial myocardium and the development of ventricular–arterial and atrio-ventricular valve systems, as well as the evolution of the conduction system and coronary vessels [7,8].
3.2. Pathogenesis of Congenital Heart Diseases
- Right-sided obstructions: pulmonary stenosis (PA), tricuspid atresia (TA), and pulmonary atresia (PA);
- Left-sided obstruction defects: including hypoplastic left heart syndrome (HLHS), mitral stenosis, aortic stenosis, aortic coarctation, and interrupted aortic arch;
- Septation defects: ineffective separation of the atria (atrial septal defects, ASDs), the ventricles (ventricular septal defects, VSDs), or both (atrioventricular septal defects, AVSDs);
- Conotruncal defects: transposition of the great arteries (TGA), double outlet right ventricle (DORV), Tetralogy of Fallot (TOF), and persistent truncus arteriosus (PTA).
3.2.1. Genetic Factors
3.2.2. Environmental Factors
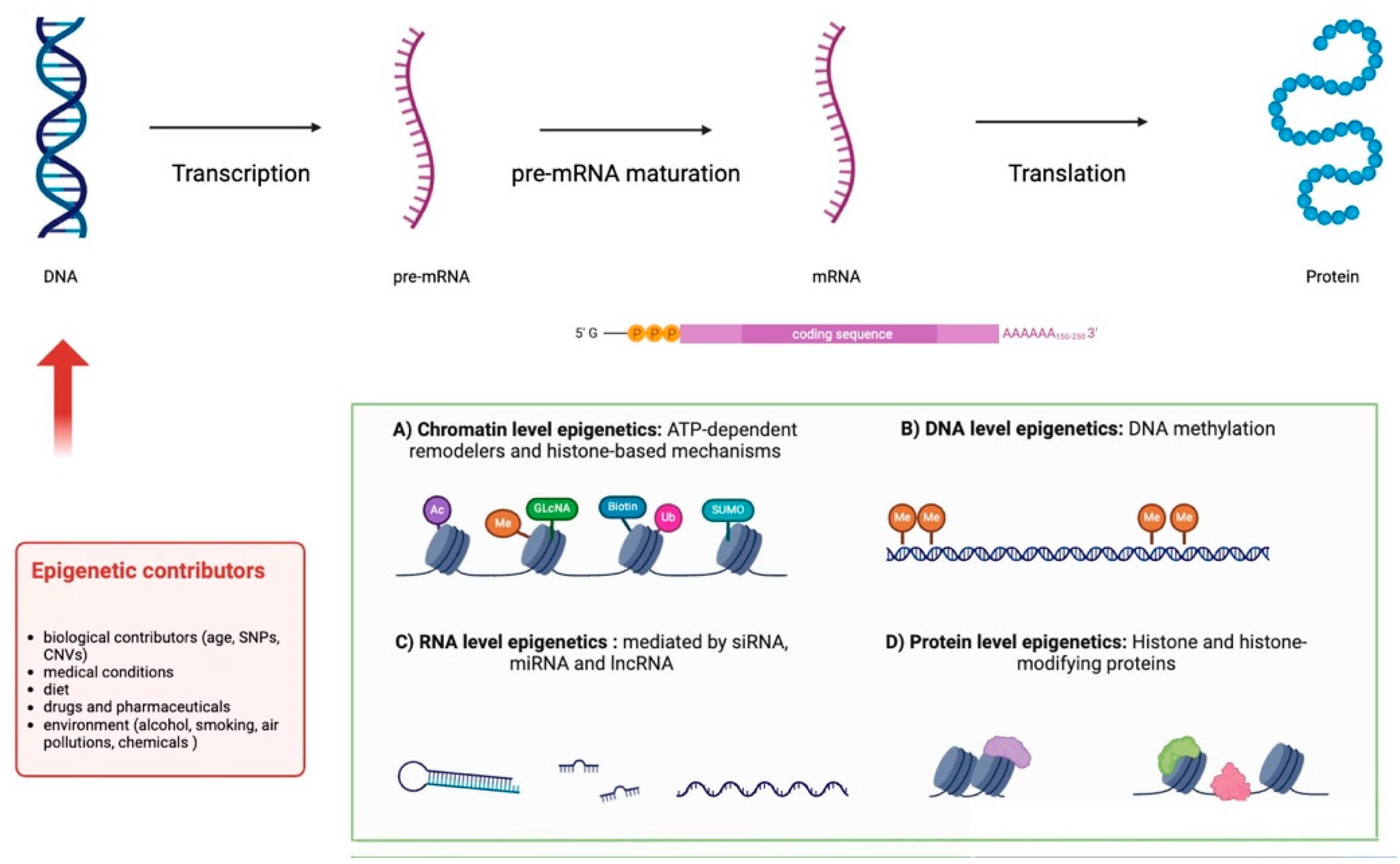
3.2.3. Gene Expression and Epigenetics
4. Modulation of mRNA Expression in Congenital Heart Diseases
4.1. Left Obstructive Heart Disease (LVOTO)
4.2. Bicuspid Aortic Valve
4.3. Septation Defects
4.3.1. Ventricular Septal Defects
4.3.2. Atrial Septal Defects
4.3.3. Atrioventricular Septal Defects
4.4. Tetralogy of Fallot and Other Conotruncal Anomalies
Tetralogy of Fallot
5. Future Perspectives on miRNAs as Biomarkers
6. Limitations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Linde, D.; Konings, E.E.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Carson, J.; Lo, C. Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, B.; Yang, P. Epigenetics in Congenital Heart Disease. J. Am. Heart Assoc. 2022, 11, e025163. [Google Scholar] [CrossRef]
- Linglart, L.; Bonnet, D. Epigenetics and Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 185. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Zubrzycki, M.; Schramm, R.; Costard-Jäckle, A.; Grohmann, J.; Gummert, J.F.; Zubrzycka, M. Cardiac Development and Factors Influencing the Development of Congenital Heart Defects (CHDs): Part I. Int. J. Mol. Sci. 2024, 25, 7117. [Google Scholar] [CrossRef]
- Sylva, M.; van den Hoff, M.J.; Moorman, A.F. Development of the human heart. Am. J. Med. Genet. Part A 2014, 164, 1347–1371. [Google Scholar] [CrossRef]
- Waldo, K.L.; Kumiski, D.H.; Wallis, K.T.; Stadt, H.A.; Hutson, M.R.; Platt, D.H.; Kirby, M.L. Conotruncal myocardium arises from a secondary heart field. Development 2001, 128, 3179–3188. [Google Scholar] [CrossRef]
- Buijtendijk, M.F.J.; Barnett, P.; van den Hoff, M.J.B. Development of the human heart. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 7–22. [Google Scholar] [CrossRef]
- Anderson, R.H.; Yanni, J.; Boyett, M.R.; Chandler, N.J.; Dobrzynski, H. The anatomy of the cardiac conduction system. Clin. Anat. 2009, 22, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Trojnarska, O.; Grajek, S.; Katarzyński, S.; Kramer, L. Predictors of mortality in adult patients with congenital heart disease. Cardiol. J. 2009, 16, 341–347. [Google Scholar] [PubMed]
- Nees, S.N.; Chung, W.K. Genetic Basis of Human Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a036749. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.W.; Chung, W.K.; Kaltman, J.R.; Miller, T.A. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J. Am. Heart Assoc. 2018, 7, e006906. [Google Scholar] [CrossRef]
- Wang, X.; Li, P.; Chen, S.; Xi, L.; Guo, Y.; Guo, A.; Sun, K. Influence of genes and the environment in familial congenital heart defects. Mol. Med. Rep. 2014, 9, 695–700. [Google Scholar] [CrossRef]
- Lien, C.L.; Wu, C.; Mercer, B.; Webb, R.; Richardson, J.A.; Olson, E.N. Control of early cardiac-specific transcription of Nkx2-5 by a GATA-dependent enhancer. Development 1999, 126, 75–84. [Google Scholar] [CrossRef]
- Tanaka, M.; Chen, Z.; Bartunkova, S.; Yamasaki, N.; Izumo, S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999, 126, 1269–1280. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Ma, Q.; Juraszek, A.L.; Moses, K.; Schwartz, R.J.; Izumo, S.; Pu, W.T. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J. Clin. Investig. 2005, 115, 1522–1531. [Google Scholar] [CrossRef]
- He, A.; Kong, S.W.; Ma, Q.; Pu, W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. USA 2011, 108, 5632–5637. [Google Scholar] [CrossRef]
- Campione, M.; Franco, D. Current Perspectives in Cardiac Laterality. J. Cardiovasc. Dev. Dis. 2016, 3, 34. [Google Scholar] [CrossRef]
- Wei, Y.; Bader, D.; Litvin, J. Identification of a novel cardiac-specific transcript critical for cardiac myocyte differentiation. Development 1996, 122, 2779–2789. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Reiter, R.S.; Lin, J.L.; Wang, Q.; Williams, H.S.; Krob, S.L.; Schultheiss, T.M.; Evans, S.; Lin, J.J. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development 1999, 126, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.M.; Barnett, P.; Moorman, A.F.; Christoffels, V.M. T-box factors determine cardiac design. Cell. Mol. Life Sci. CMLS 2007, 64, 646–660. [Google Scholar] [CrossRef]
- Cleves, M.A.; Malik, S.; Yang, S.; Carter, T.C.; Hobbs, C.A. Maternal urinary tract infections and selected cardiovascular malformations. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 464–473. [Google Scholar] [CrossRef]
- Tomanek, R.J. Formation of the coronary vasculature during development. Angiogenesis 2005, 8, 273–284. [Google Scholar] [CrossRef]
- Mamasoula, C.; Bigirumurame, T.; Chadwick, T.; Addor, M.C.; Cavero-Carbonell, C.; Dias, C.M.; Echevarría-González-de-Garibay, L.J.; Gatt, M.; Khoshnood, B.; Klungsoyr, K.; et al. Maternal age and the prevalence of congenital heart defects in Europe, 1995–2015: A register-based study. Birth Defects Res. 2023, 115, 583–594. [Google Scholar] [CrossRef]
- Moreno-Rodriguez, R.A.; Krug, E.L. Cardiovascular development. In Charlene à McQueen, Comprehensive Toxicology; Academic Press: Oxford, UK, 2010; Volume 6, pp. 3–33. [Google Scholar]
- Musson, R.; Gąsior, Ł.; Bisogno, S.; Ptak, G.E. DNA damage in preimplantation embryos and gametes: Specification, clinical relevance and repair strategies. Hum. Reprod. Update 2022, 28, 376–399. [Google Scholar] [CrossRef]
- Dolk, H.; McCullough, N.; Callaghan, S.; Casey, F.; Craig, B.; Given, J.; Loane, M.; Lagan, B.M.; Bunting, B.; Boyle, B.; et al. Risk factors for congenital heart disease: The Baby Hearts Study, a population-based case-control study. PLoS ONE 2020, 15, e0227908. [Google Scholar] [CrossRef]
- Øyen, N.; Diaz, L.J.; Leirgul, E.; Boyd, H.A.; Priest, J.; Mathiesen, E.R.; Quertermous, T.; Wohlfahrt, J.; Melbye, M. Prepregnancy Diabetes and Offspring Risk of Congenital Heart Disease: A Nationwide Cohort Study. Circulation 2016, 133, 2243–2253. [Google Scholar] [CrossRef]
- Riehle-Colarusso, T.J.; Patel, S.S. Maternal nongenetic risk factors for congenital heart defects. In Congenital Heart Disease; Karger Publishers: Basel, Switzerland, 2015; pp. 57–69. [Google Scholar]
- Stothard, K.J.; Tennant, P.W.; Bell, R.; Rankin, J. Maternal overweight and obesity and the risk of congenital anomalies: A systematic review and meta-analysis. JAMA 2009, 301, 636–650. [Google Scholar] [CrossRef]
- Dong, D.; Zhang, Y.; Reece, E.A.; Wang, L.; Harman, C.R.; Yang, P. microRNA expression profiling and functional annotation analysis of their targets modulated by oxidative stress during embryonic heart development in diabetic mice. Reprod. Toxicol. 2016, 65, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Zhu, J.Y.; LaHaye, S.; Majumdar, U.; Jiao, K.; Han, Z.; Garg, V. Epigenetic mechanisms underlying maternal diabetes-associated risk of congenital heart disease. JCI Insight 2017, 2, e95085. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Rajakumar, B.; Carreon, C.K.; Morton, S.U. Placental-Heart Axis: An Evolutionary Perspective. Int. J. Mol. Sci. 2024, 25, 11212. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, C.B.; Mangin-Heimos, K.S.; Gu, H.; Edmunds, M.; Bebbington, M.; Lee, C.K.; He, M.; Ortinau, C.M. Placental delayed villous maturation is associated with fetal congenital heart disease. Am. J. Obstet. Gynecol. 2023, 228, 231.e1–231.e11. [Google Scholar] [CrossRef]
- Mahadevan, A.; Tipler, A.; Jones, H. Shared developmental pathways of the placenta and fetal heart. Placenta 2023, 141, 35–42. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, P.; Zhang, L. Fetal Hypoxia Impacts on Proliferation and Differentiation of Sca-1+ Cardiac Progenitor Cells and Maturation of Cardiomyocytes: A Role of MicroRNA-210. Genes 2020, 11, 328. [Google Scholar] [CrossRef]
- Patel, S.S.; Burns, T.L. Nongenetic risk factors and congenital heart defects. Pediatr. Cardiol. 2013, 34, 1535–1555. [Google Scholar] [CrossRef]
- Ramakrishnan, A.; Lee, L.J.; Mitchell, L.E.; Agopian, A.J. Maternal Hypertension During Pregnancy and the Risk of Congenital Heart Defects in Offspring: A Systematic Review and Meta-analysis. Pediatr. Cardiol. 2015, 36, 1442–1451. [Google Scholar] [CrossRef]
- Edwards, M.J. Review: Hyperthermia and fever during pregnancy. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 507–516. [Google Scholar] [CrossRef]
- Liu, S.; Joseph, K.S.; Lisonkova, S.; Rouleau, J.; Van den Hof, M.; Sauve, R.; Kramer, M.S.; Canadian Perinatal Surveillance System (Public Health Agency of Canada). Association between maternal chronic conditions and congenital heart defects: A population-based cohort study. Circulation 2013, 128, 583–589. [Google Scholar] [CrossRef]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; O’Malley, C.D.; Wasserman, C.R.; Tolarova, M.M.; Lammer, E.J. Maternal periconceptional use of multivitamins and reduced risk for conotruncal heart defects and limb deficiencies among offspring. Am. J. Med. Genet. 1995, 59, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Bergström, S.; Carr, H.; Petersson, G.; Stephansson, O.; Bonamy, A.K.; Dahlström, A.; Halvorsen, C.P.; Johansson, S. Trends in Congenital Heart Defects in Infants With Down Syndrome. Pediatrics 2016, 138, e20160123. [Google Scholar] [CrossRef]
- Mao, B.; Qiu, J.; Zhao, N.; Shao, Y.; Dai, W.; He, X.; Cui, H.; Lin, X.; Lv, L.; Tang, Z.; et al. Maternal folic acid supplementation and dietary folate intake and congenital heart defects. PLoS ONE 2017, 12, e0187996. [Google Scholar] [CrossRef]
- Grewal, J.; Carmichael, S.L.; Ma, C.; Lammer, E.J.; Shaw, G.M. Maternal periconceptional smoking and alcohol consumption and risk for select congenital anomalies. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 519–526. [Google Scholar] [CrossRef]
- Yan, X.; Pan, B.; Lv, T.; Liu, L.; Zhu, J.; Shen, W.; Huang, X.; Tian, J. Inhibition of histone acetylation by curcumin reduces alcohol-induced fetal cardiac apoptosis. J. Biomed. Sci. 2017, 24, 1. [Google Scholar] [CrossRef]
- Wang, L.; Sun, H.; Pan, B.; Zhu, J.; Huang, G.; Huang, X.; Tian, J. Inhibition of histone acetylation by curcumin reduces alcohol-induced expression of heart development-related transcription factors in cardiac progenitor cells. Biochem. Biophys. Res. Commun. 2012, 424, 593–596. [Google Scholar] [CrossRef]
- Loffredo, C.A.; Silbergeld, E.K.; Ferencz, C.; Zhang, J. Association of transposition of the great arteries in infants with maternal exposures to herbicides and rodenticides. Am. J. Epidemiol. 2001, 153, 529–536. [Google Scholar] [CrossRef]
- Suhl, J.; Conway, K.M.; Rhoads, A.; Langlois, P.H.; Feldkamp, M.L.; Michalski, A.M.; Oleson, J.; Sidhu, A.; Scholz, T.D.; Kancherla, V.; et al. Prepregnancy exposure to dietary arsenic and congenital heart defects. Birth Defects Res. 2023, 115, 79–87. [Google Scholar] [CrossRef]
- Cipollone, D.; Amati, F.; Carsetti, R.; Placidi, S.; Biancolella, M.; D’Amati, G.; Novelli, G.; Siracusa, G.; Marino, B. A multiple retinoic acid antagonist induces conotruncal anomalies, including transposition of the great arteries, in mice. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2006, 15, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Süleyman, H.; Demircan, B.; Karagöz, Y. Anti-inflammatory and side effects of cyclooxygenase inhibitors. Pharmacol. Rep. PR 2007, 59, 247–258. [Google Scholar] [PubMed]
- Weber, M.; Schweitzer, M.; Andre, J.M.; Tridon, P.; Vert, P. Epilepsie, medicaments antiepileptiques et grossesse [Epilepsy, anticonvulsants and pregnancy]. Arch. Fr. De Pediatr. 1977, 34, 374–383. [Google Scholar]
- Redline, R.W.; Abramowsky, C.R. Transposition of the great vessels in an infant exposed to massive doses of oral contraceptives. Am. J. Obstet. Gynecol. 1981, 141, 468–469. [Google Scholar] [CrossRef]
- Ferencz, C.; Loffredo, C.A.; Corea-Villasenor, A.; Wilson, P.D. Genetic and environmental risk factors of major cardiovascular malformations: The Baltimore-Washington Infant Study, 1981–1989. In Perspectives in Pediatric Cardiology, 1st ed.; Ferencz, C., Loffredo, C.A., Corea-Villasenor, A., Wilson, P.D., Eds.; Futura Publishing Co. Inc.: Armonk, NY, USA, 1997; Volume 5, pp. 867–868. [Google Scholar]
- Tararbit, K.; Houyel, L.; Bonnet, D.; De Vigan, C.; Lelong, N.; Goffinet, F.; Khoshnood, B. Risk of congenital heart defects associated with assisted reproductive technologies: A population-based evaluation. Eur. Heart J. 2011, 32, 500–508. [Google Scholar] [CrossRef]
- Giorgione, V.; Parazzini, F.; Fesslova, V.; Cipriani, S.; Candiani, M.; Inversetti, A.; Sigismondi, C.; Tiberio, F.; Cavoretto, P. Congenital heart defects in IVF/ICSI pregnancy: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2018, 51, 33–42. [Google Scholar] [CrossRef]
- Liang, Y.; Chen, L.; Yu, H.; Wang, H.; Li, Q.; Yu, R.; Qin, J. Which type of congenital malformations is significantly increased in singleton pregnancies following after in vitro fertilization/intracytoplasmic sperm injection: A systematic review and meta-analysis. Oncotarget 2017, 9, 4267–4278. [Google Scholar] [CrossRef]
- Sargisian, N.; Petzold, M.; Furenäs, E.; Gissler, M.; Spangmose, A.L.; Malchau Lauesgaard, S.; Opdahl, S.; Pinborg, A.; Henningsen, A.A.; Westvik-Johari, K.; et al. Congenital heart defects in children born after assisted reproductive technology: A CoNARTaS study. Eur. Heart J. 2024, 45, 4840–4858. [Google Scholar] [CrossRef]
- Peng, J.; Meng, Z.; Zhou, S.; Zhou, Y.; Wu, Y.; Wang, Q.; Wang, J.; Sun, K. The non-genetic paternal factors for congenital heart defects: A systematic review and meta-analysis. Clin. Cardiol. 2019, 42, 684–691. [Google Scholar] [CrossRef]
- Snijder, C.A.; Vlot, I.J.; Burdorf, A.; Obermann-Borst, S.A.; Helbing, W.A.; Wildhagen, M.F.; Steegers, E.A.; Steegers-Theunissen, R.P. Congenital heart defects and parental occupational exposure to chemicals. Hum. Reprod. 2012, 27, 1510–1517. [Google Scholar] [CrossRef]
- Amaldi, F.; Benedetti, P.; Pesole, G.; Plevani, P. Biologia Molecolare; Terza; Casa Editrice Ambrosiana: Milano, Italy, 2011; pp. 554–565. Available online: www.zanichelli.it (accessed on 18 March 2025).
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl. 3), 245–254. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.L.; Yan, M.S.; Marsden, P.A. Epigenetics and cardiovascular disease. Can. J. Cardiol. 2013, 29, 46–57. [Google Scholar] [CrossRef]
- Grunert, M.; Dorn, C.; Cui, H.; Dunkel, I.; Schulz, K.; Schoenhals, S.; Sun, W.; Berger, F.; Chen, W.; Sperling, S.R. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc. Res. 2016, 112, 464–477. [Google Scholar] [CrossRef]
- Sheng, W.; Qian, Y.; Zhang, P.; Wu, Y.; Wang, H.; Ma, X.; Chen, L.; Ma, D.; Huang, G. Association of promoter methylation statuses of congenital heart defect candidate genes with Tetralogy of Fallot. J. Transl. Med. 2014, 12, 31. [Google Scholar] [CrossRef]
- Serra-Juhé, C.; Cuscó, I.; Homs, A.; Flores, R.; Torán, N.; Pérez-Jurado, L.A. DNA methylation abnormalities in congenital heart disease. Epigenetics 2015, 10, 167–177. [Google Scholar] [CrossRef]
- Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef]
- Matthis, A.L.; Zhang, B.; Denson, L.A.; Yacyshyn, B.R.; Aihara, E.; Montrose, M.H. Importance of the Evaluation of N-Acetyltransferase Enzyme Activity Prior to 5-Aminosalicylic Acid Medication for Ulcerative Colitis. Inflamm. Bowel Dis. 2016, 22, 1793–1802. [Google Scholar] [CrossRef]
- Han, Y.; Tanios, F.; Reeps, C.; Zhang, J.; Schwamborn, K.; Eckstein, H.H.; Zernecke, A.; Pelisek, J. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin. Epigenetics 2016, 8, 3. [Google Scholar] [CrossRef]
- Lewandowski, S.L.; Janardhan, H.P.; Smee, K.M.; Bachman, M.; Sun, Z.; Lazar, M.A.; Trivedi, C.M. Histone deacetylase 3 modulates Tbx5 activity to regulate early cardiogenesis. Hum. Mol. Genet. 2014, 23, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hou, Z.; Wang, C.; Wei, C.; Li, Y.; Jiang, L. Association between 5, 10-methylenetetrahydrofolate reductase (MTHFR) polymorphisms and congenital heart disease: A meta-analysis. Meta Gene 2013, 1, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Junker, R.; Kotthoff, S.; Vielhaber, H.; Halimeh, S.; Kosch, A.; Koch, H.G.; Kassenböhmer, R.; Heineking, B.; Nowak-Göttl, U. Infant methylenetetrahydrofolate reductase 677TT genotype is a risk factor for congenital heart disease. Cardiovasc. Res. 2001, 51, 251–254. [Google Scholar] [CrossRef]
- Kuehl, K.; Loffredo, C.; Lammer, E.J.; Iovannisci, D.M.; Shaw, G.M. Association of congenital cardiovascular malformations with 33 single nucleotide polymorphisms of selected cardiovascular disease-related genes. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 101–110. [Google Scholar] [CrossRef]
- Coppedè, F. The genetics of folate metabolism and maternal risk of birth of a child with Down syndrome and associated congenital heart defects. Front. Genet. 2015, 6, 223. [Google Scholar] [CrossRef]
- van Beynum, I.M.; den Heijer, M.; Blom, H.J.; Kapusta, L. The MTHFR 677C->T polymorphism and the risk of congenital heart defects: A literature review and meta-analysis. QJM Mon. J. Assoc. Physicians 2007, 100, 743–753. [Google Scholar] [CrossRef]
- van Beynum, I.M.; Mooij, C.; Kapusta, L.; Heil, S.; den Heijer, M.; Blom, H.J. Common 894G>T single nucleotide polymorphism in the gene coding for endothelial nitric oxide synthase (eNOS) and risk of congenital heart defects. Clin. Chem. Lab. Med. 2008, 46, 1369–1375. [Google Scholar] [CrossRef]
- Feng, Q.; Song, W.; Lu, X.; Hamilton, J.A.; Lei, M.; Peng, T.; Yee, S.P. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation 2002, 106, 873–879. [Google Scholar] [CrossRef]
- Lambrechts, D.; Devriendt, K.; Driscoll, D.A.; Goldmuntz, E.; Gewillig, M.; Vlietinck, R.; Collen, D.; Carmeliet, P. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: A family based association study. J. Med. Genet. 2005, 42, 519–522. [Google Scholar] [CrossRef]
- Griffin, H.R.; Hall, D.H.; Topf, A.; Eden, J.; Stuart, A.G.; Parsons, J.; Peart, I.; Deanfield, J.E.; O’Sullivan, J.; Babu-Narayan, S.V.; et al. Genetic variation in VEGF does not contribute significantly to the risk of congenital cardiovascular malformation. PLoS ONE 2009, 4, e4978. [Google Scholar] [CrossRef]
- Lalani, S.R.; Shaw, C.; Wang, X.; Patel, A.; Patterson, L.W.; Kolodziejska, K.; Szafranski, P.; Ou, Z.; Tian, Q.; Kang, S.H.; et al. Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur. J. Hum. Genet. EJHG 2013, 21, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Tomita-Mitchell, A.; Mahnke, D.K.; Struble, C.A.; Tuffnell, M.E.; Stamm, K.D.; Hidestrand, M.; Harris, S.E.; Goetsch, M.A.; Simpson, P.M.; Bick, D.P.; et al. Human gene copy number spectra analysis in congenital heart malformations. Physiol. Genom. 2012, 44, 518–541. [Google Scholar] [CrossRef]
- Soemedi, R.; Wilson, I.J.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Yang, W.J.; Yang, D.D.; Na, S.; Sandusky, G.E.; Zhang, Q.; Zhao, G. Dicer is required for embryonic angiogenesis during mouse development. J. Biol. Chem. 2005, 280, 9330–9335. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.C.; Shaw, R.M.; De Val, S.; Kang, G.; Jan, L.Y.; Black, B.L.; Stainier, D.Y. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008, 22, 734–739. [Google Scholar] [CrossRef]
- Porrello, E.R. microRNAs in cardiac development and regeneration. Clin. Sci. (1979) 2013, 125, 151–166. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2017, 26, 865–874. [Google Scholar] [CrossRef]
- Ono, K.; Kuwabara, Y.; Han, J. MicroRNAs and cardiovascular diseases. FEBS J. 2011, 278, 1619–1633. [Google Scholar] [CrossRef]
- Hoelscher, S.C.; Stich, T.; Diehm, A.; Lahm, H.; Dreßen, M.; Zhang, Z.; Neb, I.; Aherrahrou, Z.; Erdmann, J.; Schunkert, H.; et al. miR-128a Acts as a Regulator in Cardiac Development by Modulating Differentiation of Cardiac Progenitor Cell Populations. Int. J. Mol. Sci. 2020, 21, 1158. [Google Scholar] [CrossRef]
- Chiavacci, E.; Dolfi, L.; Verduci, L.; Meghini, F.; Gestri, G.; Evangelista, A.M.; Wilson, S.W.; Cremisi, F.; Pitto, L. MicroRNA 218 mediates the effects of Tbx5a over-expression on zebrafish heart development. PLoS ONE 2012, 7, e50536. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.K.S.; Phua, Q.H.; Soh, B.S. Applications of miRNAs in cardiac development, disease progression and regeneration. Stem Cell Res. Ther. 2019, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Greene, S.B.; Bonilla-Claudio, M.; Tao, Y.; Zhang, J.; Bai, Y.; Huang, Z.; Black, B.L.; Wang, F.; Martin, J.F. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a MicroRNA-mediated mechanism. Dev. Cell 2010, 19, 903–912. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, L.; Wang, Q.; You, X.; Li, Y.; Zhao, Y.; Han, X.; Chang, Z.; He, X.; Cheng, C.; et al. NEXN inhibits GATA4 and leads to atrial septal defects in mice and humans. Cardiovasc. Res. 2014, 103, 228–237. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef]
- Boettger, T.; Braun, T. A new level of complexity: The role of microRNAs in cardiovascular development. Circ. Res. 2012, 110, 1000–1013. [Google Scholar] [CrossRef]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA expression in hypoplastic left heart syndrome. J. Card. Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef]
- Nigam, V.; Sievers, H.H.; Jensen, B.C.; Sier, H.A.; Simpson, P.C.; Srivastava, D.; Mohamed, S.A. Altered microRNAs in bicuspid aortic valve: A comparison between stenotic and insufficient valves. J. Heart Valve Dis. 2010, 19, 459–465. [Google Scholar]
- Yanagawa, B.; Lovren, F.; Pan, Y.; Garg, V.; Quan, A.; Tang, G.; Singh, K.K.; Shukla, P.C.; Kalra, N.P.; Peterson, M.D.; et al. miRNA-141 is a novel regulator of BMP-2-mediated calcification in aortic stenosis. J. Thorac. Cardiovasc. Surg. 2012, 144, 256–262. [Google Scholar] [CrossRef]
- Sophocleous, F.; De Garate, E.; Bigotti, M.G.; Anwar, M.; Jover, E.; Chamorro-Jorganes, A.; Rajakaruna, C.; Mitrousi, K.; De Francesco, V.; Wilson, A.; et al. A Segmental Approach from Molecular Profiling to Medical Imaging to Study Bicuspid Aortic Valve Aortopathy. Cells 2022, 11, 3721. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Sequeira-Gross, T.; Petersen, J.; Detlef, I.; Sachse, M.; Zeller, T.; Reichenspurner, H.; Girdauskas, E. Circulating microRNAs in the prediction of BAV aortopathy: Do the expression patterns correlate between blood and aortic tissue? Rev. Cardiovasc. Med. 2022, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Borghini, A.; Foffa, I.; Pulignani, S.; Vecoli, C.; Ait-Ali, L.; Andreassi, M.G. miRNome Profiling in Bicuspid Aortic Valve-Associated Aortopathy by Next-Generation Sequencing. Int. J. Mol. Sci. 2017, 18, 2498. [Google Scholar] [CrossRef] [PubMed]
- Girdauskas, E.; Neumann, N.; Petersen, J.; Sequeira-Gross, T.; Naito, S.; von Stumm, M.; von Kodolitsch, Y.; Reichenspurner, H.; Zeller, T. Expression Patterns of Circulating MicroRNAs in the Risk Stratification of Bicuspid Aortopathy. J. Clin. Med. 2020, 9, 276. [Google Scholar] [CrossRef]
- Antequera-González, B.; Collell-Hernández, R.; Martínez-Micaelo, N.; Marimon-Blanch, C.; Carbonell-Prat, B.; Escribano, J.; Alegret, J.M. miR-130a expression is related to aortic dilation in bicuspid aortic valve children. Pediatr. Res. 2024, 95, 1741–1748. [Google Scholar] [CrossRef]
- Sanchez-Garcia, A.J.; Soule-Egea, M.; Fuentevilla-Alvarez, G.; Vargas-Alarcon, G.; Hernández-Mejia, B.I.; Martínez-Hernández, H.; Mora-Canela, S.L.; Santibanez-Escobar, F.; Ávila-Martinez, V.; Castrejón-Tellez, V.; et al. Role of miRNAs in Regulating Ascending Aortic Dilation in Bicuspid Aortic Valve Patients Operated for Aortic Stenosis. Int. J. Mol. Sci. 2025, 26, 779. [Google Scholar] [CrossRef]
- Li, J.; Cao, Y.; Ma, X.J.; Wang, H.J.; Zhang, J.; Luo, X.; Chen, W.; Wu, Y.; Meng, Y.; Zhang, J.; et al. Roles of miR-1-1 and miR-181c in ventricular septal defects. Int. J. Cardiol. 2013, 168, 1441–1446. [Google Scholar] [CrossRef]
- Zhu, S.; Cao, L.; Zhu, J.; Kong, L.; Jin, J.; Qian, L.; Zhu, C.; Hu, X.; Li, M.; Guo, X.; et al. Identification of maternal serum microRNAs as novel non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 66–72. [Google Scholar] [CrossRef]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Zhou, Z.; Jiang, J.; Zhang, Z.; Ye, L.; Hong, H. A gain-of-function ACTC1 3′UTR mutation that introduces a miR-139-5p target site may be associated with a dominant familial atrial septal defect. Sci. Rep. 2016, 6, 25404. [Google Scholar] [CrossRef]
- Yu, K.; Ji, Y.; Wang, H.; Xuan, Q.K.; Li, B.B.; Xiao, J.J.; Sun, W.; Kong, X.Q. Association of miR-196a2, miR-27a, and miR-499 polymorphisms with isolated congenital heart disease in a Chinese population. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, D.; Zhang, R.R.; Yu, L.W.; Zhao, J.Y.; Yang, X.Y.; Jiang, S.S.; Ma, D.; Qiao, B.; Zhang, F.; et al. A TBX5 3′UTR variant increases the risk of congenital heart disease in the Han Chinese population. Cell Discov. 2017, 3, 17026. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Higgins, H.; Guo, J.; Harrison, K.; Schultz, E.N.; Hales, B.J.; Moses, E.K.; Goldblatt, J.; Pachter, N.; Zhang, G. Clinical significance of circulating microRNAs as markers in detecting and predicting congenital heart defects in children. J. Transl. Med. 2018, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, W.J.; Duan, L.; Hou, Z.L.; Zeng, J.Y.; Li, L.; Jiang, L.H. MicroRNA profiling of patients with sporadic atrial septal defect. Biotechnol. Biotechnol. Equip. 2019, 33, 510–519. [Google Scholar] [CrossRef]
- Gu, H.; Chen, L.; Xue, J.; Huang, T.; Wei, X.; Liu, D.; Ma, W.; Cao, S.; Yuan, Z. Expression profile of maternal circulating microRNAs as non-invasive biomarkers for prenatal diagnosis of congenital heart defects. Biomed. Pharmacother. 2019, 109, 823–830. [Google Scholar] [CrossRef]
- Jin, Y.; Ai, L.; Chai, X.; Tang, P.; Zhang, W.; Yang, L.; Hu, Y.; Xu, Y.; Li, S. Maternal Circulating Exosomal miRNAs as Non-invasive Biomarkers for the Prediction of Fetal Ventricular Septal Defect. Front. Genet. 2021, 12, 717208. [Google Scholar] [CrossRef]
- Jia, L.; Limeng, D.; Xiaoyin, T.; Junwen, W.; Xintong, Z.; Gang, X.; Yun, B.; Hong, G. A Novel Splicing Mutation c.335-1 G > A in the Cardiac Transcription Factor NKX2-5 Leads to Familial Atrial Septal Defect Through miR-19 and PYK2. Stem Cell Rev. Rep. 2022, 18, 2646–2661. [Google Scholar] [CrossRef]
- Ramachandran, V.; Bhagavatheeswaran, S.; Shanmugam, S.; Vasudevan, M.; Ragunathan, M.; Cherian, K.M.; Munirajan, A.K.; Ravi, S.; Balakrishnan, A. Deep sequencing unveils altered cardiac miRNome in congenital heart disease. Mol. Genet. Genom. MGG 2022, 297, 1123–1139. [Google Scholar] [CrossRef]
- O’Brien, J.E., Jr.; Kibiryeva, N.; Zhou, X.G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, X.J.; Wang, H.J.; Li, W.C.; Chen, L.; Ma, D.; Huang, G.Y. Expression of Cx43-related microRNAs in patients with tetralogy of Fallot. World J. Pediatr. WJP 2014, 10, 138–144. [Google Scholar] [CrossRef]
- Zhang, J.; Chang, J.J.; Xu, F.; Ma, X.J.; Wu, Y.; Li, W.C.; Wang, H.J.; Huang, G.Y.; Ma, D. MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of fallot. Can. J. Cardiol. 2013, 29, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, P.; Jian, Z.; Li, J.; Zhu, Y.; Feng, Z.; Xiao, Y. miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem. Biophys. Res. Commun. 2013, 441, 763–769. [Google Scholar] [CrossRef]
- Bittel, D.C.; Kibiryeva, N.; Marshall, J.A.; O’Brien, J.E. MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot. Cells 2014, 3, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Xu, X.; Deng, F.; Feng, J.; Zhang, H.; Liu, Y.; Zhang, Y.; Pan, L.; Liu, Y.; Zhang, D.; et al. miRNA-940 reduction contributes to human Tetralogy of Fallot development. J. Cell. Mol. Med. 2014, 18, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Abu-Halima, M.; Meese, E.; Keller, A.; Abdul-Khaliq, H.; Rädle-Hurst, T. Analysis of circulating microRNAs in patients with repaired Tetralogy of Fallot with and without heart failure. J. Transl. Med. 2017, 15, 156. [Google Scholar] [CrossRef]
- Wang, B.; Shi, G.; Zhu, Z.; Chen, H.; Fu, Q. Sexual difference of small RNA expression in Tetralogy of Fallot. Sci. Rep. 2018, 8, 12847. [Google Scholar] [CrossRef]
- Grunert, M.; Appelt, S.; Dunkel, I.; Berger, F.; Sperling, S.R. Altered microRNA and target gene expression related to Tetralogy of Fallot. Sci. Rep. 2019, 9, 19063. [Google Scholar] [CrossRef]
- You, G.; Zu, B.; Wang, B.; Fu, Q.; Li, F. Identification of miRNA-mRNA-TFs Regulatory Network and Crucial Pathways Involved in Tetralogy of Fallot. Front. Genet. 2020, 11, 552. [Google Scholar] [CrossRef]
- Chouvarine, P.; Photiadis, J.; Cesnjevar, R.; Scheewe, J.; Bauer, U.M.M.; Pickardt, T.; Kramer, H.H.; Dittrich, S.; Berger, F.; Hansmann, G. RNA expression profiles and regulatory networks in human right ventricular hypertrophy due to high pressure load. iScience 2021, 24, 102232. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Chen, Q.; Li, S.; Yang, Y.; Lyu, G. Analyzing exosomal miRNA profiles in tetralogy of fallot fetuses’ amniotic fluid. Sci. Rep. 2025, 15, 96. [Google Scholar] [CrossRef]
- Perrot, A.; Rickert-Sperling, S. Human Genetics of Ventricular Septal Defect. Adv. Exp. Med. Biol. 2024, 1441, 505–534. [Google Scholar] [CrossRef] [PubMed]
- Catalucci, D.; Latronico, M.V.; Condorelli, G. MicroRNAs control gene expression: Importance for cardiac development and pathophysiology. Ann. N. Y. Acad. Sci. 2008, 1123, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, F.; Mi, Y.; Wang, F.; Cai, K.; Yang, X.; Zhang, R.; Liu, L.; Zhang, Y.; Wang, Y.; et al. Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif. 2020, 53, e12764. [Google Scholar] [CrossRef]
- Huan, T.; Rong, J.; Tanriverdi, K.; Meng, Q.; Bhattacharya, A.; McManus, D.D.; Joehanes, R.; Assimes, T.L.; McPherson, R.; Samani, N.J.; et al. Dissecting the roles of microRNAs in coronary heart disease via integrative genomic analyses. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1011–1021. [Google Scholar] [CrossRef]
- Liu, B.L.; Cheng, M.; Hu, S.; Wang, S.; Wang, L.; Tu, X.; Huang, C.X.; Jiang, H.; Wu, G. Overexpression of miR-142-3p improves mitochondrial function in cardiac hypertrophy. Biomed. Pharmacother. 2018, 108, 1347–1356. [Google Scholar] [CrossRef]
- Greenway, S.C.; McLeod, R.; Hume, S.; Roslin, N.M.; Alvarez, N.; Giuffre, M.; Zhan, S.H.; Shen, Y.; Preuss, C.; Andelfinger, G.; et al. Exome sequencing identifies a novel variant in ACTC1 associated with familial atrial septal defect. Can. J. Cardiol. 2014, 30, 181–187. [Google Scholar] [CrossRef]
- AmiRsardari, Z.; Gholipour, A.; Khajali, Z.; Maleki, M.; Malakootian, M. Exploring the role of non-coding RNAs in atrial septal defect pathogenesis: A systematic review. PLoS ONE 2024, 19, e0306576. [Google Scholar] [CrossRef]
- Hirayama-Yamada, K.; Kamisago, M.; Akimoto, K.; Aotsuka, H.; Nakamura, Y.; Tomita, H.; Furutani, M.; Imamura, S.; Takao, A.; Nakazawa, M.; et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. Part A 2005, 135, 47–52. [Google Scholar] [CrossRef]
- Borghini, A.; Vecoli, C.; Mercuri, A.; Turchi, S.; Andreassi, M.G. Individual and joint effects of genetic polymorphisms in microRNA-machinery genes on congenital heart disease susceptibility. Cardiol. Young 2021, 31, 965–968. [Google Scholar] [CrossRef]
- Vinci, S.; Gelmini, S.; Pratesi, N.; Conti, S.; Malentacchi, F.; Simi, L.; Pazzagli, M.; Orlando, C. Genetic variants in miR-146a, miR-149, miR-196a2, miR-499 and their influence on relative expression in lung cancers. Clin. Chem. Lab. Med. 2011, 49, 2073–2080. [Google Scholar] [CrossRef]
- Craig, B. Atrioventricular septal defect: From fetus to adult. Heart (Br. Card. Soc.) 2006, 92, 1879–1885. [Google Scholar] [CrossRef]
- Latronico, M.V.; Catalucci, D.; Condorelli, G. MicroRNA and cardiac pathologies. Physiol. Genom. 2008, 34, 239–242. [Google Scholar] [CrossRef]
- Brás, A.; Rodrigues, A.S.; Gomes, B.; Rueff, J. Down syndrome and microRNAs. Biomed. Rep. 2018, 8, 11–16. [Google Scholar] [CrossRef]
- Coppola, A.; Romito, A.; Borel, C.; Gehrig, C.; Gagnebin, M.; Falconnet, E.; Izzo, A.; Altucci, L.; Banfi, S.; Antonarakis, S.E.; et al. Cardiomyogenesis is controlled by the miR-99a/let-7c cluster and epigenetic modifications. Stem Cell Res. 2014, 12, 323–337. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, L.; Tian, J. Molecular mechanisms of congenital heart disease in down syndrome. Genes Dis. 2019, 6, 372–377. [Google Scholar] [CrossRef]
- Townsley, M.M.; Windsor, J.; Briston, D.; Alegria, J.; Ramakrishna, H. Tetralogy of Fallot: Perioperative Management and Analysis of Outcomes. J. Cardiothorac. Vasc. Anesth. 2019, 33, 556–565. [Google Scholar] [CrossRef]
- Kołcz, J.; Drukała, J.; Bzowska, M.; Rajwa, B.; Korohoda, W.; Malec, E. The expression of connexin 43 in children with Tetralogy of Fallot. Cell. Mol. Biol. Lett. 2005, 10, 287–303. [Google Scholar]
- Omran, A.; Elimam, D.; Webster, K.A.; Shehadeh, L.A.; Yin, F. MicroRNAs: A new piece in the paediatric cardiovascular disease puzzle. Cardiol. Young 2013, 23, 642–655. [Google Scholar] [CrossRef]
- Sellier, C.; Hwang, V.J.; Dandekar, R.; Durbin-Johnson, B.; Charlet-Berguerand, N.; Ander, B.P.; Sharp, F.R.; Angkustsiri, K.; Simon, T.J.; Tassone, F. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS ONE 2014, 9, e103884. [Google Scholar] [CrossRef]
- Li, Y.; Du, J.; Deng, S.; Liu, B.; Jing, X.; Yan, Y.; Liu, Y.; Wang, J.; Zhou, X.; She, Q. The molecular mechanisms of cardiac development and related diseases. Signal Transduct. Target. Ther. 2024, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Elton, T.S.; Selemon, H.; Elton, S.M.; Parinandi, N.L. Regulation of the MIR155 host gene in physiological and pathological processes. Gene 2013, 532, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18. [Google Scholar] [CrossRef]
- Goel, H.; Goel, A. MicroRNA and Rare Human Diseases. Genes 2024, 15, 1243. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Author | Type of Study | Population | Tissue Type | miRNA | Main Results |
|---|---|---|---|---|---|
| Left Obstructive Heart Disease (LVOTO) | |||||
| Sucharov et al., 2015 [102] | Case–control study | Patients with diagnosis of HLHS < 13 years who underwent cardiac transplantation | Right ventricle myocardial cells | Upregulated: miR-22, mi-R-181a, mi-R29b, mi-R130, mi-R302d, and m-R646; downregulated:mi-R577, mi-R193a-5p, andmi-R338-5P; mi-R differently regulated according to stage: niR-145a, mi-R100, mi-R99a, mi-R-204, and mi-R137-3p | Significant correlation between right ventricle and miRs. |
| Bicuspid Aortic Valve (BAV) | |||||
| Nigam et al., 2010 [103] | Case–control study | Adult patients with BAV undergoing aortic valve replacement | Valval leaflet | Downregulated: miR 26, mir 195, and miR30B; Upregulated: miR14.5 | Significant correlation between peripheral miRs and aortic replacement. |
| Yanagawa et al., 2012 [104] | Case–control study | Adult patients with BAV undergoing aortic valve replacement | Valvar leaflet | Downregulated: miR-141 and miR14 | Significant correlation between peripheral miRs and aortic replacement. |
| Sophocleus et al., 2022 [105] | Prospective study | Patients with BAV and aortopathy | Tissue biopsy | miR-128-3p, miR-210-3p, miR-150-5p, miR-199b-5p, and miR-21-5p | Correlation between miRNAs and aortic dilation. |
| Naito et al., 2022 [106] | Prospective study | Patients who received elective aortic valve repair/replacement ± proximal aortic replacement to BAV disease | Blood serum and aortic tissue | miR-21 miR-133a miR-143, and miR-145 | Significant correlation between peripheral whole blood and aortic tissue miRs. |
| Borghini et al., 2017 [107] | Prospective study | Patients with ascending TAA associated with BAV or TAV | Tissue specimens | Downregulation of miR-424-3p and miR-3688-3p in BAV patients compared to TAV patients | Correlation between miRs and aortic dilation. |
| Girdauskas et al., 2020 [108] | Prospective study | Patients who underwent aortic valve replacement | Blood serum | miR-17, miR-20a, and miR-106a | Correlation between miRs and aortic dilation. |
| Antequera-González et al., 2024 [109] | Prospective study | Patients with BAV < 17 years | Blood serum | miR-130a expression in plasma inversely correlated with ascending aorta and aortic root z scores | Significant correlation between miRs and ascending aorta and aortic root z scores. |
| Sanchez-Garcia et al., 2025 [110] | Prospective Study | Patients who underwent aortic valve replacement for aortic stenosis | Aortic tissue | miR-17-5p, hsa-let-7e, and mi-196a-5p | Significant correlation between aortic tissue miRs and aortic dilation and calcification. |
| Septation defects | |||||
| Li et al., 2013 [111] | Case–control study | Patients with CHDs who underwent repair of VSDs and 28 healthy controls | Cardiac tissue | miR-1-1 and miR-181c | Downregulation of miR-1-1 and upregulation of miR-181c in VSD patients. |
| Zhu et al., 2013 [112] | Case–control study | Pregnant women bearing a fetus with ASDs, VSDs, or TOF and 30 normal pregnancy cases | Maternal serum | miR-19b, miR-29c, and miR-375 | Upregulation of miR-19b, miR-22, miR-29c, and miR-375 in pregnant women with fetal CHDs; miR-19b and miR-29c were found to bear correlation with VSDs, whereas miR-19b, miR-29c, and miR-375 correlated with ASDs and all four miRs with TOF. |
| Li et al., 2014 [113] | Case–control study | Patients with VSDs and 15 healty controls | Serum | miR-let-7e, miR-155-5p, miR-222-3p, miR-379-5p, miR-409-3p, miR-433, miR-487b, and miR-498 | Circulating miR profile for patients with VSDs showed that miR-let-7e, miR-155-5p, miR-222-3p, miR-379-5p, miR-409-3p, miR-433, and miR-487b were downregulated and miR-498 was upregulated when matched to VSD-free controls. |
| Wang et al., 2016 [114] | Family study | Chinese family with autosomal-dominant isolated ASD; four of the five individuals in the family had a similar clinical expression and a diagnosis of ASD | Serum | miR-139-5p | c.*1784 (T>C) mutation in the 3′UTR of the ACTC1 gene in familial ASD patients entailed a new miR-139-5p target site, and miR-139-5p binding to this target site decreased ACTC1 expression. |
| Yu et al., 2016 [115] | Cross-sectional study | Chinese individuals | Serum | miR-196-a2 | c.*1784 (T>C) mutation in the 3′UTR of the ACTC1 gene in familial ASD patients entailed a new miR-139-5p target site, and miR-139-5p binding to this target site decreased ACTC1 expression. |
| Wang et al., 2017 [116] | Case–control study | CHD patients and healthy controls | Serum | miR-9 * and miR-30a | rs6489956 (C>T) single-nucleotide polymorphism in the 3′UTR of the TBX5 gene was associated with ASD and VSD occurrence, as the T allele showed a higher affinity for binding to miR-9 and miR-30a compared to the C allele, thus decreasing TBX5 expression. |
| Song et al., 2018 [117] | Case–control study | Families, each having a child with CHDs and parents without any cardiovascular disorder, and families unaffected by cardiovascular disease as controls | Serum, maternal serum | miR-486 | Upregulation of hsa-miR-let-7a, hsa-miR-let-7b, and miR-486 in children with ASDs, VSDs, and AVSDs; hsa-miR-let-7a and hsa-miR-let-7b were specifically overexpressed in ASD children and a similar expression profile was confirmed in mothers of children with ASDs. hsa-miR-486 level was significantly higher in all ASD, VSD, and AVSD groups. |
| Han et al., 2019 [118] | Case–control study | Infants with ASDs and normal fetuses obtained from pregnant women who underwent voluntary abortion as controls | Atrial septum | miR-29 *, miR-143/145 *, miR-17-92 *, miR-106b-25, and miR-503/424 * | Upregulation of miR-17-92, miR-106b-25, and miR-503/424 clusters and downregulation of miR-29 and miR-143/145 clusters in atrial septum tissues of sporadic ASD patients compared with healthy controls. |
| Gu et al., 2019 [119] | Case–control study | Pregnant women with CHD fetuses and women carrying normal fetuses | Maternal serum | miR-142-5p, miR-1275, miR-3664-3p, and miR-4666a-3p | Maternal serum of fetuses with VSDs had a higher expression of miR-1275 and miR-3664-3p and a reduced expression of miR-142-5p and miR-4666a-3p. Interestingly, these microRNAs were rapidly reduced in maternal serum after delivery as compared to before delivery. |
| Jin et al., 2021 [120] | Case–control study | Pregnant women bearing a fetus with VSDs and women carrying normal fetuses | Maternal serum | miR-146a | Reduced expression of hsa-miR-146a-5p was found to effectively distinguish cases of fetuses with VSDs from controls. |
| Jia et al., 2022 [121] | Family study | Family members with familial VSD and healthy family members | Serum | miR-146a | c.335-1 (G>A) mutation located at the splicing site of NKX2 in individuals with familial ASDs appeared to inhibit the expression of miR-19a/b which in turns upregulated PYK2, a key cytoskeletal protein and tyrosine kinase in the regulation of cell processes like cardiomyocyte proliferation, differentiation, and apoptosis. |
| Ramachandran et al., 2022 [122] | Cross-sectional study | CHD patients | Cardiac tissue | miR-218-5p, miR-221-3p, and miR-2392 | Upregulation of miR-218-5p was associated with VSDs, whereas downregulation of miR-221-3p and miR-2392 was associated with ASDs. |
| Tetralogy of Fallot and conotruncal anomalies | |||||
| O’Brien et al., 2012 [123] | Case–control study | A total of 16 infants with non-syndromic TOF and 8 healthy controls | RV outflow tract myocardium | miR-1275, miR-27b, miR-421, miR-1201, miR-122, and another +56 miRNAs | A total of 61 miRNAs were found to be significantly changed in expression in the RV myocardium of children with TOF compared to normally developing controls. Of these, miR-1275, miR-27b, miR-421, miR-1201, and miR-122 were shown to potentially target genes critical to cardiac development. |
| Wu et al., 2014 [124] | Case–control study | Myocardial samples from 30 TOF patients and 10 healthy controls; blood samples from 200 TOF patients and 200 controls | RV myocardium | miR-1 and miR-206 | Among 10 putative connexin-43-related miRNAs, miR-1 and miR-206 expression was significantly decreased in the TOF patients as compared to controls, suggesting a role of these miRNAs in the pathogenesis of the disease. |
| Zhang et al., 2013 [125] | Case–control study | A total of five infants with non-syndromic TOF and three healty controls | RV outflow tract myocardium | miR-146b-5p, miR-155, miR-19a, miR-222, miR-424, miR-337-5p, miR-363, miR-130b, miR-154, miR-708, miR-181c, miR-424 *, miR-181d, miR-192, miR-660, miR-29c, miR-720, and miR-181a * | A total of 18 miRNAs had significantly altered expression, and it was found that 16 of these targeted several genes involved in heart development. miR-424 targeted the NF1 and HAS2 genes, whose expression was decreased in RVOT myocardial tissues from patients with TOF, suggesting a pathogenetic role. |
| Zhu et al., 2013 [112] | Case–control study | A total of 30 pregnant women bearing a fetus with ASDs, VSDs, or TOF and 30 normal pregnancy controls | Maternal serum | miR-19b, miR-22, miR-29c, and miR-375 | Upregulation of miR-19b, miR-22, miR-29c, and miR-375 in pregnant women with fetal CHDs; miR-22 appeared to be specifically upregulated in TOF patients. |
| He at al., 2013 [126] | Experimental study | A total of 21 CHD patients, 10 with cyanotic CHDs (9 TOF, 1 PA + VSD) and 11 with acyanotic CHDs (VSD + RVOTO) | Right ventricular myocardium | miR-138 | Hypoxia induced upregulation of miR-138, which decreased the protein level of its target MLK3 and attenuated hypoxia-induced apoptosis in cardiomyocytes. |
| Bitttel et al., 2014 [127] | Experimental study | Primary cells from right ventricular tissue of 16 infants with TOF and of 8 healthy controls | Right ventricular myocardium | miR-421 | miR-421 modulated the expression of genes of importance to heart development such as SOX4 and could play a role in the pathogenesis of cardiac defects. |
| Liang et al., 2014 [128] | Case–control study | A total of 26 TOF patients and 15 healthy individuals | Right ventricular myocardium | miR-940 and another + 74 miRNAs | A total of 75 miRNAs were found to be differentially expressed between TOF patients and healthy controls. miR-940 was the most downregulated miRNA in the myocardium from patients with TOF and was the only one to be most highly expressed in the normal human RVOT compared to other chambers within the heart, suggesting a potential pathogenetic role. |
| Abu-Halima et al., 2017 [129] | Case–control study | A total of 37 long-term post-repair TOF patients and 15 healthy controls | Serum | miR-421, miR-1233-3p, and miR-625-5p | Expression levels of miR-421, miR-1233-3p, and miR-625-5p were lower in TOF patients with symptomatic right heart failure, potentially indicating disease progression in these patients. |
| Wang et al., 2018 [130] | Observational study | A total of five female TOF patients and five male TOF patients | Right ventricular myocardium | miR-1/miR-133 * | Significant sexual differences in small RNA expression in TOF patients; miR-1/miR-133 cluster accounted for the greatest variance in sRNA expression between the sexes. |
| Grunert et al., 2019 [131] | Case–control study | A total of 22 isolated TOF patients and 3 healthy controls | Right ventricular myocardium | miR-1, miR-133b, miR-139-5p, miR-140-5p, miR-146b-5p, and another +167 miRNAs | A total of 172 miRNAs were significantly differentially expressed in TOF patients. Among these, the Authors highlighted the potential pathogenetic role of five miRNAs—miR-1, miR-133b, miR-139-5p, miR-140-5p, and miR-146b-5p—targeting 18 genes essential to cardiac development and function—including KCNJ2, FBN2, SLC38A3, and TNNI1. |
| You et al., 2020 [132] | Cross-sectional study | NCBI Gene Expression Omnibus (GEO) database, consisting at the time of the study of 75 TOF patients and 32 matched healthy controls | miRNA expression profile datasets | miR-499, miR-23b, miR-222, miR-1275, miR-93, miR-155, and miR-187 | Seven miRNAs were significantly upregulated in TOF patients compared to healthy controls—miR-499, miR-23b, miR-222, miR-1275, miR-93, miR-155, and miR-187. These miRNAs have been demonstrated to participate in cardiac development and function and their dysregulation is supposed to contribute to abnormal cardiac cell division, differentiation, and apoptosis in TOF pathogenesis. |
| Chouvarine et al., 2021 [133] | Case–control study | A total of 19 infants with TOF of PS and 8 controls affected by VSDs without PS | Right ventricular myocardium | miR-31, miR-216a, miR-372, and miR-5008 | Four miRNAs (miR-31, miR-216a, miR-372, and miR-5008) were shown to be potentially involved in the epigenetic regulation of RVH in TOF/PS. Interestingly, miR-31 was more prominent in male patients, while miR-372 was predominant in females. |
| Yang et al., 2025 [134] | Case–control study | Five pregnant women carrying fetuses diagnosed with TOF through fetal echocardiography and five pregnant women carrying healthy fetuses | Amniotic fluid | miR-3064-5p, miR-206, miR-193b-3p, miR-205-5p, miR-10a-5p, miR-338-3p, miR-106b-5p, miR-25-3p, miR-223-3p, miR-20b-5p, miR-let-7e-5p, miR-34c-5p, miR-125b-5p, miR-200a-3p, miR-125a-5p, miR-let-7c-5p, miR-30b-5p, miR-16-5p, miR-331-3p, miR-27b-3p, miR-27a-3p, miR-let-7f-5p, miR-let-7a-5p, miR-let-7g-5p, and miR-449a | There were a total of 257 significantly dysregulated miRNAs, 25 of which targeted genes involved in Tetralogy of Fallot and congenital heart diseases. The upregulation of miR-10a-5p was found to directly inhibit the expression of the TBX5 gene, which is crucial for cardiomyocyte differentiation and heart development. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannarino, S.; Calcaterra, V.; Puricelli, F.; Cecconi, G.; Chillemi, C.; Raso, I.; Cordaro, E.; Zuccotti, G. The Role of miRNA Expression in Congenital Heart Disease: Insights into the Mechanisms and Biomarker Potential. Children 2025, 12, 611. https://doi.org/10.3390/children12050611
Mannarino S, Calcaterra V, Puricelli F, Cecconi G, Chillemi C, Raso I, Cordaro E, Zuccotti G. The Role of miRNA Expression in Congenital Heart Disease: Insights into the Mechanisms and Biomarker Potential. Children. 2025; 12(5):611. https://doi.org/10.3390/children12050611
Chicago/Turabian StyleMannarino, Savina, Valeria Calcaterra, Filippo Puricelli, Giulia Cecconi, Claudia Chillemi, Irene Raso, Erika Cordaro, and Gianvincenzo Zuccotti. 2025. "The Role of miRNA Expression in Congenital Heart Disease: Insights into the Mechanisms and Biomarker Potential" Children 12, no. 5: 611. https://doi.org/10.3390/children12050611
APA StyleMannarino, S., Calcaterra, V., Puricelli, F., Cecconi, G., Chillemi, C., Raso, I., Cordaro, E., & Zuccotti, G. (2025). The Role of miRNA Expression in Congenital Heart Disease: Insights into the Mechanisms and Biomarker Potential. Children, 12(5), 611. https://doi.org/10.3390/children12050611