
Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin–Siris Syndromic Congenital Heart Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval and Informed Consent
2.2. Human Research Participants
2.3. Genetic Investigation of Human SOX4
2.4. Preparation of Gene Expression Constructs
2.5. Functional Characterization of Glu111*-Mutant SOX4 with Dual Report Genes
2.6. Statistics
3. Results
3.1. Baseline Clinical Data of the Research Participants
3.2. Discovery of a New SOX4 Mutation Accountable for CHD
3.3. Failure of Glu111*-Mutant SOX4 to Induce NKX2.5 Expression
3.4. No Synergistic Activation of GATA4 by Glu111*-Mutant SOX4 with TBX20
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [CrossRef] [PubMed]
- Abhinav, P.; Li, Y.J.; Huang, R.T.; Liu, X.Y.; Gu, J.N.; Yang, C.X.; Xu, Y.J.; Wang, J.; Yang, Y.Q. Somatic GATA4 mutation contributes to tetralogy of Fallot. Exp. Ther. Med. 2024, 27, 91. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, X.Y.; Yang, C.X.; Zhou, H.M.; Li, Y.J.; Qiu, X.B.; Huang, R.T.; Cen, S.S.; Wang, Y.; Xu, Y.J.; et al. Discovery and functional investigation of BMP4 as a new causative gene for human congenital heart disease. Am. J. Transl. Res. 2024, 16, 2034–2048. [Google Scholar] [CrossRef] [PubMed]
- Karas Kuželički, N.; Doljak, B. Congenital Heart Disease and Genetic Changes in Folate/Methionine Cycles. Genes 2024, 15, 872. [Google Scholar] [CrossRef]
- Ly, R.; Karsenty, C.; Amedro, P.; Cohen, S.; Domanski, O.; Godart, F.; Radojevic, J.; Vaksmann, G.; Naccache, N.; Boubrit, A.; et al. Health-Related Quality of Life and Its Association With Outcomes in Adults With Congenital Heart Disease and Heart Failure: Insight From FRESH-ACHD Registry. J. Am. Heart Assoc. 2023, 12, e027819. [Google Scholar] [CrossRef]
- Hock, J.; Brudy, L.; Willinger, L.; Hager, A.; Ewert, P.; Oberhoffer-Fritz, R.; Müller, J. Cardiopulmonary Exercise Test and Daily Physical Activity in Pediatric Congenital Heart Disease: An Exploratory Analysis. Am. J. Cardiol. 2024, 225, 84–88. [Google Scholar] [CrossRef]
- Luca, A.C.; Țarcă, E.; Tănase, V.G.; Pădureț, I.A.; Dragoiu, T.S.; Butnariu, L.I.; Roșu, S.T.; Roca, I.C.; Mîndru, D.E. Benefits of Physical Activity in Children with Cardiac Diseases-A Concise Summary for Pediatricians. Children 2024, 11, 1432. [Google Scholar] [CrossRef]
- Tobias, D.; Helm, P.C.; Bauer, U.M.M.; Niessner, C.; Hahn, S.; Siaplaouras, J.; Apitz, C. Trends in Nutritional Status and Dietary Behavior in School-Aged Children with Congenital Heart Defects. Children 2024, 11, 1264. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, S.; Rollins, C. Fetal Brain Development in Congenital Heart Disease. Can. J. Cardiol. 2023, 39, 115–122. [Google Scholar] [CrossRef]
- Nijman, M.; van der Meeren, L.E.; Nikkels, P.G.J.; Stegeman, R.; Breur, J.M.P.J.; Jansen, N.J.G.; ter Heide, H.; Steenhuis, T.J.; de Heus, R.; Bekker, M.N.; et al. Placental Pathology Contributes to Impaired Volumetric Brain Development in Neonates with Congenital Heart Disease. J. Am. Heart Assoc. 2024, 13, e033189. [Google Scholar] [CrossRef]
- Angerbjörn, M.; Johansson, B.; Eriksson, M.; Rinnström, D.; Sandberg, C.; Christersson, C.; Sörensson, P.; Trzebiatowska-Krzynska, A.; Mandalenakis, Z.; Thilén, U.; et al. Ischemic Stroke in Adults with Congenital Heart Disease: Cumulative Incidence and Associated Factors. J. Am. Heart Assoc. 2024, 13, e034206. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Ye, L. Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases. Biology 2024, 13, 234. [Google Scholar] [CrossRef]
- Jone, P.-N.; Ivy, D.D.; Hauck, A.; Karamlou, T.; Truong, U.; Coleman, R.D.; Sandoval, J.P.; Marín, M.J.d.C.; Eghtesady, P.; Tillman, K.; et al. Pulmonary Hypertension in Congenital Heart Disease: A Scientific Statement From the American Heart Association. Circ. Heart Fail. 2023, 16, e00080. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, G.; Kanakis, M.; Samanidis, G.; Tzannis, K.; Bobos, D.; Kousi, T.; Apostolopoulou, S.; Kakava, F.; Kyriakoulis, K.; Bounta, S.; et al. Acute Kidney Injury Predictors and Outcomes after Cardiac Surgery in Children with Congenital Heart Disease: An Observational Cohort Study. Diagnostics 2022, 12, 2397. [Google Scholar] [CrossRef]
- Reiter, F.P.; Hadjamu, N.J.; Nagdyman, N.; Zachoval, R.; Mayerle, J.; De Toni, E.N.; Kaemmerer, H.; Denk, G. Congenital heart disease-associated liver disease: A narrative review. Cardiovasc. Diagn. Ther. 2021, 11, 577–590. [Google Scholar] [CrossRef]
- Moschino, L.; Guiducci, S.; Duci, M.; Meggiolaro, L.; Nardo, D.; Bonadies, L.; Salvadori, S.; Verlato, G.; Baraldi, E. Noninvasive Tools to Predict Necrotizing Enterocolitis in Infants with Congenital Heart Diseases: A Narrative Review. Children 2024, 11, 1343. [Google Scholar] [CrossRef]
- Havers-Borgersen, E.; Butt, J.H.; Østergaard, L.; Petersen, J.K.; Torp-Pedersen, C.; Køber, L.; Fosbøl, E.L. Long-term incidence of infective endocarditis among patients with congenital heart disease. Am. Heart J. 2023, 259, 9–20. [Google Scholar] [CrossRef]
- Abramczyk, U.; Cześniewicz, P.; Kusa, J. Infective Endocarditis in Children as an Increasing Clinical Problem—A Case Series. Children 2024, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Făgărășan, A.; Ghiragosian-Rusu, S.E.; Ghiragosian, C.; Gozar, L.; Suteu, C.; Toma, D.; Al-Akel, F.C.; Cucerea, M. Speckle Strain Analysis of Left Ventricular Dysfunction in Paediatric Patients with Bicuspid Aortic Valve-A Pilot Study. Children 2024, 11, 1514. [Google Scholar] [CrossRef]
- Amdani, S.; Conway, J.; George, K.; Martinez, H.R.; Asante-Korang, A.; Goldberg, C.S.; Davies, R.R.; Miyamoto, S.D.; Hsu, D.T.; American Heart Association Council on Lifelong Congenital Heart Disease and Heart Health in the Young; et al. Evaluation and Management of Chronic Heart Failure in Children and Adolescents With Congenital Heart Disease: A Scientific Statement From the American Heart Association. Circulation 2024, 150, e33–e50. [Google Scholar] [CrossRef]
- Wu, M.H.; Chiu, S.N.; Tseng, W.C.; Lu, C.W.; Kao, F.Y.; Huang, S.K. Atrial fibrillation in adult congenital heart disease and the general population. Heart Rhythm. 2023, 20, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Schamroth Pravda, N.; Kalter-Leibovici, O.; Nir, A.; Lorber, A.; Dadashev, A.; Hirsch, R.; Benderly, M.; Israeli Congenital Heart Disease Research Group. Arrhythmia Burden Among Adult Patients With Congenital Heart Disease: A Population-Based Study. J. Am. Heart Assoc. 2024, 13, e031760. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Khaliq, H.; Gomes, D.; Meyer, S.; von Kries, R.; Wagenpfeil, S.; Pfeifer, J.; Poryo, M. Trends of mortality rate in patients with congenital heart defects in Germany-analysis of nationwide data of the Federal Statistical Office of Germany. Clin. Res. Cardiol. 2024, 113, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, R.; Omann, C.; Ekelund, C.K.; Gaynor, J.W.; Hjortdal, V.E. Impact of an Impaired Maternal-Fetal Environment on Death in Children With Congenital Heart Defects Undergoing Surgery in Denmark From 1994 to 2018. J. Am. Heart Assoc. 2024, 13, e031575. [Google Scholar] [CrossRef]
- Kawai, S.; Pak, K.; Iwamoto, S.; Kawakami, C.; Inuzuka, R.; Maeda, J.; Furutani, Y.; Kamisago, M.; Takatsuki, S.; Uyeda, T.; et al. Association Between Maternal Factors in Early Pregnancy and Congenital Heart Defects in Offspring: The Japan Environment and Children’s Study. J. Am. Heart Assoc. 2023, 12, e029268. [Google Scholar] [CrossRef]
- Lv, Y.; Gao, R.F.; Yang, C.X.; Xu, Y.J.; Yang, Y.Q. Increased gestational palmitic acid predisposes offspring to congenital heart disease. Cell Rep. Med. 2023, 4, 100984. [Google Scholar] [CrossRef]
- Zhang, M.; Han, Y. MicroRNAs in chronic pediatric diseases (Review). Exp. Ther. Med. 2024, 27, 100. [Google Scholar] [CrossRef]
- Walton, N.A.; Nguyen, H.H.; Procknow, S.S.; Johnson, D.; Anzelmi, A.; Jay, P.Y. Repurposing Normal Chromosomal Microarray Data to Harbor Genetic Insights into Congenital Heart Disease. Biology 2023, 12, 1290. [Google Scholar] [CrossRef]
- Gabriel, G.C.; Ganapathiraju, M.; Lo, C.W. The Role of Cilia and the Complex Genetics of Congenital Heart Disease. Annu. Rev. Genom. Hum. Genet. 2024, 25, 309–327. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, W.; Wang, Z.; Wang, Y.; Zheng, H.; Zhou, K.; Xu, Q.; Bai, L.; Liu, H.; Ren, Z.; et al. Genetics of congenital heart disease. Clin. Chim. Acta 2024, 552, 117683. [Google Scholar] [CrossRef]
- Bolunduț, A.C.; Lazea, C.; Mihu, C.M. Genetic Alterations of Transcription Factors and Signaling Molecules Involved in the Development of Congenital Heart Defects-A Narrative Review. Children 2023, 10, 812. [Google Scholar] [CrossRef] [PubMed]
- Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Liu, X.; Wang, C.; Wu, Z.; Sun, Z.; Su, J.; Yan, R.; Peng, Y.; Yang, Y.; Wang, C.; et al. Assessment of evidence on reported non-genetic risk factors of congenital heart defects: The updated umbrella review. BMC Pregnancy Childbirth 2022, 22, 371. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, L.; Rong, H.; Liu, X.; Yang, L.; Li, N.; Shi, H. ENU-based dominant genetic screen identifies contractile and neuronal gene mutations in congenital heart disease. Genome Med. 2024, 16, 97. [Google Scholar] [CrossRef]
- Xiao, F.; Zhang, X.; Morton, S.U.; Kim, S.W.; Fan, Y.; Gorham, J.M.; Zhang, H.; Berkson, P.J.; Mazumdar, N.; Cao, Y.; et al. Functional dissection of human cardiac enhancers and noncoding de novo variants in congenital heart disease. Nat. Genet. 2024, 56, 420–430. [Google Scholar] [CrossRef]
- Mansoorshahi, S.; Yetman, A.T.; Bissell, M.M.; Kim, Y.Y.; Michelena, H.I.; De Backer, J.; Mosquera, L.M.; Hui, D.S.; Caffarelli, A.; Andreassi, M.G.; et al. Whole-exome sequencing uncovers the genetic complexity of bicuspid aortic valve in families with early-onset complications. Am. J. Hum. Genet. 2024, 111, 2219–2231. [Google Scholar] [CrossRef]
- Zuo, J.Y.; Chen, H.X.; Yang, Q.; He, G.W. Variants of the promoter of MYH6 gene in congenital isolated and sporadic patent ductus arteriosus: Case-control study and cellular functional analyses. Hum. Mol. Genet. 2024, 33, 884–893. [Google Scholar] [CrossRef]
- Inoue, T.; Takase, R.; Uchida, K.; Kodo, K.; Suda, K.; Watanabe, Y.; Yoshiura, K.-I.; Kunimatsu, M.; Ishizaki, R.; Azuma, K.; et al. The c.1617del variant of TMEM260 is identified as the most frequent single gene determinant for Japanese patients with a specific type of congenital heart disease. J. Hum. Genet. 2024, 69, 215–222. [Google Scholar] [CrossRef]
- Tabrizi, F.; Khatami, M.; Heidari, M.M.; Bragança, J.; Tatari, H.; Namnabat, M.; Hadadzadeh, M.; Navabi Shirazi, M.A. Novel and deleterious nucleotide variations in the HAND1 gene probably affect miRNA target sites and protein function in pediatric patients with congenital heart disease. Mol. Biol. Rep. 2024, 51, 468. [Google Scholar] [CrossRef]
- Dong, B.B.; Li, Y.J.; Liu, X.Y.; Huang, R.T.; Yang, C.X.; Xu, Y.J.; Lv, H.T.; Yang, Y.Q. Discovery of BMP10 as a new gene underpinning congenital heart defects. Am. J. Transl. Res. 2024, 16, 109–125. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, J.; Guo, Q.; Dong, X.; Li, J.; Wang, J.; Li, S.; Shen, Y.; Lin, K.; Yang, Z.; et al. Exploring Genetic Diversity of SOD2 and POU5F1 for Congenital Heart Disease in the Southwest Chinese Population. Int. Heart J. 2024, 65, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, K.; Watt, K.E.; Ide, S.; Baltrunaite, K.; Brunswick, C.; Inskeep, K.; Capannari, C.; Adam, M.P.; Begtrup, A.; Bertola, D.R.; et al. POLR1A variants underlie phenotypic heterogeneity in craniofacial, neural, and cardiac anomalies. Am. J. Hum. Genet. 2023, 110, 809–825. [Google Scholar] [CrossRef]
- Hays, T.; Hernan, R.; Disco, M.; Griffin, E.L.; Goldshtrom, N.; Vargas, D.; Krishnamurthy, G.; Bomback, M.; Rehman, A.U.; Wilson, A.T.; et al. Implementation of Rapid Genome Sequencing for Critically Ill Infants With Complex Congenital Heart Disease. Circ. Genom. Precis. Med. 2023, 16, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiao, F.; Qian, Y.; Wu, B.; Dong, X.; Lu, Y.; Cheng, G.; Wang, L.; Yan, K.; Chen, L.; et al. Genetic architecture in neonatal intensive care unit patients with congenital heart defects: A retrospective study from the China Neonatal Genomes Project. J. Med. Genet. 2023, 60, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Padua, M.B.; Helm, B.M.; Wells, J.R.; Smith, A.M.; Bellchambers, H.M.; Sridhar, A.; Ware, S.M. Congenital heart defects caused by FOXJ1. Hum. Mol. Genet. 2023, 32, 2335–2346. [Google Scholar] [CrossRef]
- Li, Y.-J.; Wang, J.; Ye, W.G.; Liu, X.-Y.; Li, L.; Qiu, X.-B.; Chen, H.; Xu, Y.-J.; Yang, Y.-Q.; Bai, D.; et al. Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias. Biology 2023, 12, 346. [Google Scholar] [CrossRef]
- Deng, Q.; Wang, X.; Gao, J.; Xia, X.; Wang, Y.; Zhang, Y.; Chen, Y. Growth restriction and congenital heart disease caused by a novel TAB2 mutation: A case report. Exp. Ther. Med. 2023, 25, 258. [Google Scholar] [CrossRef]
- Wu, H.; Wu, H.; He, Y.; Sun, W.; Meng, Y.; Wen, B.; Chu, M. Functional characterization of GATA6 genetic variants associated with mild congenital heart defects. Biochem. Biophys. Res. Commun. 2023, 641, 77–83. [Google Scholar] [CrossRef]
- Taha, M.; Awny, N.; Ismail, S.; Ashaat, E.A.; Senousy, M.A. Screening and evaluation of TBX20 and CITED2 mutations in children with congenital cardiac septal defects: Correlation with cardiac troponin T and caspase-3. Gene 2023, 882, 147660. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.J.; Yang, C.X.; Huang, R.T.; Xue, S.; Yuan, F.; Yang, Y.Q. SMAD4 loss-of-function mutation predisposes to congenital heart disease. Eur. J. Med. Genet. 2023, 66, 104677. [Google Scholar] [CrossRef]
- Grippa, M.; Graziano, C. Landscape of Constitutional SOX4 Variation in Human Disorders. Genes 2024, 15, 158. [Google Scholar] [CrossRef] [PubMed]
- Schilham, M.W.; Oosterwegel, T.A.; Moerer, P.; Ya, J.; de Boer, P.A.J.; van de Wetering, M.; Verbeek, S.; Lamers, W.H.; Kruisbeek, A.M.; Cumano, A.; et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature 1996, 380, 711–714. [Google Scholar] [CrossRef]
- Ya, J.; Schilham, M.W.; de Boer, P.A.; Moorman, A.F.; Clevers, H.; Lamers, W.H. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ. Res. 1998, 83, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Angelozzi, M.; Karvande, A.; Molin, A.N.; Ritter, A.L.; Leonard, J.M.M.; Savatt, J.M.; Douglass, K.; Myers, S.M.; Grippa, M.; Tolchin, D.; et al. Consolidation of the clinical and genetic definition of a SOX4-related neurodevelopmental syndrome. J. Med. Genet. 2022, 59, 1058–1068. [Google Scholar] [CrossRef]
- Jiang, W.F.; Sun, Y.M.; Qiu, X.B.; Wu, S.H.; Ding, Y.Y.; Li, N.; Yang, C.X.; Xu, Y.J.; Jiang, T.B.; Yang, Y.Q. Identification and Functional Investigation of SOX4 as a Novel Gene Underpinning Familial Atrial Fibrillation. Diagnostics 2024, 14, 2376. [Google Scholar] [CrossRef]
- Li, N.; Li, Y.-J.; Guo, X.-J.; Wu, S.-H.; Jiang, W.-F.; Zhang, D.-L.; Wang, K.-W.; Li, L.; Sun, Y.-M.; Xu, Y.-J.; et al. Discovery of TBX20 as a Novel Gene Underlying Atrial Fibrillation. Biology 2023, 12, 1186. [Google Scholar] [CrossRef]
- Shi, H.Y.; Xie, M.S.; Yang, C.X.; Huang, R.T.; Xue, S.; Liu, X.Y.; Xu, Y.J.; Yang, Y.Q. Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease. Diagnostics 2022, 12, 1917. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Rajakumar, G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016, 7, 6. [Google Scholar] [CrossRef]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Yu, Y.; Lei, W.; Yang, J.; Wei, Y.C.; Zhao, Z.L.; Zhao, Z.A.; Hu, S. Functional mutant GATA4 identification and potential application in preimplantation diagnosis of congenital heart diseases. Gene 2018, 641, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.P.; Sunde, M.; Costa, M.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xiao, D.; Zhang, L.; Cai, C.L.; Li, B.Y.; Liu, Y. The Role of Tbx20 in Cardiovascular Development and Function. Front. Cell Dev. Biol. 2021, 9, 638542. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Ni, Y.; Thachil, V.; Morley, M.; Moravec, C.S.; Tang, W.H.W. Differential expression of members of SOX family of transcription factors in failing human hearts. Transl. Res. 2022, 242, 66–78. [Google Scholar] [CrossRef]
- Cheng, C.K.; Lin, X.; Pu, Y.; Tse, J.K.Y.; Wang, Y.; Zhang, C.-L.; Cao, X.; Lau, C.W.; Huang, J.; He, L.; et al. SOX4 is a novel phenotypic regulator of endothelial cells in atherosclerosis revealed by single-cell analysis. J. Adv. Res. 2023, 43, 187–203. [Google Scholar] [CrossRef]
- Boogerd, C.J.; Wong, L.Y.; van den Boogaard, M.; Bakker, M.L.; Tessadori, F.; Bakkers, J.; ‘t Hoen, P.A.; Moorman, A.F.; Christoffels, V.M.; Barnett, P. Sox4 mediates Tbx3 transcriptional regulation of the gap junction protein Cx43. Cell. Mol. Life Sci. 2011, 68, 3949–3961. [Google Scholar] [CrossRef]
- Britz-Cunningham, S.; Shah, M.; Zuppan, C.; Fletcher, W. Mutations of the connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N. Engl. J. Med. 1995, 332, 1323–1329. [Google Scholar] [CrossRef]
- Liu, N.; Schoch, K.; Luo, X.; Pena, L.D.M.; Bhavana, V.H.; Kukolich, M.K.; Stringer, S.; Powis, Z.; Radtke, K.; Mroske, C.; et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum. Mol. Genet. 2018, 27, 2454–2465. [Google Scholar] [CrossRef]
- Xie, H.; Zhang, E.; Hong, N.; Fu, Q.; Li, F.; Chen, S.; Yu, Y.; Sun, K. Identification of TBX2 and TBX3 variants in patients with conotruncal heart defects by target sequencing. Hum. Genom. 2018, 12, 44. [Google Scholar] [CrossRef]
- Chen, D.; Qiao, Y.; Meng, H.; Pang, S.; Huang, W.; Zhang, H.; Yan, B. Genetic analysis of the TBX3 gene promoter in ventricular septal defects. Gene 2013, 512, 185–188. [Google Scholar] [CrossRef]
- Li, Q.Y.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.; Yi, C.H.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Møller Nielsen, A.K.; Dehn, A.M.; Hjortdal, V.; Larsen, L.A. TBX5 variants and cardiac phenotype: A systematic review of the literature and a novel variant. Eur. J. Med. Genet. 2024, 68, 104920. [Google Scholar] [CrossRef] [PubMed]
- Penzo-Méndez, A.; Dy, P.; Pallavi, B.; Lefebvre, V. Generation of mice harboring a Sox4 conditional null allele. Genesis 2007, 45, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.H.; Harvey, R.P.; Wegner, M.; Sock, E. Cardiac outflow tract development relies on the complex function of Sox4 and Sox11 in multiple cell types. Cell. Mol. Life Sci. 2014, 71, 2931–2945. [Google Scholar] [CrossRef]
- Vasko, A.; Drivas, T.G.; Schrier Vergano, S.A. Genotype-Phenotype Correlations in 208 Individuals with Coffin-Siris Syndrome. Genes 2021, 12, 937. [Google Scholar] [CrossRef]
- Ghaffar, A.; Rasheed, F.; Rashid, M.; van Bokhoven, H.; Ahmed, Z.M.; Riazuddin, S.; Riazuddin, S. Biallelic in-frame deletion of SOX4 is associated with developmental delay, hypotonia and intellectual disability. Eur. J. Hum. Genet. 2022, 30, 243–247. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, F.; Dai, Q.; Dou, J.; Wu, Y.; Zhu, Y. Identification of a novel de novo mutation in SOX4 for syndromic tooth agenesis. Clin. Oral Investig. 2024, 28, 287. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Number or Mean | Percentage or Range |
|---|---|---|
| Demographic characteristics | ||
| Male probands | 131 | 52.82 |
| Female probands | 117 | 47.18 |
| Age at the time of enrollment (year) | 3.86 ± 2.51 | 0.25–7.17 |
| A positive family history of CHD | 51 | 20.56 |
| Distribution of various forms of CHD | ||
| VSD | 63 | 25.40 |
| ASD | 56 | 22.58 |
| PDA | 38 | 15.32 |
| DORV | 12 | 4.84 |
| TOF | 10 | 4.03 |
| PTA | 5 | 2.02 |
| TGA | 4 | 1.61 |
| HLV | 2 | 0.81 |
| AS | 2 | 0.81 |
| PS | 1 | 0.40 |
| APVC | 1 | 0.40 |
| SV | 1 | 0.40 |
| PCAC | 1 | 0.40 |
| VSD + PDA | 16 | 6.46 |
| VSD + ASD | 12 | 4.84 |
| DORV + VSD | 8 | 3.23 |
| ASD + PDA | 6 | 2.42 |
| TGA + VSD | 5 | 2.02 |
| TOF + ASD | 3 | 1.21 |
| PTA + VSD | 2 | 0.81 |
| Incidence of cardiac dysrhythmias | ||
| AVB | 12 | 4.84 |
| AF | 5 | 2.02 |
| Medical management | ||
| Catheter-based therapy | 139 | 56.05 |
| Surgical treatment | 87 | 35.08 |
| Follow-up observation | 22 | 8.87 |
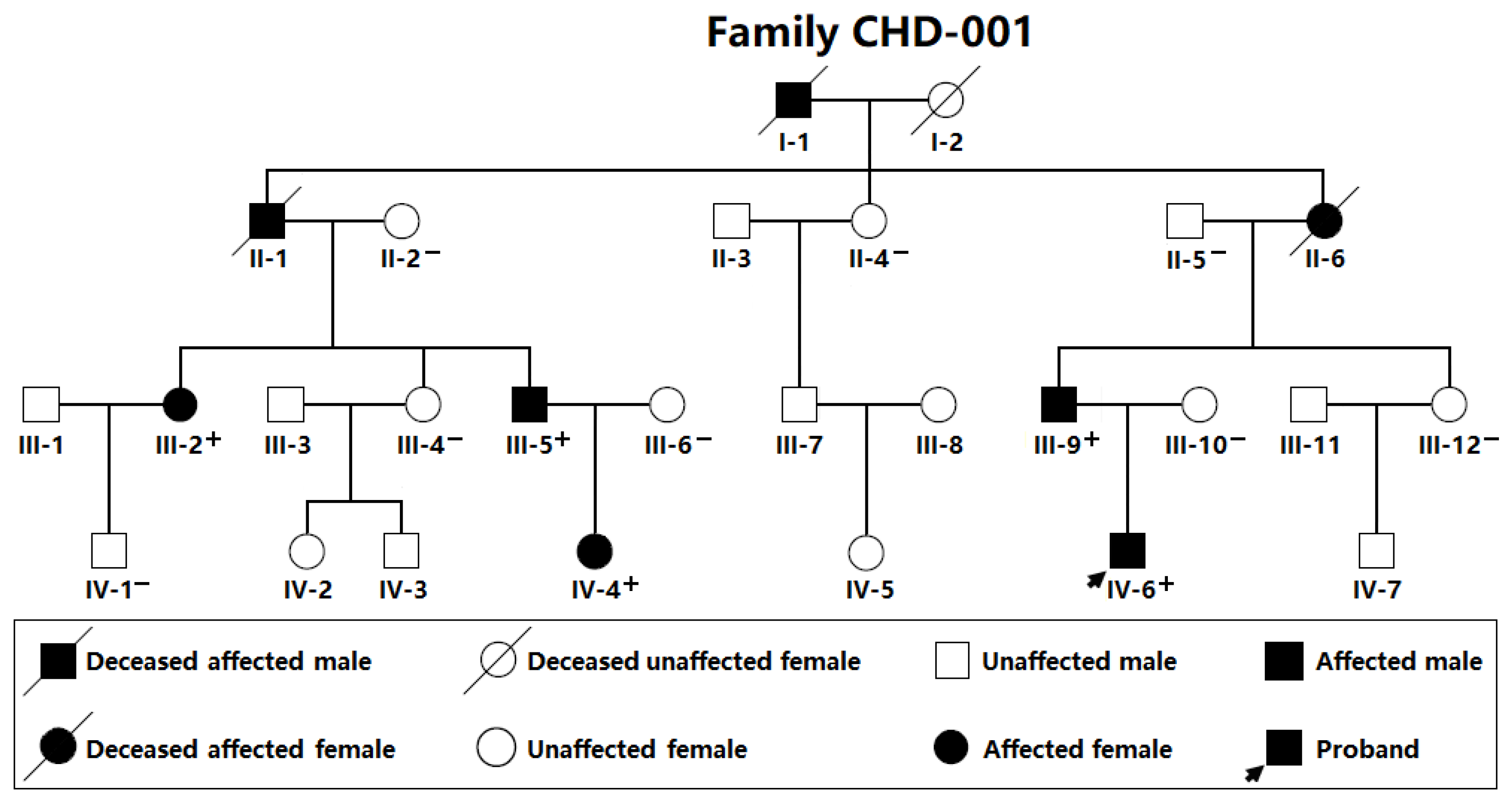
| Individual (Family CHD-001) | Sex | Age (Years) | Cardiovascular Structural Malformations |
|---|---|---|---|
| III-2 | Female | 31 | PDA |
| III-5 | Male | 27 | PDA, VSD |
| III-9 | Male | 29 | PDA |
| IV-4 | Female | 2 | PDA, VSD |
| IV-6 | Male | 4 | PDA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Dong, B.-B.; Li, Y.-J.; Yang, C.-X.; Xu, Y.-J.; Huang, R.-T.; Liu, X.-Y.; Yang, Y.-Q. Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin–Siris Syndromic Congenital Heart Disease. Children 2025, 12, 608. https://doi.org/10.3390/children12050608
Yan Z, Dong B-B, Li Y-J, Yang C-X, Xu Y-J, Huang R-T, Liu X-Y, Yang Y-Q. Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin–Siris Syndromic Congenital Heart Disease. Children. 2025; 12(5):608. https://doi.org/10.3390/children12050608
Chicago/Turabian StyleYan, Zi, Bin-Bin Dong, Yan-Jie Li, Chen-Xi Yang, Ying-Jia Xu, Ri-Tai Huang, Xing-Yuan Liu, and Yi-Qing Yang. 2025. "Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin–Siris Syndromic Congenital Heart Disease" Children 12, no. 5: 608. https://doi.org/10.3390/children12050608
APA StyleYan, Z., Dong, B.-B., Li, Y.-J., Yang, C.-X., Xu, Y.-J., Huang, R.-T., Liu, X.-Y., & Yang, Y.-Q. (2025). Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin–Siris Syndromic Congenital Heart Disease. Children, 12(5), 608. https://doi.org/10.3390/children12050608