

Prospective Monitoring of Lyso-Gb1 on DBS Sample in Three Children Recognized at Newborn Screening for Gaucher Disease and Untreated

 ,
,  ,
,  ,
, 

Abstract
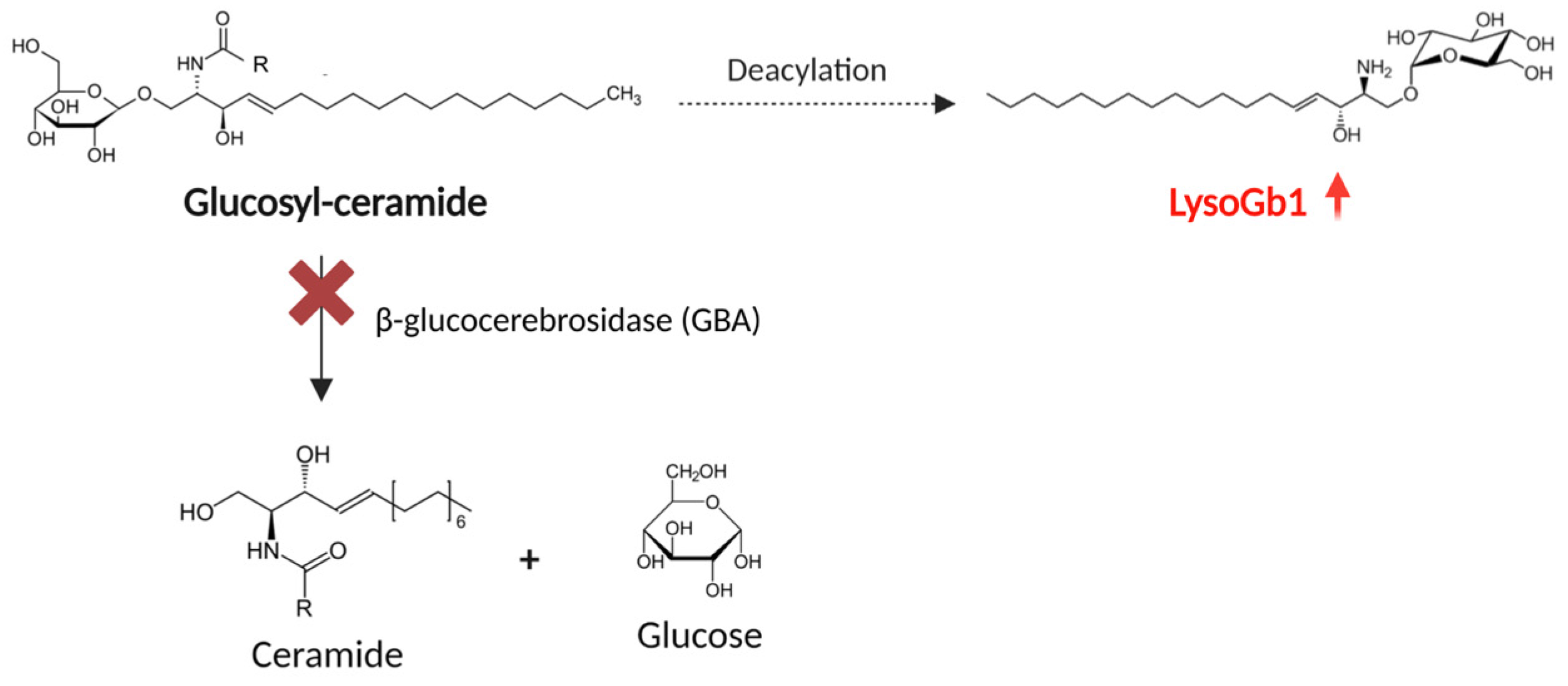
1. Introduction
2. Materials and Methods
2.1. Workflow for LSDs: First and Second-Tier Testing
2.2. Sanger Sequencing of GBA Gene
2.3. Follow-Up Protocol
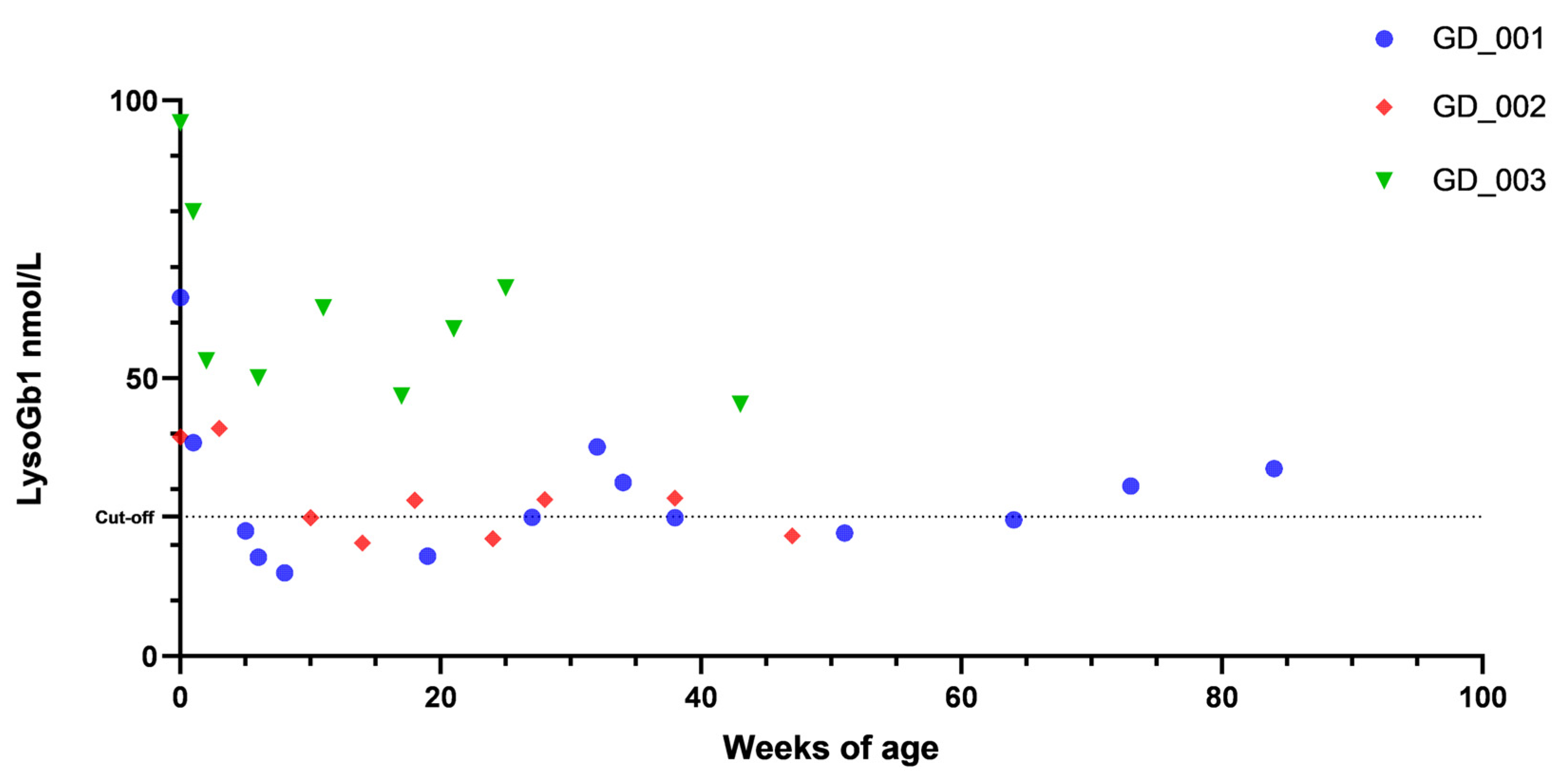
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef]
- Zimran, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Fernandez-Sasso, D.; Giraldo, P.; Goker-Alpan, O.; Lau, H.; Lukina, E.; et al. Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am. J. Hematol. 2018, 93, 205–212. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Curado, F.; Rösner, S.; Zielke, S.; Westphal, G.; Grittner, U.; Skrahina, V.; Alasel, M.; Malik, A.M.; Beetz, C.; Böttcher, T.; et al. Insights into the Value of Lyso-Gb1 as a Predictive Biomarker in Treatment-Naïve Patients with Gaucher Disease Type 1 in the LYSO-PROOF Study. Diagnostics 2023, 13, 2812. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. New perspectives in type 2 Gaucher disease. Adv. Pediatr. 1997, 44, 73–107. [Google Scholar] [CrossRef] [PubMed]
- Daykin, E.C.; Ryan, E.; Sidransky, E. Diagnosing neuronopathic Gaucher disease: New considerations and challenges in assigning Gaucher phenotypes. Mol. Genet. Metab. 2021, 132, 49–58. [Google Scholar] [CrossRef]
- Tylki-Szymańska, A.; Vellodi, A.; El-Beshlawy, A.; Cole, J.A.; Kolodny, E. Neuronopathic Gaucher disease: Demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J. Inherit. Metab. Dis. 2010, 33, 339–346. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Schiffmann, R.; Park, J.K.; Stubblefield, B.K.; Tayebi, N.; Sidransky, E. Phenotypic continuum in neuronopathic Gaucher disease: An intermediate phenotype between type 2 and type 3. J. Pediatr. 2003, 143, 273–276. [Google Scholar] [CrossRef]
- Mhatre, S.P.; Muranjan, M.; Gogtay, N.J. Economic Burden of Gaucher Disease at a Tertiary Care Public Hospital in Mumbai. Indian J. Pediatr. 2024, 91, 463–469. [Google Scholar] [CrossRef]
- Farahbakhshian, S.; Inocencio, T.J.; Poorman, G.; Wright, E.; Pathak, R.R.; Bullano, M. The budget impact of enzyme replacement therapy in type 1 Gaucher disease in the United States. J. Med. Econ. 2022, 25, 755–761. [Google Scholar] [CrossRef]
- Richardson, J.S.; Kemper, A.R.; Grosse, S.D.; Lam WK, K.; Rose, A.M.; Ahmad, A.; Gebremariam, A.; Prosser, L.A. Health and economic outcomes of for infantile-onset Pompe disease. Genet. Med. 2021, 23, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef]
- Rossi, C.; Ferrante, R.; Valentinuzzi, S.; Zucchelli, M.; Buccolini, C.; Di Rado, S.; Trotta, D.; Stuppia, L.; Federici, L.; Aricò, M. Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease. Biomedicines 2023, 11, 2672. [Google Scholar] [CrossRef] [PubMed]
- Gayed, M.M.; Jung, S.H.; Huggins, E.; Rodriguez-Rassi, E.; DeArmey, S.; Kishnani, P.S.; Stiles, A.R. Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease. Int. J. Mol. Sci. 2022, 23, 14938. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Al-Hertani, W.; Balwani, M.; Göker-Alpan, Ö.; Lau, H.A.; Wasserstein, M.; Weinreb, N.J.; Grabowski, G. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: Results from a Delphi consensus. Mol. Genet. Metab. 2022, 135, 154–162. [Google Scholar] [CrossRef]
- Ida, H.; Watanabe, Y.; Sagara, R.; Inoue, Y.; Fernandez, J. An observational study to investigate the relationship between plasma glucosylsphingosine (lyso-Gb1) concentration and treatment outcomes of patients with Gaucher disease in Japan. Orphanet J. Rare Dis. 2022, 17, 401. [Google Scholar] [CrossRef]
- Malinová, V.; Poupětová, H.; Řeboun, M.; Dvořáková, L.; Reichmannová, S.; Švandová, I.; Murgašová, L.; Kasper, D.C.; Magner, M. Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients. Int. J. Mol. Sci. 2023, 24, 14440. [Google Scholar] [CrossRef]
- Dinur, T.; Bauer, P.; Beetz, C.; Cozma, C.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Skrahina, V.; Zimran, A.; Revel-Vilk, S. Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease. Int. J. Mol. Sci. 2023, 24, 3945. [Google Scholar] [CrossRef]
- Viall, S. PO-450: “After 5+ years of for Gaucher, Fabry, Pompe and Mucopolysaccharidosis Type 1, does a “true positive” provide more questions or answers?” Abstracts. J. Inherit. Metab. Dis. 2024, 47, 1–477. [Google Scholar]
- Available online: https://franklin.genoox.com/clinical-db/variant/snp/chr1-155208334-G-A (accessed on 8 October 2024).
- Stiles, A.R.; Huggins, E.; Fierro, L.; Jung, S.H.; Balwani, M.; Kishnani, P.S. The role of glucosylsphingosine as an early indicator of disease progression in early symptomatic type 1 Gaucher disease. Mol. Genet. Metab. Rep. 2021, 27, 100729. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Second-Tier Test | Number of Positive | Recall Rate (%) | Genetic Confirmation | PPV % (Positive/Recalls) | |
|---|---|---|---|---|---|
| Gaucher disease | 255 | 3 | 0.022 | 3 | 100 |
| Newborn | GBA1 Genotype | GBA Activity (Cut-off > 3.89 μmol/L/h) |
|---|---|---|
| GD_001 | c.562C>T; p.Leu88Phe c.1226A>G; p.Asn409Ser | 0.26 |
| GD_002 | c.1226A>G; p.Asn409Ser c.1226A>G; p.Asn409Ser | 1.21 |
| GD_003 | c.1226A>G; p.Asn409Ser c.1226A>G; p.Asn409Ser | 0.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, C.; Trotta, D.; Ferrante, R.; Pieragostino, D.; Valentinuzzi, S.; Federici, L.; Stuppia, L.; De Laurenzi, V.; Aricò, M. Prospective Monitoring of Lyso-Gb1 on DBS Sample in Three Children Recognized at Newborn Screening for Gaucher Disease and Untreated. Children 2025, 12, 350. https://doi.org/10.3390/children12030350
Rossi C, Trotta D, Ferrante R, Pieragostino D, Valentinuzzi S, Federici L, Stuppia L, De Laurenzi V, Aricò M. Prospective Monitoring of Lyso-Gb1 on DBS Sample in Three Children Recognized at Newborn Screening for Gaucher Disease and Untreated. Children. 2025; 12(3):350. https://doi.org/10.3390/children12030350
Chicago/Turabian StyleRossi, Claudia, Daniela Trotta, Rossella Ferrante, Damiana Pieragostino, Silvia Valentinuzzi, Luca Federici, Liborio Stuppia, Vincenzo De Laurenzi, and Maurizio Aricò. 2025. "Prospective Monitoring of Lyso-Gb1 on DBS Sample in Three Children Recognized at Newborn Screening for Gaucher Disease and Untreated" Children 12, no. 3: 350. https://doi.org/10.3390/children12030350
APA StyleRossi, C., Trotta, D., Ferrante, R., Pieragostino, D., Valentinuzzi, S., Federici, L., Stuppia, L., De Laurenzi, V., & Aricò, M. (2025). Prospective Monitoring of Lyso-Gb1 on DBS Sample in Three Children Recognized at Newborn Screening for Gaucher Disease and Untreated. Children, 12(3), 350. https://doi.org/10.3390/children12030350