
Clinical Outcomes in Patients with Cystic Fibrosis Receiving CFTR Modulators: A Comparison of Childhood Versus Adolescent Initiation

 ,
,  , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Data Source
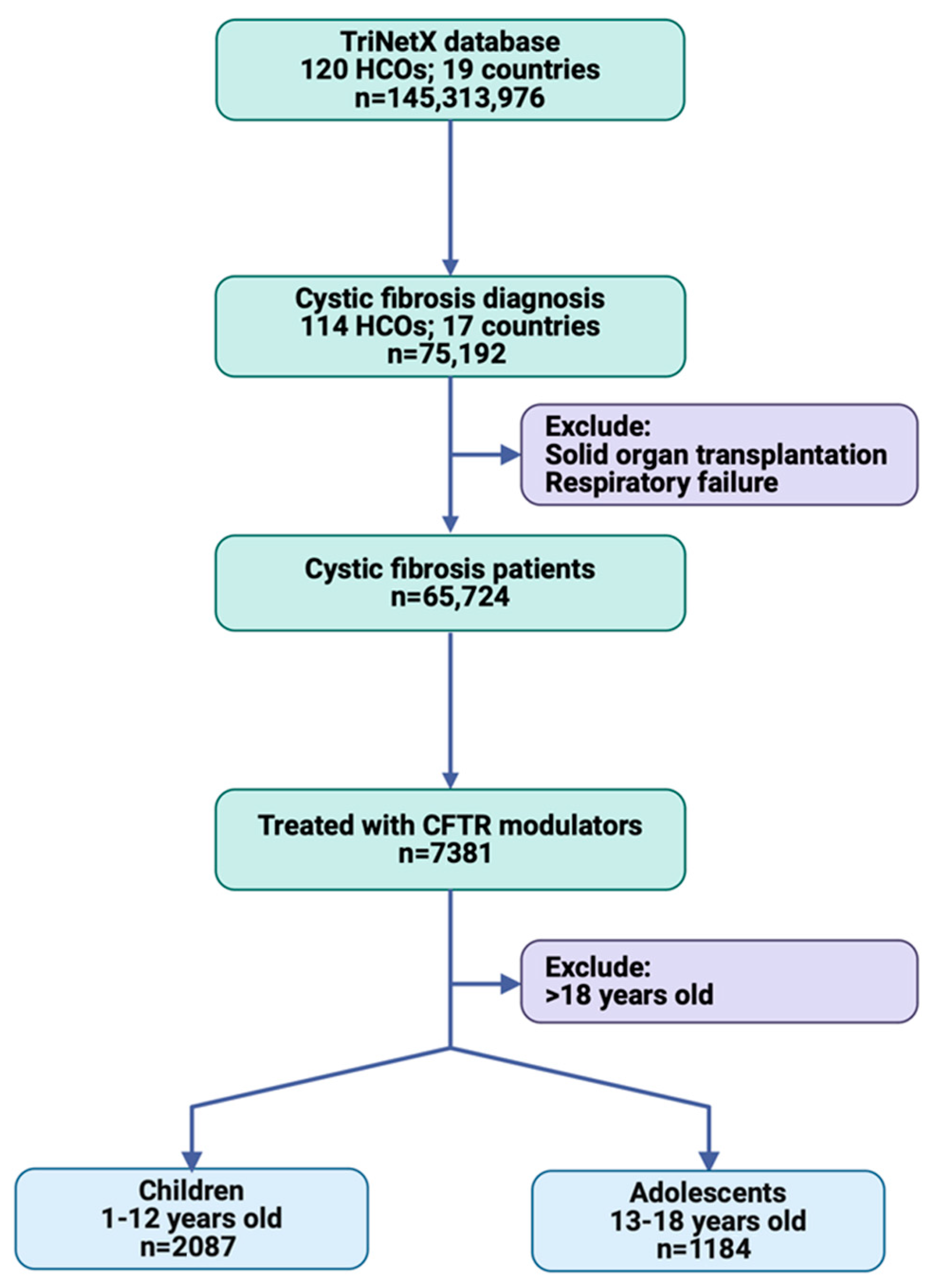
2.2. Study Population
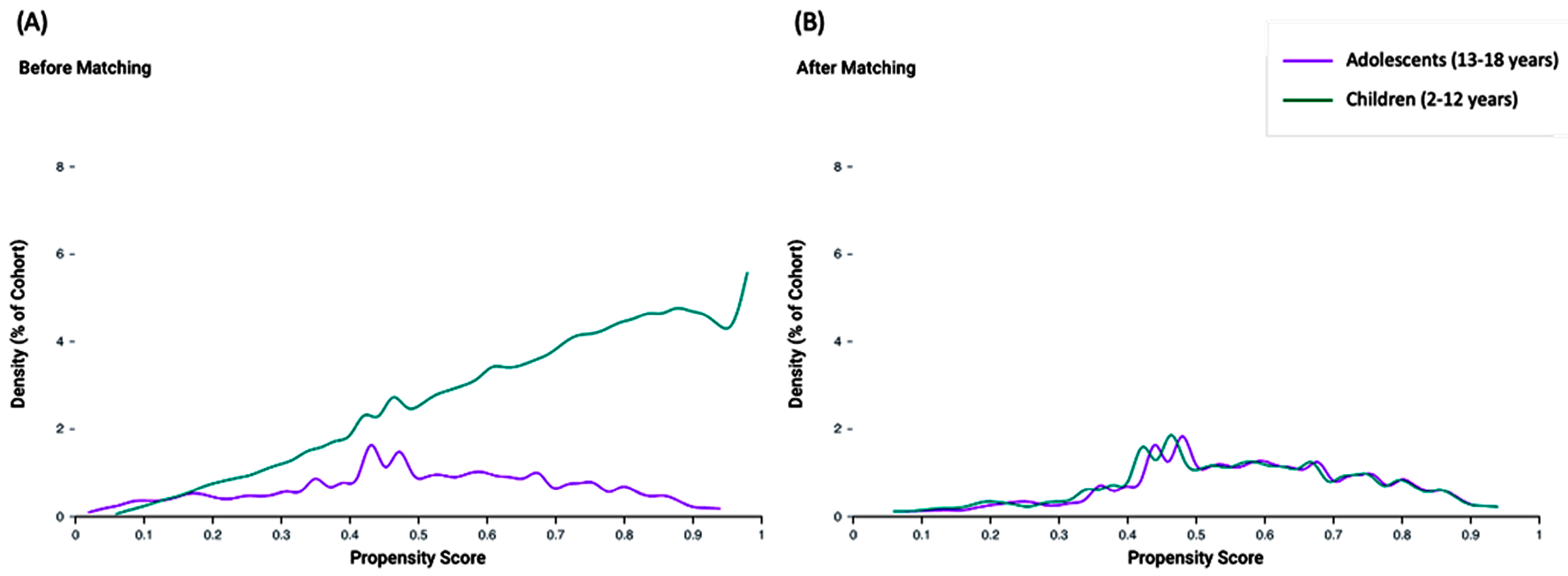
2.3. Propensity Score Matching
2.4. Outcomes
2.5. Statistical Analysis
3. Results
3.1. Study Population
3.2. Baseline Characteristics
3.3. Propensity Score Matching
3.4. Pulmonary Outcomes
3.5. Hospitalization Rate
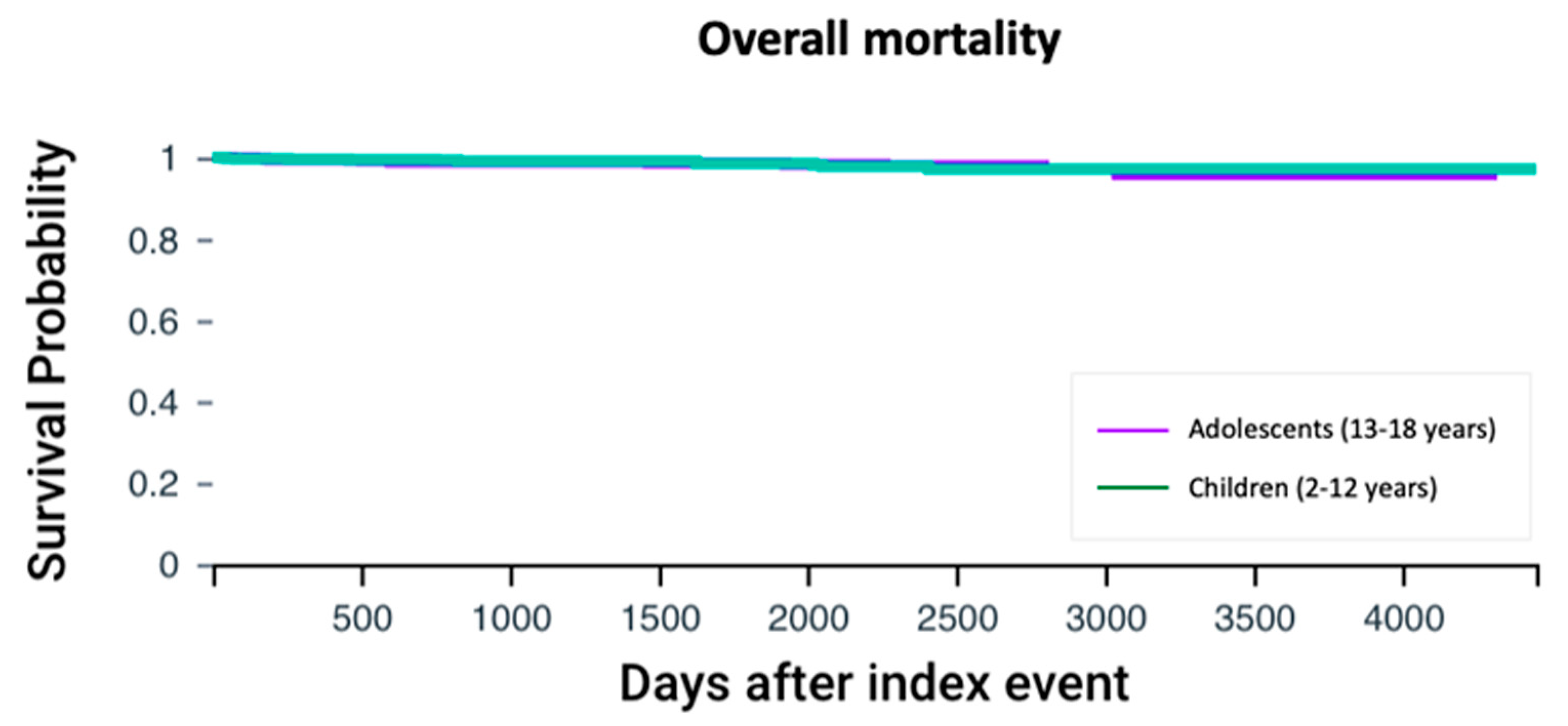
3.6. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zampoli, M.; Morrow, B.M.; Paul, G. Real-world disparities and ethical considerations with access to CFTR modulator drugs: Mind the gap! Front. Pharmacol. 2023, 14, 1163391. [Google Scholar] [CrossRef] [PubMed]
- King, J.A.; Nichols, A.-L.; Bentley, S.; Carr, S.B.; Davies, J.C. An Update on CFTR Modulators as New Therapies for Cystic Fibrosis. Pediatr. Drugs 2022, 24, 321–333. [Google Scholar] [CrossRef]
- Mariotti Zani, E.; Grandinetti, R.; Cunico, D.; Torelli, L.; Fainardi, V.; Pisi, G.; Esposito, S. Nutritional Care in Children with Cystic Fibrosis. Nutrients 2023, 15, 479. [Google Scholar] [CrossRef]
- Alenazi, S.A. Cystic fibrosis: Saudi arabia current situation and perspectives. Ann. Clin. Anal. Med. 2019, 10, 775–780. [Google Scholar]
- Hodson, M.; Bush, A.; Geddes, D. Cystic Fibrosis; StatPearls: St. Petersburg, FL, USA, 2012; NCBI Bookshelf. [Google Scholar]
- Khan, M.A.; Ali, Z.S.; Sweezey, N.; Grasemann, H.; Palaniyar, N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes 2019, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Sellers, Z.M. Pancreatic complications in children with cystic fibrosis. Curr. Opin. Pediatr. 2020, 32, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Turcios, N.L. Cystic Fibrosis Lung Disease: An Overview. Respir. Care 2020, 65, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.; Ramsey, B.W. Cystic Fibrosis: A Review. JAMA 2023, 329, 1859–1871. [Google Scholar] [CrossRef]
- Burgener, E.B.; Moss, R.B. Cystic fibrosis transmembrane conductance regulator modulators: Precision medicine in cystic fibrosis. Curr. Opin. Pediatr. 2018, 30, 372–377. [Google Scholar] [CrossRef]
- Dwight, M.; Marshall, B. CFTR modulators: Transformative therapies for cystic fibrosis. J. Manag. Care Spec. Pharm. 2021, 27, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Kuek, S.; McCullagh, A.; Paul, E.; Armstrong, D. Real world outcomes of CFTR modulator therapy in Australian adults and children. Pulm. Pharmacol. Ther. 2023, 82, 102247. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.J.; Nahata, M.C. Therapeutic management of cystic fibrosis. Clin. Pharm. 1985, 4, 555–565. [Google Scholar]
- Choong, E.; Sauty, A.; Koutsokera, A.; Blanchon, S.; André, P.; Decosterd, L. Therapeutic Drug Monitoring of Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor in Cystic Fibrosis: Where Are We Now? Pharmaceutics 2022, 14, 1674. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.; McColley, S.A.; McNally, P.; Powers, M.; Ratjen, F.; Rayment, J.H.; Retsch-Bogart, G.; Roesch, E.; Ahluwalia, N.; Chin, A.; et al. Long-Term Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged ≥ 6 Years with Cystic Fibrosis and at Least One F508del Allele: A Phase 3, Open-Label Clinical Trial. Am. J. Respir. Crit. Care Med. 2023, 208, 68–78. [Google Scholar] [CrossRef]
- Mall, M.A.; Brugha, R.; Gartner, S.; Legg, J.; Moeller, A.; Mondejar-Lopez, P.; Prais, D.; Pressler, T.; Ratjen, F.; Reix, P.; et al. Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 Through 11 Years of Age with Cystic Fibrosis Heterozygous for F508del and a Minimal Function Mutation: A Phase 3b, Randomized, Placebo-controlled Study. Am. J. Respir. Crit. Care Med. 2022, 206, 1361–1369. [Google Scholar] [CrossRef]
- Aoyama, B.C.; Mogayzel, P.J. Ivacaftor for the treatment of cystic fibrosis in children under six years of age. Expert Rev. Respir. Med. 2020, 14, 547–557. [Google Scholar] [CrossRef]
- Segal, T.Y. Adolescence: What the cystic fibrosis team needs to know. J. R. Soc. Med. 2008, 101, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar Jennifer, L.; Munck, A.; McKone Edward, F.; van der Ent Cornelis, K.; Moeller, A.; Simard, C.; Wang Linda, T.; Ingenito Edward, P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Middleton Peter, G.; Mall Marcus, A.; Dřevínek, P.; Lands Larry, C.; McKone Edward, F.; Polineni, D.; Ramsey Bonnie, W.; Taylor-Cousar Jennifer, L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Allen, L.; Allen, L.; Carr, S.B.; Davies, G.; Downey, D.; Egan, M.; Forton, J.T.; Gray, R.; Haworth, C.; Horsley, A.; et al. Future therapies for cystic fibrosis. Nat. Commun. 2023, 14, 693. [Google Scholar] [CrossRef] [PubMed]
- Muilwijk, D.; Bierlaagh, M.; van Mourik, P.; Kraaijkamp, J.; van der Meer, R.; van den Bor, R.; Heijerman, H.; Eijkemans, R.; Beekman, J.; van der Ent, K. Prediction of Real-World Long-Term Outcomes of People with CF Homozygous for the F508del Mutation Treated with CFTR Modulators. J. Pers. Med. 2021, 11, 1376. [Google Scholar] [CrossRef] [PubMed]
- Muilwijk, D.; Zomer-van Ommen, D.D.; Gulmans, V.A.M.; Eijkemans, M.J.C.; van der Ent, C.K. Long-term effectiveness of dual CFTR modulator treatment of cystic fibrosis. ERJ Open Res. 2022, 8, 00204–02022. [Google Scholar] [CrossRef]
- Kramer-Golinkoff, E.; Camacho, A.; Kramer, L.; Taylor-Cousar, J.L. A survey: Understanding the health and perspectives of people with CF not benefiting from CFTR modulators. Pediatr. Pulmonol. 2022, 57, 1253–1261. [Google Scholar] [CrossRef]
- van Gool, K.; Norman, R.; Delatycki, M.B.; Hall, J.; Massie, J. Understanding the costs of care for cystic fibrosis: An analysis by age and health state. Value Health 2013, 16, 345–355. [Google Scholar] [CrossRef]
- Marshall, L.Z.; Espinosa, R.; Starner, C.I.; Gleason, P.P. Real-world outcomes and direct care cost before and after elexacaftor/tezacaftor/ivacaftor initiation in commercially insured members with cystic fibrosis. J. Manag. Care Spec. Pharm. 2023, 29, 599–606. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Levels | Adolescents (n = 1184) | Children (n = 2087) | p-Value |
|---|---|---|---|---|
| Demographics | ||||
| Age at onset | Mean ± SD, years | 15.7 ± 1.8 | 7.0 ± 3.4 | <0.001 |
| Sex | Female | 607 (51.3%) | 989 (47.4%) | 0.033 |
| Male | 576 (48.6%) | 1096 (52.5%) | ||
| Race | White | 0.98 | 1791 (85.8%) | |
| Black | 26 (2.2%) | 82 (3.9%) | ||
| Other race | 27 (2.3%) | 58 (2.8%) | ||
| Ethnicity | Not Hispanic or Latino | 924 (78%) | 1745 (83.6%) | 0.76 |
| Hispanic or Latino | 79 (6.7%) | 145 (6.9%) | ||
| Clinical data | ||||
| Comorbidities | Diabetes mellitus | 219 (18.5%) | 83 (4%) | <0.001 |
| Diseases of liver | 135 (11.4%) | 136 (6.5%) | <0.001 | |
| Gastro-esophageal reflux disease | 299 (25.3%) | 741 (35.5%) | <0.001 | |
| Vitamin D deficiency | 287 (24.2%) | 463 (22.2%) | 0.18 | |
| Other vitamin deficiencies | 75 (6.3%) | 107 (5.1%) | 0.15 | |
| Presentation | Exocrine pancreatic insufficiency | 284 (24%) | 772 (37%) | <0.001 |
| Cystic fibrosis with pulmonary manifestations | 812 (68.6%) | 1447 (69.3%) | 0.65 | |
| Cystic fibrosis with intestinal manifestations | 509 (43%) | 1117 (53.5%) | <0.001 | |
| Cystic fibrosis with other manifestations | 493 (41.6%) | 908 (43.5%) | 0.30 | |
| Sodium chloride in sweat ≥ 60 mmol/L | 681 (57.5%) | 1368 (65.5%) | <0.001 | |
| Lab data | ||||
| Complete blood picture | Blood leukocytes, 103/uL | 8.7 ± 3.6 | 9.1 ± 3.4 | 0.014 |
| Lymphocytes/100 leukocytes, % | 29.5 ± 10.7 | 42.3 ± 14.8 | <0.001 | |
| Neutrophils, 103/uL | 142.1 ± 1019.1 | 89.4 ± 627.6 | 0.17 | |
| Eosinophils/100 leukocytes, % | 2.9 ± 2.8 | 2.6 ± 2.6 | 0.07 | |
| Glycemia | Hemoglobin A1c, % | 5.8 ± 1.5 | 5.5 ± 1.0 | <0.001 |
| Liver function test | Total bilirubin, mg/dL | 0.5 ± 0.3 | 0.3 ± 0.3 | <0.001 |
| Serum albumin, g/dL | 4.2 ± 0.5 | 4.3 ± 0.4 | <0.001 | |
| Aspartate aminotransferase, U/L | 28.5 ± 18.6 | 35.5 ± 15.6 | <0.001 | |
| Alanine aminotransferase, U/L | 31.7 ± 24.9 | 31.3 ± 20.9 | 0.69 | |
| Alkaline phosphatase, U/L | 183.3 ± 105.3 | 256.7 ± 85.5 | <0.001 | |
| Renal function test | Blood urea nitrogen, mg/dL | 12.8 ± 5.8 | 12.4 ± 3.9 | 0.09 |
| Serum creatinine, mg/dL | 0.7 ± 0.4 | 0.5 ± 3.0 | 0.24 | |
| Electrolytes | Sodium, mmol/L | 139.4 ± 2.4 | 139.0 ± 2.1 | 0.002 |
| Potassium, mmol/L | 4.2 ± 0.4 | 4.2 ± 0.5 | 0.05 | |
| Chloride, mmol/L | 103.9 ± 3.0 | 104.2 ± 5.5 | 0.25 | |
| Bicarbonate, mmol/L | 25.0 ± 2.8 | 23.4 ± 2.6 | <0.001 | |
| Inflammatory markers | C-reactive protein, mh/L | 15.6 ± 22.9 | 15.3 ± 30.1 | 0.92 |
| Erythrocyte sedimentation rate, mm/h | 17.8 ± 19.2 | 13.0 ± 15.4 | 0.005 | |
| Medications | ||||
| Respiratory medications | Antiasthma/bronchodilators | 847 (71.5%) | 1752 (83.9%) | <0.001 |
| Nasal anti-inflammatories | 630 (53.2%) | 1106 (53%) | 0.91 | |
| Adrenal corticosteroids | 537 (45.4%) | 1059 (50.7%) | 0.003 | |
| Nasal decongestants | 115 (9.7%) | 212 (10.2%) | 0.68 | |
| Nasal antihistamines | 48 (4.1%) | 64 (3.1%) | 0.14 | |
| Antitussives/expectorants | 59 (5%) | 42 (2%) | <0.001 | |
| Mucolytics | 34 (2.9%) | 70 (3.4%) | 0.45 | |
| Digestive drugs | Digestants | 696 (58.8%) | 1506 (72.2%) | <0.001 |
| Nutritional supplements | Therapeutic nutrients/minerals/electrolytes | 715 (60.4%) | 1431 (68.6%) | <0.001 |
| Vitamins | 571 (48.2%) | 1043 (50%) | 0.34 |
| Characteristics | Levels | Adolescents (n = 946) | Children (n = 946) | p-Value |
|---|---|---|---|---|
| Demographics | ||||
| Sex | Female | 462 (48.8%) | 487 (51.5%) | 0.25 |
| Male | 483 (51.1%) | 458 (48.4%) | ||
| Race | White | 809 (85.5%) | 815 (86.2%) | 0.69 |
| Black | 26 (2.7%) | 22 (2.3%) | ||
| Other race | 22 (2.3%) | 28 (3%) | ||
| Ethnicity | Not Hispanic or Latino | 732 (77.4%) | 755 (79.8%) | 0.19 |
| Hispanic or Latino | 60 (6.3%) | 81 (8.6%) | ||
| Clinical data | ||||
| Comorbidities | Diabetes mellitus | 72 (7.6%) | 76 (8%) | 0.73 |
| Diseases of liver | 85 (9%) | 84 (8.9%) | 0.94 | |
| Gastro-esophageal reflux disease | 228 (24.1%) | 238 (25.2%) | 0.59 | |
| Vitamin D deficiency | 208 (22%) | 200 (21.1%) | 0.66 | |
| Other vitamin deficiencies | 54 (5.7%) | 42 (4.4%) | 0.21 | |
| Presentation | Exocrine pancreatic insufficiency | 242 (25.6%) | 212 (22.4%) | 0.11 |
| Cystic fibrosis with pulmonary manifestations | 620 (65.5%) | 631 (66.7%) | 0.59 | |
| Cystic fibrosis with intestinal manifestations | 404 (42.7%) | 425 (44.9%) | 0.33 | |
| Cystic fibrosis with other manifestations | 352 (37.2%) | 374 (39.5%) | 0.30 | |
| Sodium chloride in sweat ≥ 60 mmol/L | 539 (57%) | 563 (59.5%) | 0.26 | |
| Medications | ||||
| Respiratory medications | Antiasthma/bronchodilators | 687 (72.6%) | 696 (73.6%) | 0.64 |
| Nasal anti-inflammatories | 491 (51.9%) | 511 (54%) | 0.36 | |
| Adrenal corticosteroids | 422 (44.6%) | 439 (46.4%) | 0.43 | |
| Nasal decongestants | 83 (8.8%) | 90 (9.5%) | 0.58 | |
| Nasal antihistamines | 41 (4.3%) | 33 (3.5%) | 0.34 | |
| Antitussives/expectorants | 35 (3.7%) | 31 (3.3%) | 0.62 | |
| Mucolytics | 25 (2.6%) | 26 (2.7%) | 0.89 | |
| Digestive drugs | Digestants | 546 (57.7%) | 599 (63.3%) | 0.13 |
| Nutritional supplements | Therapeutic nutrients/minerals/electrolytes | 566 (59.8%) | 586 (61.9%) | 0.35 |
| Vitamins | 437 (46.2%) | 420 (44.4%) | 0.43 |
| Outcomes | Adolescents (n = 946) | Children (n = 946) | p-Value |
|---|---|---|---|
| Lung transplantation | 0 | 0 | NA |
| Respiratory failure | 36 (3.81%) | 10 (1.06%) | <0.001 |
| Respiratory infections | 593 (62.7%) | 544 (57.5%) | 0.021 |
| Hospitalization rate | 280 (29.6%) | 192 (20.3%) | <0.001 |
| Hospital visits per person | 8.8 ± 23.9 (median = 3) | 3.77 ± 8.1 (median = 2) | 0.004 |
| Hospital length of stay, days | 32.7 ± 55.5 (median = 16) | 20.4 ± 36.9 (median = 10) | 0.006 |
| All-cause mortality | 15 (1.58%) | 12 (1.26%) | 0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toraih, E.A.; Malik, H.A.; Islam, R.K.; Pirzadah, H.A.; Abdelmaksoud, A.; Elshazli, R.M.; Boasiako, P.A.; Alenazi, S.A.; Dabel, A.; Jishu, J.A.; et al. Clinical Outcomes in Patients with Cystic Fibrosis Receiving CFTR Modulators: A Comparison of Childhood Versus Adolescent Initiation. Children 2025, 12, 157. https://doi.org/10.3390/children12020157
Toraih EA, Malik HA, Islam RK, Pirzadah HA, Abdelmaksoud A, Elshazli RM, Boasiako PA, Alenazi SA, Dabel A, Jishu JA, et al. Clinical Outcomes in Patients with Cystic Fibrosis Receiving CFTR Modulators: A Comparison of Childhood Versus Adolescent Initiation. Children. 2025; 12(2):157. https://doi.org/10.3390/children12020157
Chicago/Turabian StyleToraih, Eman A., Hassan A. Malik, Rahib K. Islam, Humza A. Pirzadah, Ahmed Abdelmaksoud, Rami M. Elshazli, Paul Antwi Boasiako, Shehab Ahmed Alenazi, Angelique Dabel, Jessan A. Jishu, and et al. 2025. "Clinical Outcomes in Patients with Cystic Fibrosis Receiving CFTR Modulators: A Comparison of Childhood Versus Adolescent Initiation" Children 12, no. 2: 157. https://doi.org/10.3390/children12020157
APA StyleToraih, E. A., Malik, H. A., Islam, R. K., Pirzadah, H. A., Abdelmaksoud, A., Elshazli, R. M., Boasiako, P. A., Alenazi, S. A., Dabel, A., Jishu, J. A., Alenezi, B. T., Aiash, H., & Fawzy, M. S. (2025). Clinical Outcomes in Patients with Cystic Fibrosis Receiving CFTR Modulators: A Comparison of Childhood Versus Adolescent Initiation. Children, 12(2), 157. https://doi.org/10.3390/children12020157