
Prenatal Features of MIRAGE Syndrome—Case Report and Review of the Literature

 ,
,  , ,
, , 
Abstract
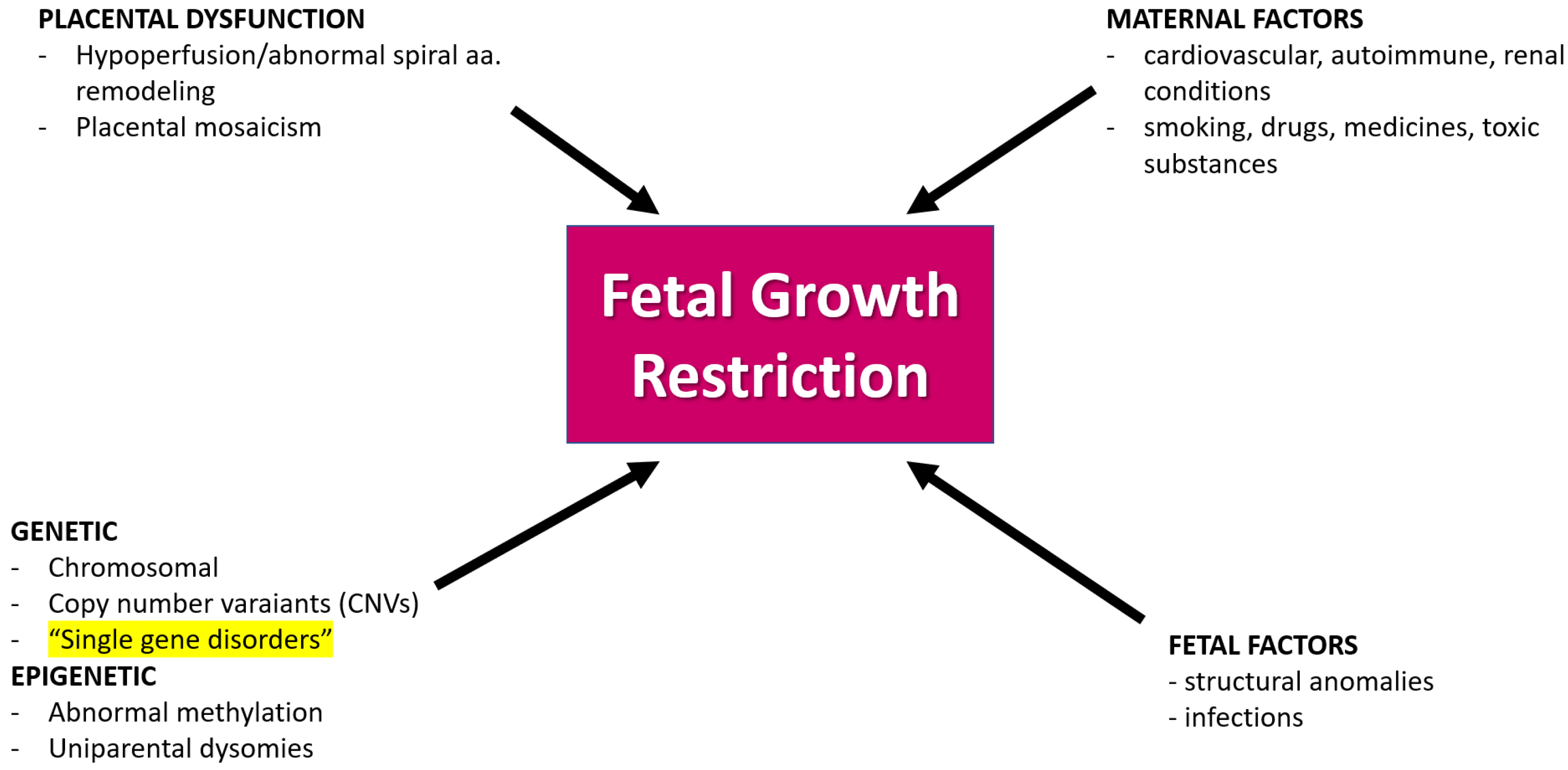
1. Introduction
- (1)
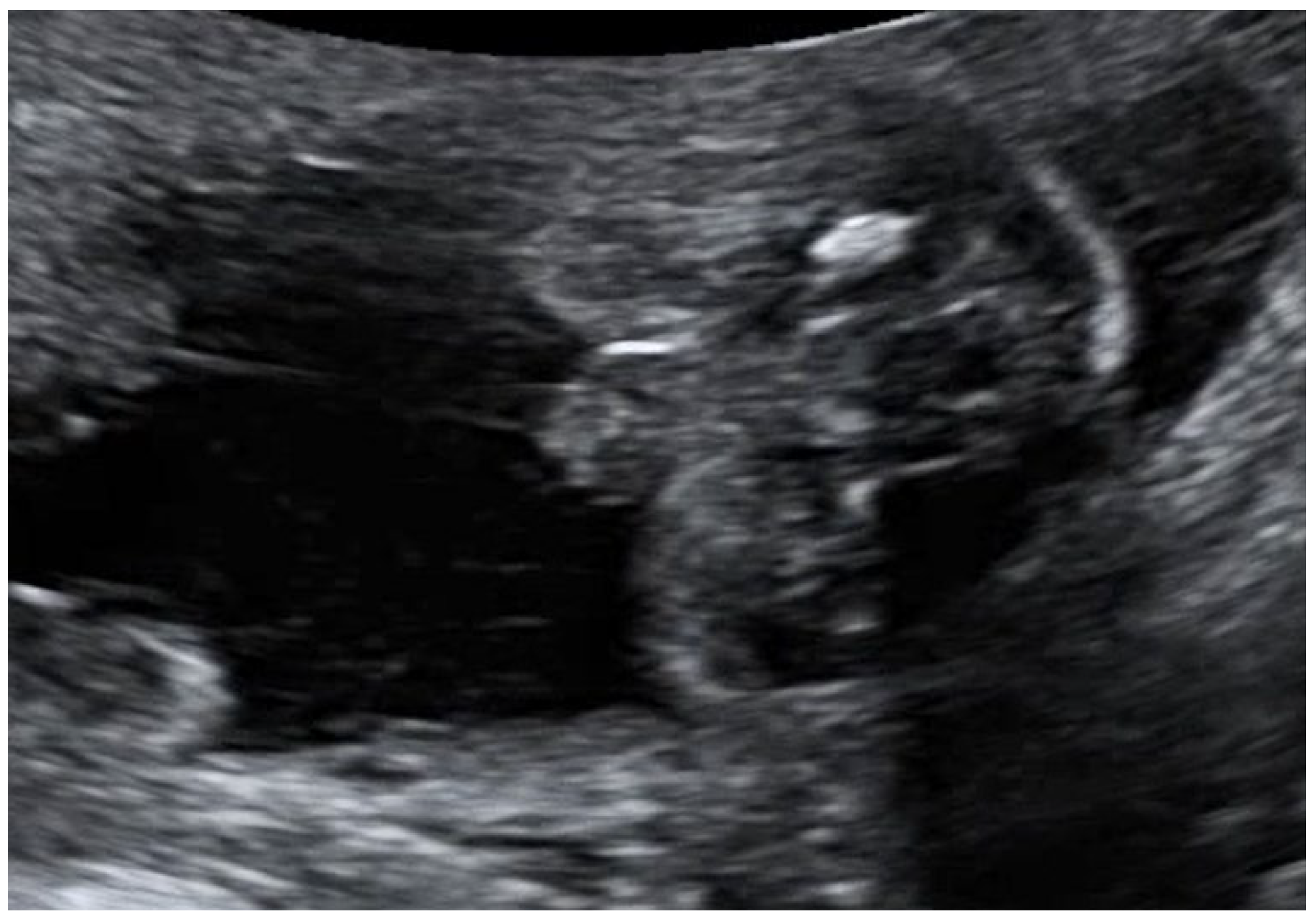
- To present prenatal features in a case of MIRAGE syndrome diagnosed prenatally in a fetus with severe growth restriction, ambiguous genitalia, hyperechogenic bowel, and oligohydramnios at the mid-gestation routine anomaly scan, at 22 weeks of pregnancy.
- (2)
- To review the literature in search of reports on prenatal features of MIRAGE syndrome.
2. Materials and Methods
3. Results
3.1. Case Presentation
3.2. Literature Review
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buonocore, F.; McGlacken-Byrne, S.M.; Del Valle, I.; Achermann, J.C. Current Insights into Adrenal Insufficiency in the Newborn and Young Infant. Front. Pediatr. 2020, 8, 619041. [Google Scholar] [CrossRef] [PubMed]
- Tanase-Nakao, K.; Olson, T.S.; Narumi, S. MIRAGE Syndrome. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, F.; Kühnen, P.; Suntharalingham, J.P.; Del Valle, I.; Digweed, M.; Stachelscheid, H.; Khajavi, N.; Didi, M.; Brady, A.F.; Blankenstein, O.; et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J. Clin. Investig. 2017, 127, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, K.H. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat. Diagn. 2011, 31, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Veduta, A.; Vayna, A.M.; Duta, S.; Panaitescu, A.; Popescu, F.; Bari, M.; Peltecu, G.; Nedelea, F. The first trimester combined test for aneuploidies—A single center experience. J. Matern. Fetal Neonatal Med. 2018, 31, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Salomon, L.J.; Alfirevic, Z.; Da Silva Costa, F.; Deter, R.L.; Figueras, F.; Ghi, T.; Glanc, P.; Khalil, A.; Lee, W.; Napolitano, R.; et al. ISUOG Practice Guidelines: Ultrasound assessment of fetal biometry and growth. Ultrasound Obstet. Gynecol. 2019, 53, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Hadlock, F.P.; Harrist, R.B.; Sharman, R.S.; Deter, R.L.; Park, S.K. Estimation of fetal weight with the use of head, body, and femur measurements—A prospective study. Am. J. Obstet. Gynecol. 1985, 151, 333–337. [Google Scholar] [CrossRef]
- Ciobanu, A.; Rouvali, A.; Syngelaki, A.; Akolekar, R.; Nicolaides, K.H. Prediction of small for gestational age neonates: Screening by maternal factors, fetal biometry, and biomarkers at 35–37 weeks’ gestation. Am. J. Obstet. Gynecol. 2019, 220, 486-e1. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, X.C.; Lv, G.J.; Liu, J.H.; Sun, C.; Mu, K. Amniotic fluid karyotype analysis and prenatal diagnosis strategy of 3117 pregnant women with amniocentesis indication. J. Comp. Eff. Res. 2023, 12, e220168. [Google Scholar] [CrossRef]
- Cigăran, R.G.; Botezatu, R.; Mînecan, E.M.; Gică, C.; Panaitescu, A.M.; Peltecu, G.; Gică, N. The Psychological Impact of the COVID-19 Pandemic on Pregnant Women. Healthcare 2021, 9, 725. [Google Scholar] [CrossRef]
- Lemos de Matos, A.; Liu, J.; McFadden, G.; Esteves, P.J. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC Evol. Biol. 2013, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingham, J.P.; Ishida, M.; Del Valle, I.; Stalman, S.E.; Solanky, N.; Wakeling, E.; Moore, G.E.; Achermann, J.C.; Buonocore, F. Emerging phenotypes linked to variants in SAMD9 and MIRAGE syndrome. Front. Endocrinol. 2022, 13, 953707. [Google Scholar] [CrossRef] [PubMed]
- Go, A.; Lee, B.H.; Choi, J.H.; Jeong, J.; Jung, E.; Lee, B.S. Case report: A premature infant with severe intrauterine growth restriction, adrenal insufficiency, and inflammatory diarrhea: A genetically confirmed case of MIRAGE syndrome. Front. Endocrinol. 2023, 14, 1242387. [Google Scholar] [CrossRef] [PubMed]
- Onuma, S.; Wada, T.; Araki, R.; Wada, K.; Tanase-Nakao, K.; Narumi, S.; Fukui, M.; Shoji, Y.; Etani, Y.; Ida, S.; et al. MIRAGE syndrome caused by a novel missense variant (p.Ala1479Ser) in the SAMD9 gene. Hum. Genome Var. 2020, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, K.; Hachiya, R.; Tomobe, Y.; Kaku, U.; Akiba, K.; Shima, H.; Narumi, S.; Hasegawa, Y. MIRAGE syndrome with recurrent pneumonia probably associated with gastroesophageal reflux and achalasia: A case report. Clin. Pediatr. Endocrinol. 2019, 28, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Janjua, D.; Shankar, S.; AlMaazmi, M.; Jadhav, D.V. MIRAGE Syndrome Enteropathy Responding to Pancrelipase Despite Normal Pancreatic Fecal Elastase: A Case Report. Am. J. Case Rep. 2022, 23, e937057-1. [Google Scholar] [CrossRef]
- Roucher-Boulez, F.; Mallet, D.; Chatron, N.; Dijoud, F.; Gorduza, D.B.; Bretones, P.; Morel, Y. Reversion SAMD9 Mutations Modifying Phenotypic Expression of MIRAGE Syndrome and Allowing Inheritance in a Usually De Novo Disorder. Front. Endocrinol. 2019, 10, 625. [Google Scholar] [CrossRef]
- Mengen, E.; Küçükçongar Yavaş, A.; Uçaktürk, S.A. A Rare Etiology of 46,XY Disorder of Sex Development and Adrenal Insufficiency: A Case of MIRAGE Syndrome Caused by Mutations in the SAMD9 Gene. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 206. [Google Scholar] [CrossRef]
- Baquedano-Lobera, I.; Romero-Salas, Y.; Ros-Arnal, I.; Miramar-Gallart, M.D.; López-Pisón, J.; Corona-Bellostas, C.; García-Romero, R. Achalasia as a symptom guide in MIRAGE syndrome: A novel case with p.R1293Q and p.R902W variants in the SAMD9 gene. Clin. Genet. 2021, 99, 740–741. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Zhang, V.W.; Zhang, C.; Ding, H.; Yin, A. Mutations in both SAMD9 and SLC19A2 genes caused complex phenotypes characterized by recurrent infection, dysphagia and profound deafness—A case report for dual diagnosis. BMC Pediatr. 2019, 19, 364. [Google Scholar] [CrossRef]
- Perisa, M.P.; Rose, M.J.; Varga, E.; Kamboj, M.K.; Spencer, J.D.; Bajwa, R.P.S. A novel SAMD9 variant identified in patient with MIRAGE syndrome: Further defining syndromic phenotype and review of previous cases. Pediatr. Blood Cancer 2019, 66, e27726. [Google Scholar] [CrossRef] [PubMed]
- Panaitescu, A.M.; Nicolaides, K. Maternal autoimmune disorders and fetal defects. J. Matern. Fetal Neonatal Med. 2018, 31, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Meler, E.; Sisterna, S.; Borrell, A. Genetic syndromes associated with isolated fetal growth restriction. Prenat. Diagn. 2020, 40, 432–446. [Google Scholar] [CrossRef]
- Pauta, M.; Martinez-Portilla, R.J.; Meler, E.; Otaño, J.; Borrell, A. Diagnostic yield of exome sequencing in isolated fetal growth restriction: Systematic review and meta-analysis. Prenat. Diagn. 2023, 43, 596–604. [Google Scholar] [CrossRef]
- Vena, F.; Mazza, A.; Bartolone, M.; Vasta, A.; D’Alberti, E.; Di Mascio, D.; D’Ambrosio, V.; Volpe, G.; Signore, F.; Pizzuti, A.; et al. Hyperechogenic fetal bowel: Current evidence-based prenatal diagnosis and management. J. Clin. Ultrasound 2023, 51, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Rabie, N.; Magann, E.; Steelman, S.; Ounpraseuth, S. Oligohydramnios in complicated and uncomplicated pregnancy: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 49, 442–449. [Google Scholar] [CrossRef] [PubMed]
- van Bever, Y.; Groenenberg, I.A.L.; Knapen, M.F.C.M.; Dessens, A.B.; Hannema, S.E.; Wolffenbuttel, K.P.; Diderich, K.E.M.; Hoefsloot, L.H.; Srebniak, M.I.; Bruggenwirth, H.T. Prenatal ultrasound finding of atypical genitalia: Counseling, genetic testing and outcomes. Prenat. Diagn. 2023, 43, 162–182. [Google Scholar] [CrossRef]
- Reyes, A.P.; León, N.Y.; Frost, E.R.; Harley, V.R. Genetic control of typical and atypical sex development. Nat. Rev. Urol. 2023, 20, 434–451. [Google Scholar] [CrossRef]
- Alimussina, M.; Diver, L.A.; McGowan, R.; Ahmed, S.F. Genetic testing of XY newborns with a suspected disorder of sex development. Curr. Opin. Pediatr. 2018, 30, 548–557. [Google Scholar] [CrossRef]
- Leitao Braga, B.; Lisboa Gomes, N.; Nishi, M.Y.; Freire, B.L.; Batista, R.L.; DFaria Junior, J.A.; Funari, M.F.A.; Figueredo Benedetti, A.F.; de Moraes Narcizo, A.; Cavalca Cardoso, L.; et al. Variants in 46,XY DSD-Related Genes in Syndromic and Non-Syndromic Small for Gestational Age Children with Hypospadias. Sex. Dev. 2022, 16, 27–33. [Google Scholar] [CrossRef]
- Shima, H.; Hayashi, M.; Tachibana, T.; Oshiro, M.; Amano, N.; Ishii, T.; Haruna, H.; Igarashi, M.; Kon, M.; Fukuzawa, R.; et al. MIRAGE syndrome is a rare cause of 46,XY DSD born SGA without adrenal insufficiency. PLoS ONE 2018, 13, e0206184. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Koehler, K.; Nomura, Y.; Sugimoto, K.; Satoh, A.; Ogata, T.; Fukami, M.; Jühlen, R.; Schuelke, M.; Mohnike, K.; et al. Two patients with MIRAGE syndrome lacking haematological features: Role of somatic second-site reversion SAMD9 mutations. J. Med. Genet. 2018, 55, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, L.; Shima, H.; Ji, W.; Panisello-Manterola, D.; McGrath, J.; Bird, L.M.; Konstantino, M.; Narumi, S.; Lakhani, S. A novel SAMD9 mutation causing MIRAGE syndrome: An expansion and review of phenotype, dysmorphology, and natural history. Am. J. Med. Genet. A 2018, 176, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.R.; Wang, S.; Ma, J.; Lamprecht, T.; Walsh, M.; Song, G.; Raimondi, S.C.; Wu, G.; Walsh, M.F.; McGee, R.B.; et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 2017, 31, 1827–1830. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author, Year | GA at Delivery, Indication | Prenatal Features | Postnatal Features |
|---|---|---|---|
| Go et al., 2023 [14] | 29 weeks + 6 days Iatrogenic—CS for fetal distress and oligohydramnios | FGR Lung hypoplasia Pericardial effusion Bilateral renal hypoplasia | Hyperpigmentation Adrenal insufficiency Dysplastic Kidneys Normal genitalia Transient thrombocytopenia Severe developmental delay at 5–6 months |
| Onuma et al., 2020 [15] | 31 weeks Iatrogenic—CS for fetal distress | FGR | Micropenis Hypospadias Bifid scrotum Hyperpigmentation Adrenal insufficiency Bowel dysfunction Transient thrombocytopenia |
| Yoshizaki et al., 2019 [16] | 32 weeks + 2 days Iatrogenic—CS fetal distress | FGR | Hyperpigmentation Adrenal insufficiency Transient thrombocytopenia Normal genitalia |
| Janjua et al., 2022 [17] | 34 weeks + 5 days Iatrogenic—CS | FGR Right hydroureter Female external genitalia NIPT male genotype. | Prominent clitoris Small vaginal opening Small lumps in the groin No fusion of the labia Normal appearance of the lower vagina ending blindly with no visible cervix Normal female urethra No ovaries |
| Roucher-Boulez et al., 2019 [18] | 36 weeks + 5 days Iatrogenic—CS for fetal distress | FGR | Very small, inguinal palpable testesUrogenital sinus Blind-ending vagina Genital tubercle had the appearance of a normal clitoris The karyotype was 46, XY Transient thrombocytopenia |
| Mengen et al., 2020 [19] | 31 weeks Iatrogenic—CS for fetal distress and FGR | FGR | Micropenis Transient thrombocytopenia Adrenal insufficiency due to bilateral adrenal hypoplasia Bowel dysfunction |
| Baquedano-Lobera et al., 2021 [20] | 31 weeks | FGR | |
| Zhang et al., 2019 [21] | 31 weeks | FGR | Hyperpigmentation Dysmorphic features Hypoplastic genitalia Visible penis, no visible testis and scrotum. Left testicle in the inguinal canal, right testicle in the right lower pelvic cavity Undetected bilateral epididymis No solid mass found in bilateral scrotum |
| Perisa et al. (2019) [22] | 32 weeks CS—Oligohydramnios and FGR | FGR | Microcephaly Cryptorchidism Hypospadias Unilateral vesicoureteral reflux Mild facial dysmorphism Brachydactyly of bilateral fifth digits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panaitescu, A.M.; Huluță, I.; Gorecki, G.-P.; Cima, L.N.; Voiculescu, V.M.; Nedelea, F.M.; Gică, N. Prenatal Features of MIRAGE Syndrome—Case Report and Review of the Literature. Children 2024, 11, 310. https://doi.org/10.3390/children11030310
Panaitescu AM, Huluță I, Gorecki G-P, Cima LN, Voiculescu VM, Nedelea FM, Gică N. Prenatal Features of MIRAGE Syndrome—Case Report and Review of the Literature. Children. 2024; 11(3):310. https://doi.org/10.3390/children11030310
Chicago/Turabian StylePanaitescu, Anca Maria, Iulia Huluță, Gabriel-Petre Gorecki, Luminita Nicoleta Cima, Vlad M. Voiculescu, Florina Mihaela Nedelea, and Nicolae Gică. 2024. "Prenatal Features of MIRAGE Syndrome—Case Report and Review of the Literature" Children 11, no. 3: 310. https://doi.org/10.3390/children11030310
APA StylePanaitescu, A. M., Huluță, I., Gorecki, G.-P., Cima, L. N., Voiculescu, V. M., Nedelea, F. M., & Gică, N. (2024). Prenatal Features of MIRAGE Syndrome—Case Report and Review of the Literature. Children, 11(3), 310. https://doi.org/10.3390/children11030310