
Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles

 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Patients
- (i)
- Simple heterozygosis: subjects with a mutation in a single allele of the GJB2 gene.
- (ii)
- Compound heterozygosis: patients who had two different mutations in the respective alleles of the same gene (in the case of this study, GJB2).
- (iii)
- Homozygosis: patients who had the same mutation in both alleles of the same gene (in the case of this study, GJB2).
- (iv)
- Digenic: patient who had a mutation in both GJB2 and GJB6.
2.2. Audiological Investigations
- (i)
- From birth to 3 months of age: Auditory Brainstem Response (ABR), Transient Evoked Otoacoustic Emissions (TEOAE), Distortion Product OAEs (DPOAE), and Tympanometry and Acoustic Reflex (impedancemetry).
- (ii)
- From 3 to 8 months of age: BOA (Behavioral Observation Audiometry) and ABR/OAEs according to collaboration.
- (iii)
- From 8 to 24 months of age: Visual Reinforcement Audiometry (VRA).
- (iv)
- From 2 to 3 years of age: Play Audiometry and Peep Show.
2.3. Genetic Investigations
2.4. Statistical Analysis
3. Results
3.1. General Clinical Outcomes
3.2. Association between Genotype and Clinical Features
4. Discussion
4.1. Genotype-to-Phenotype Association in GJB2 and GJB6 Genes Mutations
4.2. Open Issues in Congenital Hearing Loss Genetics
4.3. Limitations and Strengths of This Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 54, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Walker, E.; Dave, I.; Dedhia, K. Genetic testing for congenital non-syndromic sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2019, 124, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Shearer, A.E.; Hildebrand, M.S.; Van Camp, G. Deafness and Hereditary Hearing Loss Overview. In GeneReviews®; University of Washington: Seattle WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 3 May 2016).
- Kenneson, A.; Van Naarden Braun, K.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Nickel, R.; Forge, A. Gap junctions and connexins in the inner ear: Their roles in homeostasis and deafness. Curr. Opin. Otolaryngol. Head Neck Surg. 2008, 16, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Adams, J.C.; Paul, D.L.; Kimura, R.S. Gap junction systems in the rat vestibular labyrinth: Immunohistochemical and ultrastructural analysis. Acta Otolaryngol. 1994, 114, 520–528. [Google Scholar] [CrossRef]
- Erbe, C.B.; Harris, K.C.; Runge-Samuelson, C.L.; Flanary, V.A.; Wackym, P.A. Connexin 26 and connexin 30 mutations in children with nonsyndromic hearing loss. Laryngoscope 2004, 114, 607–611. [Google Scholar] [CrossRef]
- Meena, R.; Ayub, M. Genetics of human hereditary hearing impairment. J. Ayub Med. Coll. Abbottabad 2017, 29, 671–676. [Google Scholar]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic hearing impairment: Diversity of mutations and associated phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Mao, L.; Wang, Y.; An, L.; Zeng, B.; Wang, Y.; Frishman, D.; Liu, M.; Chen, Y.; Tang, W.; Xu, H. Molecular mechanisms and clinical phenotypes of GJB2 missense variants. Biology 2023, 12, 505. [Google Scholar] [CrossRef]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Milá, M.; Della Monica, M.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef]
- Gasparini, P.; Rabionet, R.; Barbujani, G.; Melçhionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; Fortina, P.; et al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- Batissoco, A.C.; Abreu-Silva, R.S.; Braga, M.C.; Lezirovitz, K.; Della-Rosa, V.; Alfredo, T., Jr.; Otto, P.A.; Mingroni-Netto, R.C. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: Implications for diagnosis and genetic counseling. Ear Hear. 2009, 30, 1–7. [Google Scholar] [CrossRef]
- Mei, L.; Chen, J.; Zong, L.; Zhu, Y.; Liang, C.; Jones, R.O.; Zhao, H.B. A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: Reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol. Dis. 2017, 108, 195–203. [Google Scholar] [CrossRef]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Mueller, R.F.; Nehammer, A.; Middleton, A.; Houseman, M.; Taylor, G.R.; Bitner-Glindzciz, M.; Van Camp, G.; Parker, M.; Young, I.D.; Davis, A.; et al. Congenital non-syndromal sensorineural hearing impairment due to connexin 26 gene mutations--molecular and audiological findings. Int. J. Pediatr. Otorhinolaryngol. 1999, 50, 3–13. [Google Scholar] [CrossRef]
- Sobe, T.; Vreugde, S.; Shahin, H.; Berlin, M.; Davis, N.; Kanaan, M.; Yaron, Y.; Orr-Urtreger, A.; Frydman, M.; Shohat, M.; et al. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum. Genet. 2000, 106, 50–57. [Google Scholar] [CrossRef]
- Orzan, E.; Polli, R.; Martella, M.; Vinanzi, C.; Leonardi, M.; Murgia, A. Molecular genetics applied to clinical practice: The Cx26 hearing impairment. Br. J. Audiol. 1999, 33, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, L.; Zhao, X.; Wang, X.; Cheng, X.; Du, Y.; Liu, D. Children with GJB2 gene mutations have various audiological phenotypes. Biosci. Trends 2018, 12, 419–425. [Google Scholar] [CrossRef]
- Lin, Y.F.; Lin, H.C.; Tsai, C.L.; Hsu, Y.C. GJB2 mutation spectrum in the Taiwanese population and genotype-phenotype comparisons in patients with hearing loss carrying GJB2 c.109G>A and c.235delC mutations. Hear. Res. 2022, 413, 108135. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 mutations and degree of hearing loss: A multicenter study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef]
- The Joint Committee on Infant Hearing. Year 2019 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs. J. Early Hear. Detect. Interv. 2019, 4, 1–44. [Google Scholar]
- British Society of Audiology. Recommended Procedure: Visual Reinforcement Audiometry; British Society of Audiology: Berkshire, UK, 2014; pp. 2–27. [Google Scholar]
- Arslan, E.; Genovese, E.; Orzan, E.; Turrini, M. Valutazione della Percezione Verbale nel Bambino Ipoacusico; Ecumenica Edition: Bari, Italy, 1997. [Google Scholar]
- del Castillo, F.J.; Rodríguez-Ballesteros, M.; Alvarez, A.; Hutchin, T.; Leonardi, E.; de Oliveira, C.A.; Azaiez, H.; Brownstein, Z.; Avenarius, M.R.; Marlin, S.; et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://databases.lovd.nl/shared/genes/GJB2 (accessed on 3 December 2023).
- Available online: https://deafnessvariationdatabase.org/gene/GJB2 (accessed on 3 December 2023).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 3 December 2023).
- Available online: https://varsome.com (accessed on 3 December 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kremer, H. Hereditary hearing loss; about the known and the unknown. Hear. Res. 2019, 376, 58–68. [Google Scholar] [CrossRef]
- Esteves, M.C.; de Lima Isaac, M.; Francisco, A.M.; da Silva Junior, W.A.; Ferreira, C.A.; Dell’Aringa, A.H. Analysis of the presence of the GJB6 mutations in patients heterozygous for GJB2 mutation in Brazil. Eur. Arch. Otorhinolaryngol. 2014, 271, 695–699. [Google Scholar] [CrossRef]
- Santos, R.L.; Aulchenko, Y.S.; Huygen, P.L.; van der Donk, K.P.; de Wijs, I.J.; Kemperman, M.H.; Admiraal, R.J.; Kremer, H.; Hoefsloot, L.H.; Cremers, C.W. Hearing impairment in Dutch patients with connexin 26 ((GJB2) and connexin 30 (GJB6) mutations. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 165–174. [Google Scholar] [CrossRef]
- Dai, Z.Y.; Sun, B.C.; Huang, S.S.; Yuan, Y.Y.; Zhu, Y.H.; Su, Y.; Dai, P. Correlation analysis of phenotype and genotype of GJB2 in patients with non-syndromic hearing loss in China. Gene 2015, 570, 272–276. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, X.; Liu, X.; Chen, S.; Kong, W.; He, M.; Sun, Y. Genotypic and allelic frequencies of GJB2 variants and features of hearing phenotypes in the Chinese population of the Dongfeng-Tongji Cohort. Genes 2023, 14, 2007. [Google Scholar] [CrossRef]
- Reda Del Barrio, S.; García Fernández, A.; Quesada-Espinosa, J.F.; Sánchez-Calvín, M.T.; Gómez-Manjón, I.; Sierra-Tomillo, O.; Juárez-Rufián, A.; de Vergas Gutiérrez, J. Genetic diagnosis of childhood sensorineural hearing loss. Acta Otorrinolaringol. Engl. Ed. 2024, in press. [CrossRef]
- Likar, T.; Hasanhodžić, M.; Teran, N.; Maver, A.; Peterlin, B.; Writzl, K. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss. PLoS ONE. 2018, 13, e0188578. [Google Scholar] [CrossRef]
- Lin, Y.H.; Wu, P.C.; Tsai, C.Y.; Lin, Y.H.; Lo, M.Y.; Hsu, S.J.; Lin, P.H.; Erdenechuluun, J.; Wu, H.P.; Hsu, C.J.; et al. Hearing impairment with monoallelic GJB2 variants: A GJB2 cause or non-GJB2 cause? J. Mol. Diagn. 2021, 23, 1279–1291. [Google Scholar] [CrossRef]
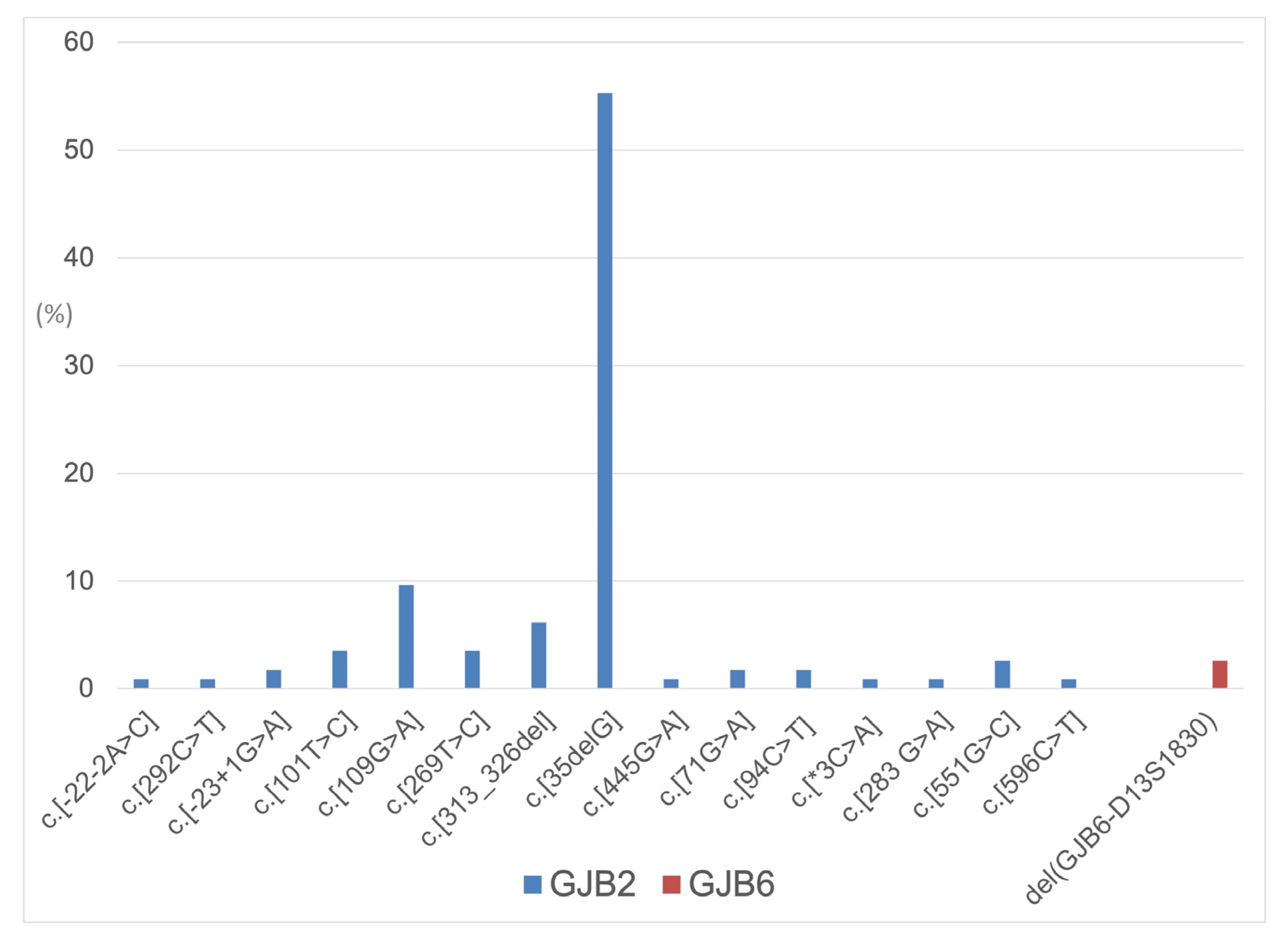



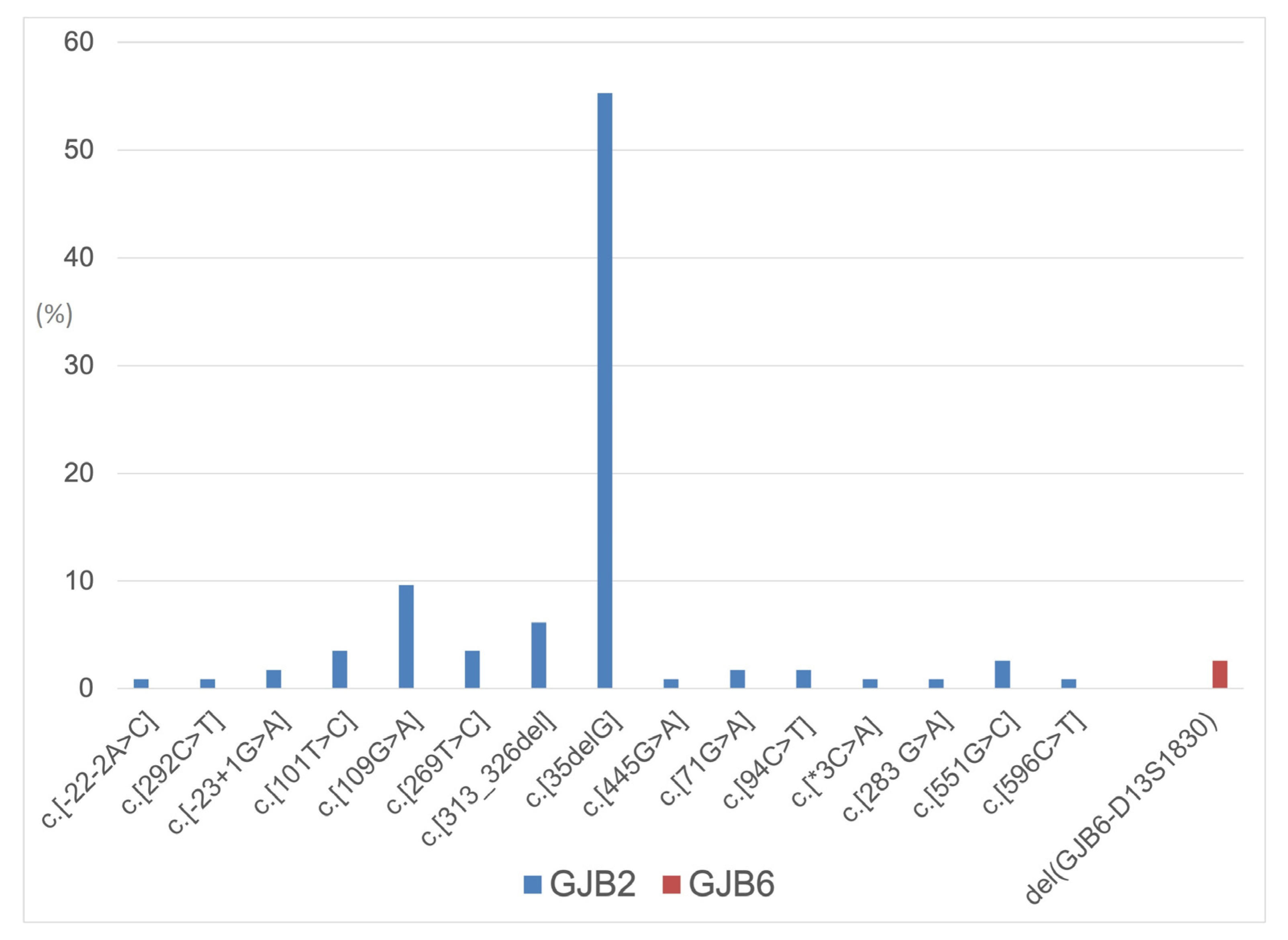
{kind=link}
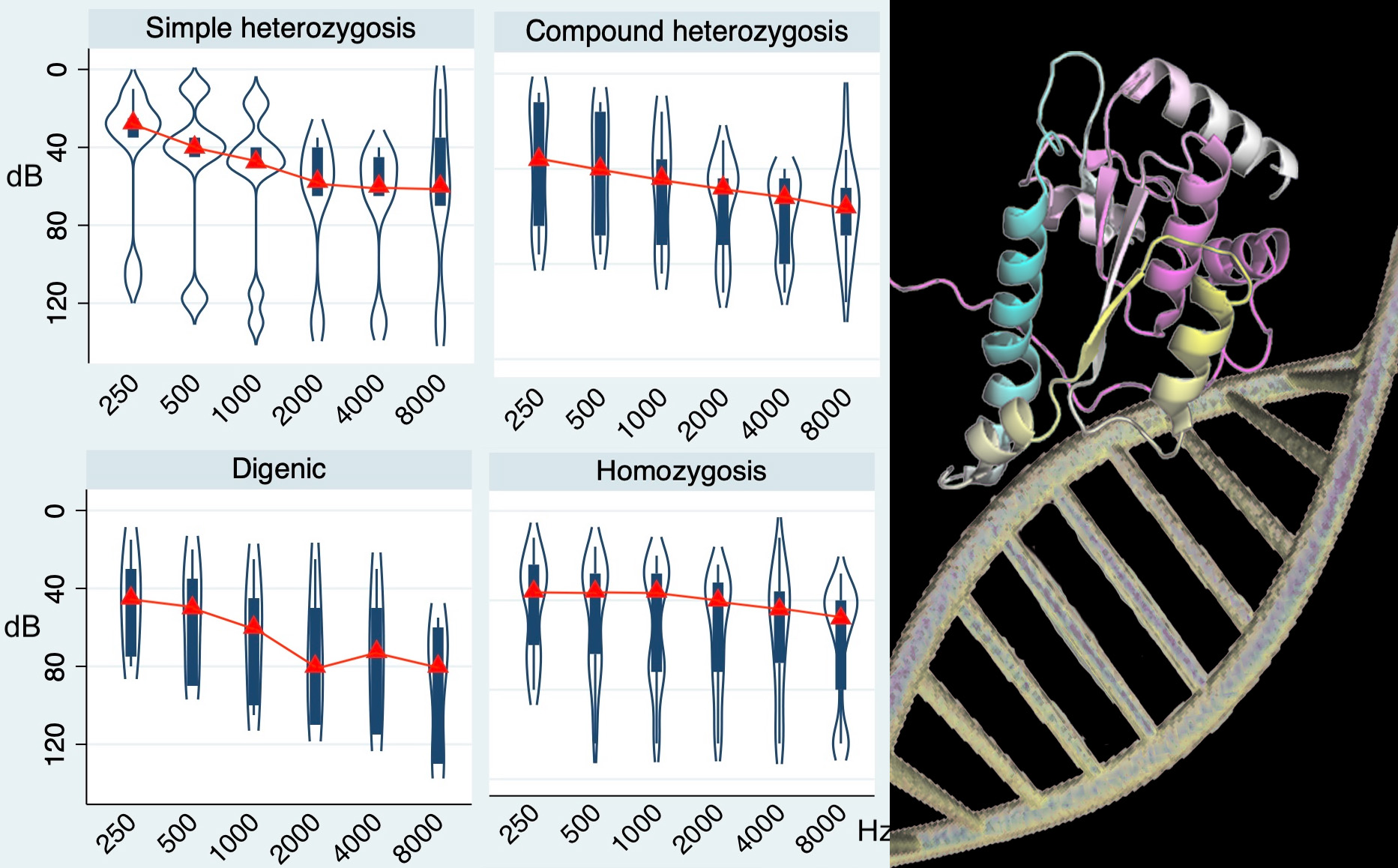
{kind=link}
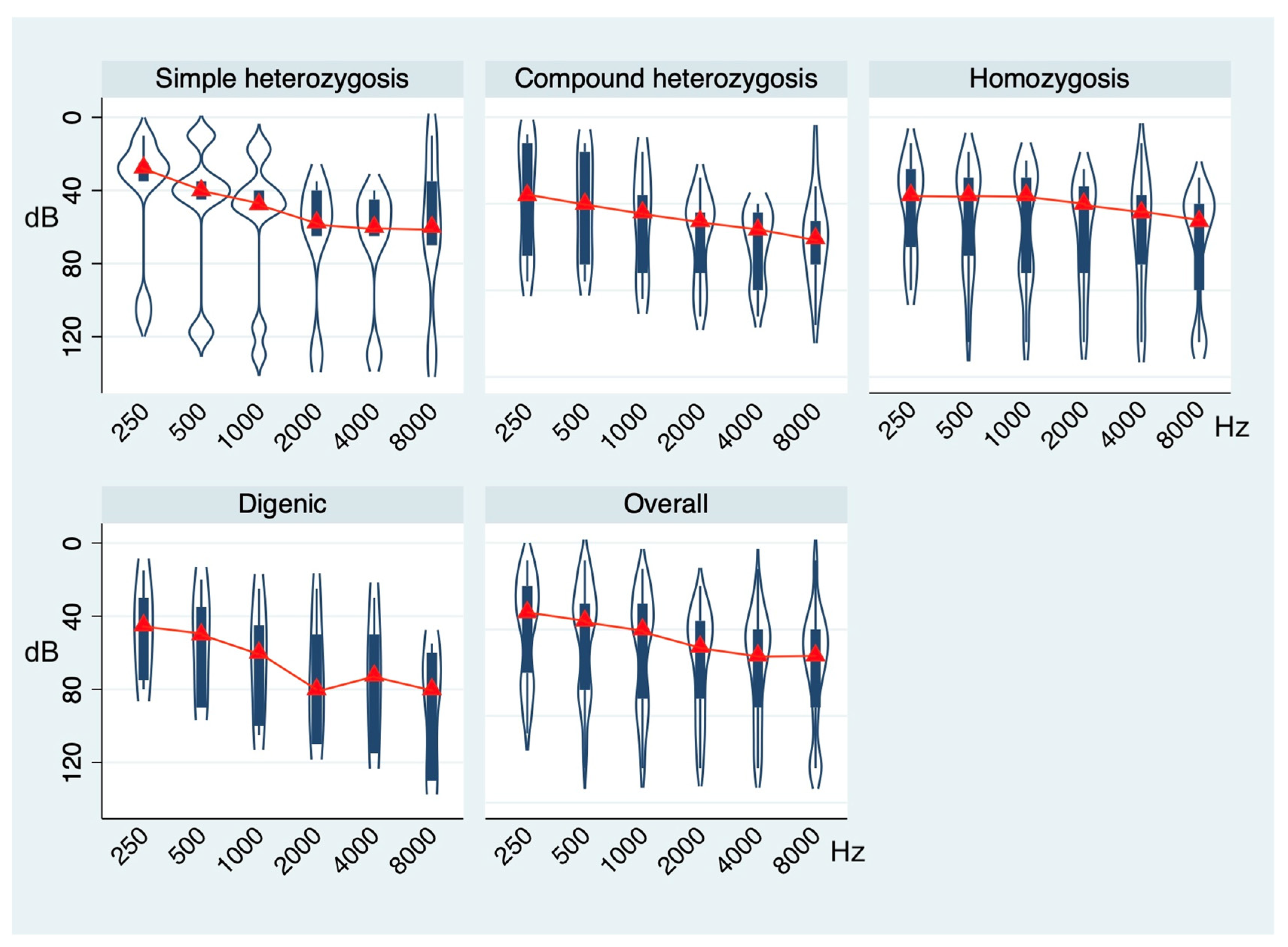{kind=link}
{kind=link}
| Variable | Total (N = 57) | Simple Heterozygosis (N = 5) | Compound Heterozygosis (N = 15) | Homozygosis (N = 34) | Digenic (N = 3) | p-Value |
|---|---|---|---|---|---|---|
| Age at diagnosis (months) | 5.37 | 5.6 | 6.88 | 5.03 | 5.67 | 0.8441 * |
| Mean (SD) | (3.49) | (3.44) | (4.22) | (3.22) | (4.04) | |
| Gender | 0.849 ** | |||||
| Male No. (%) | 26 (45.61) | 3 (60.00) | 6 (40.00) | 16 (47.06) | 1 (33.33) | |
| Female No. (%) | 31 (54.39) | 2 (40.00) | 9 (60.00) | 18 (52.94) | 2 (66.67) | |
| Familiarity | 0.439 ** | |||||
| Familial No. (%) | 19 (33.33) | 2 (40.00) | 7 (46.67) | 10 (29.41) | 0 (0.00) | |
| Non-familial No. (%) | 38 (66.67) | 3 (60.00) | 8 (53.33) | 24 (70.59) | 3 (100.00) | |
| Place of birth | 1.000 ** | |||||
| Italy No. (%) | 45 (80.00) | 4 (80.00) | 12 (80.00) | 26 (76.47) | 3 (100.0) | |
| Abroad No. (%) | 12 (20.00) | 1 (20.00) | 3 (20.00) | 8 (23.53) | 0 (0.00) | |
| Screening status | 0.124 ** | |||||
| Pass No. (%) | 5 (8.77) | 1 (20.00) | 1 (6.67) | 3 (8.82) | 0 (0.00) | |
| Fail No. (%) | 21 (36.84) | 0 (0.00) | 3 (20.00) | 17 (50.00) | 1 (33.33) | |
| No data No. (%) | 31 (54.39) | 4 (80.00) | 11 (73.33) | 14 (41.18) | 2 (66.67) | |
| Natural history | 0.391 ** | |||||
| Progressive No. (%) | 9 (15.79) | 2 (40.00) | 2 (13.33) | 5 (14.71) | 0 (0.00) | |
| Stable No. (%) | 48 (84.21) | 3 (60.00) | 13 (86.67) | 29 (85.29) | 3 (100.00) | |
| PTA § open field | 58.64 | 58.75 | 56.75 | 59.14 | 60.00 | 0.9923 * |
| Mean (SD) | (14.80) | (-) | (18.87) | (15.00) | (-) | |
| PTA § air conduction | 61.98 | 50.38 | 64.04 | 63.70 | 68.33 | 0.5785 * |
| Mean (SD) | (29.28) | (27.59) | (25.57) | (32.09) | (31.93) | |
| Air–bone gap † | 5.92 | 4.83 | 8.12 | 5.09 | 4.38 | 0.7987 * |
| Mean (SD) | (6.57) | (0.80) | (11.63) | (0.91) | (1.25) | |
| Follow-up (months) | 72.89 | 29.15 | 146.87 | 37.35 | 29.39 | 0.5147 * |
| Mean (SD) | (160.37) | (30.31) | (264.01) | (20.91) | (24.51) | |
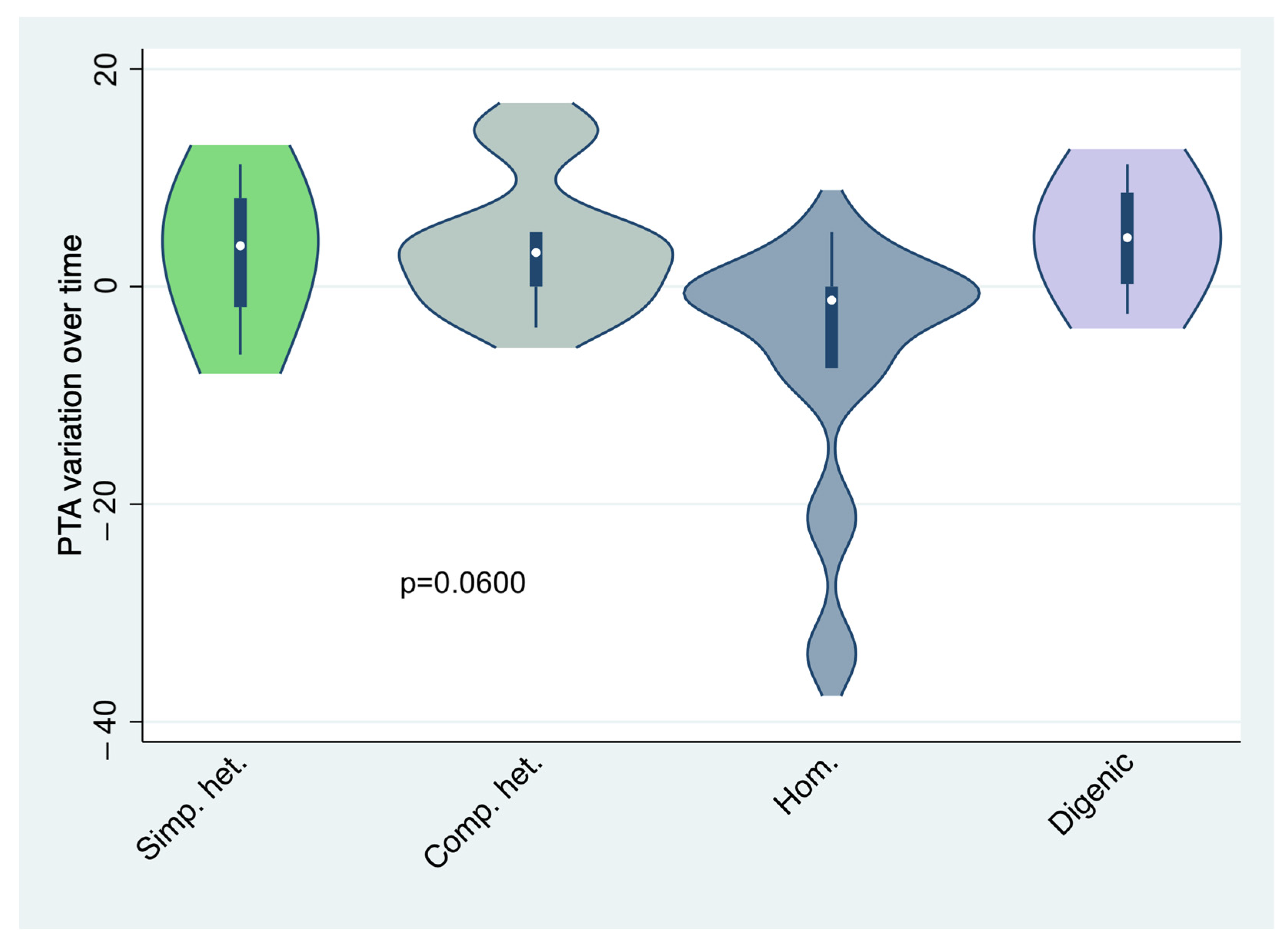
| Mean PTA difference between diagnosis and last follow-up (dB) | 0.01 | 3.13 | 3.88 | −6.25 | 4.44 | 0.0600 *** |
| Mean (SD) | (9.66) | (7.25) | (6.28) | (11.42) | (5.75) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franz, L.; Incognito, A.; Gallo, C.; Turolla, L.; Scquizzato, E.; Cenedese, R.; Matarazzo, A.; Savegnago, D.; Zanatta, P.; Genovese, E.; et al. Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles. Children 2024, 11, 194. https://doi.org/10.3390/children11020194
Franz L, Incognito A, Gallo C, Turolla L, Scquizzato E, Cenedese R, Matarazzo A, Savegnago D, Zanatta P, Genovese E, et al. Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles. Children. 2024; 11(2):194. https://doi.org/10.3390/children11020194
Chicago/Turabian StyleFranz, Leonardo, Alessandro Incognito, Chiara Gallo, Licia Turolla, Elisa Scquizzato, Roberta Cenedese, Alessandro Matarazzo, Daniel Savegnago, Paolo Zanatta, Elisabetta Genovese, and et al. 2024. "Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles" Children 11, no. 2: 194. https://doi.org/10.3390/children11020194
APA StyleFranz, L., Incognito, A., Gallo, C., Turolla, L., Scquizzato, E., Cenedese, R., Matarazzo, A., Savegnago, D., Zanatta, P., Genovese, E., de Filippis, C., & Marioni, G. (2024). Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles. Children, 11(2), 194. https://doi.org/10.3390/children11020194