
Arrhythmias May Hide a Genetic Cardiomyopathy in Left Ventricular Hypertrabeculation in Children: A Single-Center Experience

 , , , ,
, , , , 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Follow-Up
2.2. Statistical Analysis
3. Results
3.1. Study Population
3.2. Arrhythmias and Follow-Up
3.3. Tachyarrhythmias
- 4 atrioventricular reentry tachycardia;
- 1 atypical atrioventricular nodal reentry tachycardia;
- 1 atrial fibrillation;
- 3 ectopic atrial tachycardia (one was a multifocal form);
- 2 very frequent premature atrial contractions;
- 2 asymptomatic ventricular pre-excitations which were revealed to be low-risk accessory pathways by a transesophageal electrophysiology study (TE-EPS);
- 5 frequent PVCs (usually asymptomatic, monomorphic, with high arrhythmic burden at ECG Holter monitoring and variable response to exercise stress test);
- 8 sustained or non-sustained VT;
- 1 VF.
3.4. Bradyarrhythmias
3.5. Genetic Testing
4. Discussion
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AVB | Atrio-ventricular block |
| CHD | Congenital heart disease |
| CMR | Cardiac magnetic resonance |
| DCM | Dilated cardiomyopathy |
| HCM | Hypertrophic cardiomyopathy |
| LVHT | Left ventricular hypertrabeculation |
| PVC | Premature ventricular contraction |
| SCD | Sudden cardiac death |
References
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left Ventricular Non-Compaction Cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart. 2001, 86, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef]
- Børresen, M.F.; Blixenkrone-Møller, E.; Kock, T.O.; Sillesen, A.S.; Vøgg, R.O.B.; Pihl, C.A.; Norsk, J.B.; Vejlstrup, N.G.; Christensen, A.H.; Iversen, K.K.; et al. Prevalence of Left Ventricular Noncompaction in Newborns. Circ. Cardiovasc. Imaging 2022, 15, e014159. [Google Scholar] [CrossRef]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef]
- van Waning, J.I.; Caliskan, K.; Michels, M.; Schinkel, A.F.L.; Hirsch, A.; Dalinghaus, M.; Hoedemaekers, Y.M.; Wessels, M.W.; Ijpma, A.S.; Hofstra, R.M.; et al. Cardiac phenotypes, genetics, and risks in familial noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2019, 73, 1601–1611. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical gen- etics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Gati, S.; Papadakis, M.; Papamichael, N.D.; Zaidi, A.; Sheikh, N.; Reed, M.; Sharma, R.; Thilaganathan, B.; Sharma, S. Reversible de novo left ventricular trabeculations in pregnant women: Implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation 2014, 130, 475–483. [Google Scholar] [CrossRef]
- Gati, S.; Chandra, N.; Bennett, R.L.; Reed, M.; Kervio, G.; Panoulas, V.F.; Ghani, S.; Sheikh, N.; Zaidi, A.; Wilson, M.; et al. Increased left ventricular trabeculation in highly trained athletes: Do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 2013, 99, 401–408. [Google Scholar] [CrossRef]
- de la Chica, J.A.; Gomez-Talavera, S.; Garcia-Ruiz, J.M.; Garcia-Lunar, I.; Oliva, B.; Fernandez-Alvira, J.M.; López-Melgar, B.; Sánchez-González, J.; de la Pompa, J.L.; Mendiguren, J.M.; et al. Association between left ventricular noncompaction and vigorous physical activity. J. Am. Coll. Cardiol. 2020, 76, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Jones, K.; Blanch, B.; Puranik, R.; McGeechan, K.; Barratt, A.; Semsarian, C. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults. Eur. Heart J. 2020, 41, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yang, Y.; Zhou, L.; Luo, F.; Li, Y.; Fan, P.; Dong, X.; Liu, Y.; Cui, J.; Zhou, X.; et al. Left ventricular non-compaction: A cardiomyopathy with acceptable prognosis in children. Heart Lung Circ. 2018, 27, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Brescia, S.T.; Rossano, J.W.; Pignatelli, R.; Jefferies, J.L.; Price, J.F.; Decker, J.A.; Denfield, S.W.; Dreyer, W.J.; Smith, O.; Towbin, J.A.; et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation 2013, 127, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Jensen, B.; Aung, N.; Friedrich, M.G.; McMahon, C.J.; Mohiddin, S.A.; Pignatelli, R.H.; Ricci, F.; Anderson, R.H.; Bluemke, D.A. Excessive Trabeculation of the Left Ventricle: JACC: Cardiovascular Imaging Expert Panel Paper. JACC Cardiovasc. Imaging. 2023, 16, 408–425. [Google Scholar] [CrossRef]
- Łuczak-Woźniak, K.; Werner, B. Left Ventricular Noncompaction-A Systematic Review of Risk Factors in the Pediatric Population. J. Clin. Med. 2021, 10, 1232. [Google Scholar] [CrossRef]
- Rohde, S.; Muslem, R.; Kaya, E.; Dalinghaus, M.; van Waning, J.I.; Majoor-Krakauer, D.; Towbin, J.; Caliskan, K. State-of-the art review: Noncompaction cardiomyopathy in pediatric patients. Heart Fail. Rev. 2022, 27, 15–28. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef]
- De Lazzari, M.; Brunetti, G.; Frasson, E.; Zorzi, A.; Cipriani, A.; Migliore, F.; De Conti, G.; Motta, R.; Perazzolo Marra, M.; Corrado, D. Thinning of compact layer and systolic dysfunction in isolated left ventricular non-compaction: A cardiac magnetic resonance study. Int. J. Cardiol. 2024, 397, 131614. [Google Scholar] [CrossRef]
- Pignatelli, R.H.; McMahon, C.J.; Dreyer, W.J.; Denfield, S.W.; Price, J.; Belmont, J.W.; Craigen, W.J.; Wu, J.; El Said, H.; Bezold, L.I.; et al. Clinical characterization of left ventricular noncompaction in children: A relatively common form of cardiomyopathy. Circulation 2003, 108, 2672–2678. [Google Scholar] [CrossRef]
- Oechslin, E.N.; Attenhofer Jost, C.H.; Rojas, J.R.; Kaufmann, P.A.; Jenni, R. Long-Term Follow-up of 34 Adults with Isolated Left Ventricular Noncompaction: A Distinct Cardiomyopathy with Poor Prognosis. J. Am. Coll. Cardiol. 2000, 36, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Daubeney, P.E.; Nugent, A.W.; Chondros, P.; Carlin, J.B.; Colan, S.D.; Cheung, M.; Davis, A.M.; Chow, C.W.; Weintraub, R.G. Clinical Features and Outcomes of Childhood Dilated Cardiomyopathy: Results from a National Population-Based Study. Circulation 2006, 114, 2671–2678. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Wilkinson, J.D.; Sleeper, L.A.; Colan, S.D.; Lu, M.; Pahl, E.; Kantor, P.F.; Everitt, M.D.; Webber, S.A.; Kaufman, B.D.; et al. Pediatric Cardiomyopathy Registry Investigators. Cardiomyopathy Phenotypes and Outcomes for Children with Left Ventricular Myocardial Noncompaction: Results from the Pediatric Cardiomyopathy Registry. J. Card. Fail. 2015, 21, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef]
- Oechslin, E.; Jenni, R.; Klaassen, S. Left Ventricular Noncompaction Is a Myocardial Phenotype: Cardiomyopathy—Yes or No? Can. J. Cardiol. 2021, 37, 366–369. [Google Scholar] [CrossRef]
- Schultze-Berndt, A.; Kühnisch, J.; Herbst, C.; Seidel, F.; Al-Wakeel-Marquard, N.; Dartsch, J.; Theisen, S.; Knirsch, W.; Jenni, R.; Greutmann, M.; et al. Reduced Systolic Function and Not Genetic Variants Determine Outcom in Pediatric and Adult Left Ventricular Noncompaction Cardiomyopathy. Front. Pediatr. 2021, 9, 722926. [Google Scholar] [CrossRef]
- van Waning, J.I.; Moesker, J.; Heijsman, D.; Boersma, E.; Majoor-Krakauer, D. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012993. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef]
- Andreini, D.; Pontone, G.; Bogaert, J.; Roghi, A.; Barison, A.; Schwitter, J.; Mushtaq, S.; Vovas, G.; Sormani, P.; Aquaro, G.D.; et al. Long-Term Prognostic Value of Cardiac Magnetic Resonance in Left Ventricle Noncompaction: A Prospective Multicenter Study. J. Am. Coll. Cardiol. 2016, 68, 2166–2181. [Google Scholar] [CrossRef]
- Fitzsimons, L.A.; Kneeland-Barber, D.M.; Hannigan, G.C.; Karpe, D.A.; Wu, L.; Colon, M.; Randall, J.; Tucker, K.L. Electrophysiological phenotyping of left ventricular noncompaction cardiomyopathy in pediatric populations: A systematic review. Physiol. Rep. 2024, 12, e16029. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Muñoz, J.J.; Muñoz-Esparza, C.; Verdú, P.P.; Sánchez, J.M.; Almagro, F.G.; Ruiz, G.E.; Blanes, J.R.G.; Alberola, A.G. Catheter ablation of ventricular arrhythmias in left ventricular noncompaction cardiomyopathy. Heart Rhythm. 2021, 18, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Bazoukis, G.; Tyrovolas, K.; Letsas, K.P.; Vlachos, K.; Radford, D.; Chung, C.T.; Liu, T.; Efremidis, M.; Tse, G.; Baranchuk, A. Predictors of fatal arrhythmic events in patients with non-compaction cardiomyopathy: A systematic review. Heart Fail. Rev. 2022, 27, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Czosek, R.J.; Spar, D.S.; Khoury, P.R.; Anderson, J.B.; Wilmot, I.; Knilans, T.K.; Jefferies, J.L. Outcomes, arrhythmic burden and ambulatory monitoring of pediatric patients with left ventricular non-compaction and preserved left ventricular function. Am. J. Cardiol. 2015, 115, 962–966. [Google Scholar] [CrossRef]
- Hirono, K.; Hata, Y.; Miyao, N.; Okabe, M.; Takarada, S.; Nakaoka, H.; Ibuki, K.; Ozawa, S.; Origasa, H.; Nishida, N.; et al. ncreased Burden of Ion Channel Gene Variants Is Related to Distinct Phenotypes in Pediatric Patients with Left Ventricular Noncompaction. Circ. Genom. Precis. Med. 2020, 13, e002940. [Google Scholar] [CrossRef]
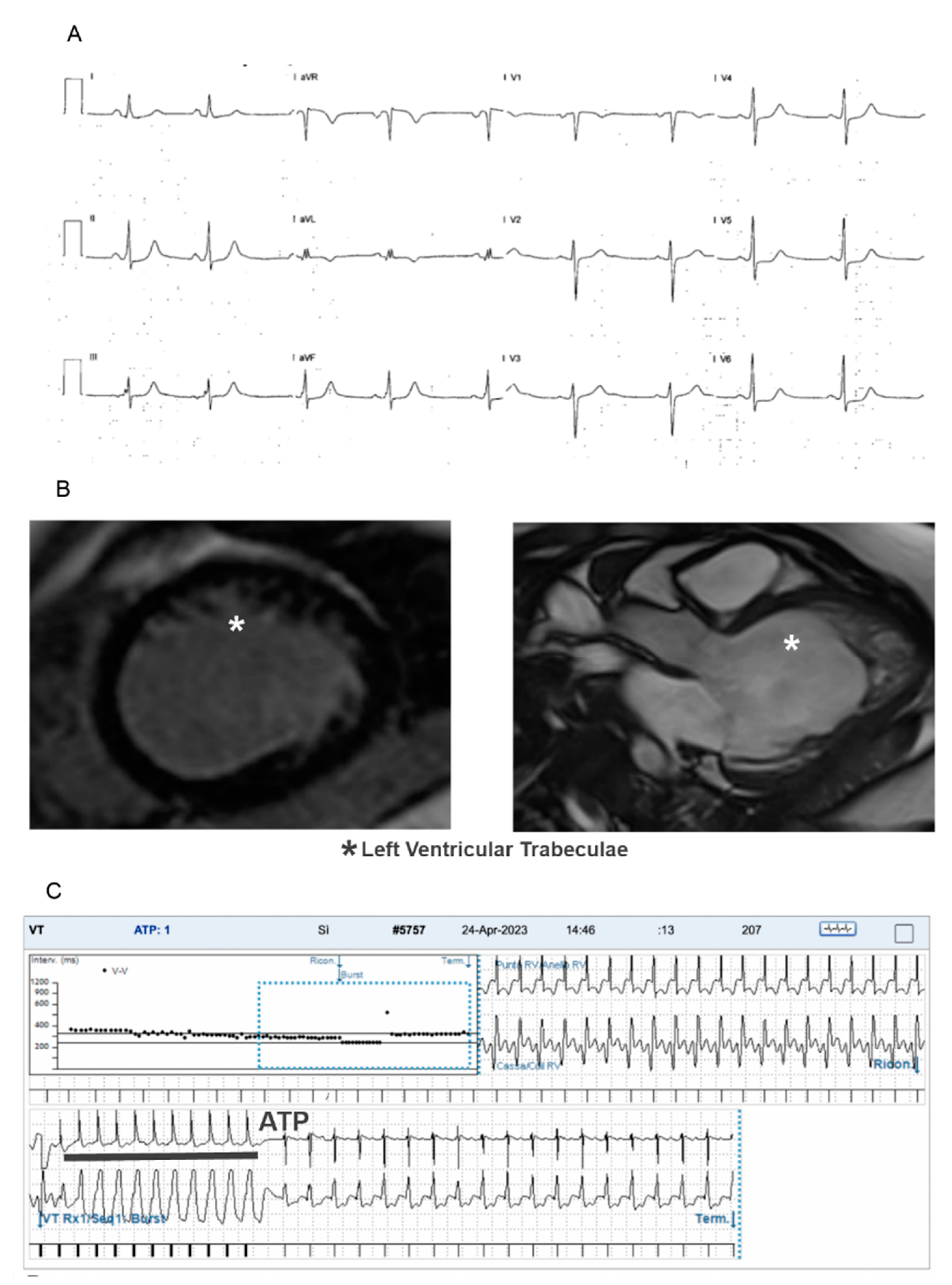

{kind=link}
| Total Patients 140 | Number of Patients | Other |
|---|---|---|
| Males/Females | 70/70 | Mean age 13.4 ± 5.9 yrs |
| Reason for diagnosis | ||
| screening | 71 (51%) | |
| symptoms | 52 (37%) | |
| arrhythmias | 17 (12%) | |
| Mean LV EF (%) | 59.5% | |
| Family history of LVHT/cardiomyopathy | 50 (29.4%) | |
| Phenotypes | ||
| Isolated LVHT | 113 (80.7%) | |
| HCM/LVHT | 4 (2.8%) | |
| DCM/LVHT | 10 (7.1%) | |
| LVHT + congenital heart disease | 13 (9.3%) | ASD, VSD, aortic coarctation, patent ductus arteriosus, coronary sinus diverticulum, bicuspid aortic valve, Ebstein anomaly, right coronary anomaly |
| Arrhythmia | Pts | Sex | Phenotype | Family History of LVHT/CMP | Genetics (Involved Genes) | EF% (Median (IQR)) | LGE at CMR |
|---|---|---|---|---|---|---|---|
| AVRT | 4 | 2 M | 3 LVHT, 1 LVHT + CHD | 0 | 1 TNNT2: c.460-1G>C (class 4) 1 TTN: c.53978_53979insA (class 4) | 58% (7.3) | 0 |
| EAT | 3 | 3 M | 1 LVHT, 1 DCM/LVHT, 1 LVHT + CHD | 2 | 1 NKX2.5: c.554G>T (class 4) | 54% (2.6) | 0 |
| AVNRT | 1 | F | LVHT + CHD | 0 | 0 | 66% | 0 |
| PACs | 2 | 2 M | 3 LVHT | 2 | 1 CTNNA3: c.2638dupA (class 4) | 56.3% (5) | 0 |
| AF | 1 | M | LVHT + CHD | 1 | LMNA: c.1201C>T (class 5) | 48% | 0 |
| VP | 2 | 1 M | LVHT | 1 | 0 | 63.5% (8.5) | 0 |
| PVCs | 5 | 3 M | 5 LVHT | 3 | 1 TTN: c.76114C>T (class 4) | 63.2% (5) | 1 |
| VTs/VF | 9 | 3 M | 6 LVHT, 1 DCM/LVHT, 2 LVHT + CHD | 4 | 1 LMNA: c.1201C>T (class 5), 1 NDUFA1: c.152_155delATAG (class 4), 1 PRDM16: c.370delA (class 4), 1 SMARCD1: c.592G>A (class 4), 1 Microdel. 1p36.33p36.32+ Microdupl. Xp22.33p22.32 (class 5) | 59.3% (7) | 0 |
| SND | 2 | 2 F | 2 LVHT | 1 | 1 KCNJ8: c.2dupT (class 5) | 66.5% (10) | 0 |
| III degree AVB | 2 | 2 M | 2 LVHT + CHD | 1 | 0 | 54% (2.5) | NP |
| Paroxysmal III degree AVB | 3 | 3 F | 3 LVHT + CHD | 2 | 2 NKX2.5: c.554G>T (class 4) | 60% (2.6) | 0 |
| I degree AVB | 1 | M | DCM/LVHT | 0 | 0 | 55% (6) | NP |
| No. Patients | Arrhythmic LVHT | Non-Arrhythmic LVHT | p Value |
|---|---|---|---|
| Genetic test result positive | 13 (43% of tested patients) | 14 (22% of tested patients) | 0.037 |
| CHD associated with LVHT | 9 (27%) | 4 (3.7%) | 0.91 |
| Familiar history positive | 15 (45%) | 31 (29%) | 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battipaglia, I.; Cantarutti, N.; Cicenia, M.; Adorisio, R.; Battista, V.; Baban, A.; Silvetti, M.S.; Drago, F. Arrhythmias May Hide a Genetic Cardiomyopathy in Left Ventricular Hypertrabeculation in Children: A Single-Center Experience. Children 2024, 11, 1233. https://doi.org/10.3390/children11101233
Battipaglia I, Cantarutti N, Cicenia M, Adorisio R, Battista V, Baban A, Silvetti MS, Drago F. Arrhythmias May Hide a Genetic Cardiomyopathy in Left Ventricular Hypertrabeculation in Children: A Single-Center Experience. Children. 2024; 11(10):1233. https://doi.org/10.3390/children11101233
Chicago/Turabian StyleBattipaglia, Irma, Nicoletta Cantarutti, Marianna Cicenia, Rachele Adorisio, Virginia Battista, Anwar Baban, Massimo Stefano Silvetti, and Fabrizio Drago. 2024. "Arrhythmias May Hide a Genetic Cardiomyopathy in Left Ventricular Hypertrabeculation in Children: A Single-Center Experience" Children 11, no. 10: 1233. https://doi.org/10.3390/children11101233
APA StyleBattipaglia, I., Cantarutti, N., Cicenia, M., Adorisio, R., Battista, V., Baban, A., Silvetti, M. S., & Drago, F. (2024). Arrhythmias May Hide a Genetic Cardiomyopathy in Left Ventricular Hypertrabeculation in Children: A Single-Center Experience. Children, 11(10), 1233. https://doi.org/10.3390/children11101233