
Identification of Extremely Rare Pathogenic CNVs by Array CGH in Saudi Children with Developmental Delay, Congenital Malformations, and Intellectual Disability
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Ethical Approval
2.2. Cytogenetics Analyses
2.3. DNA Preparation and Whole-Genome Array CGH
2.4. Interpretation of CNVs
2.5. Quantitative Real-Time PCR
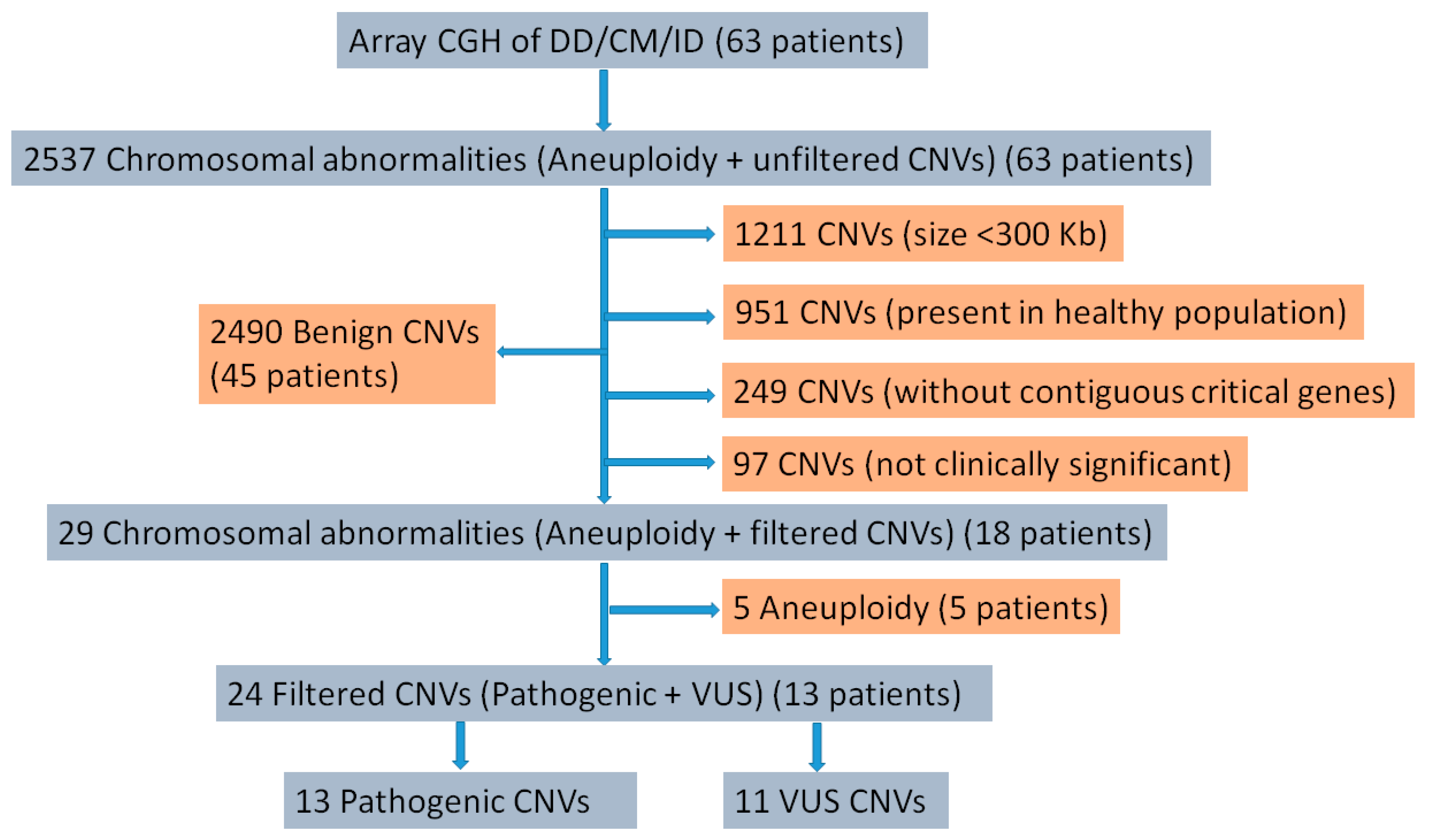
3. Results
3.1. Clinical Finding
3.2. Cytogenetic Abnormalities
3.3. Pathogenic CNVs
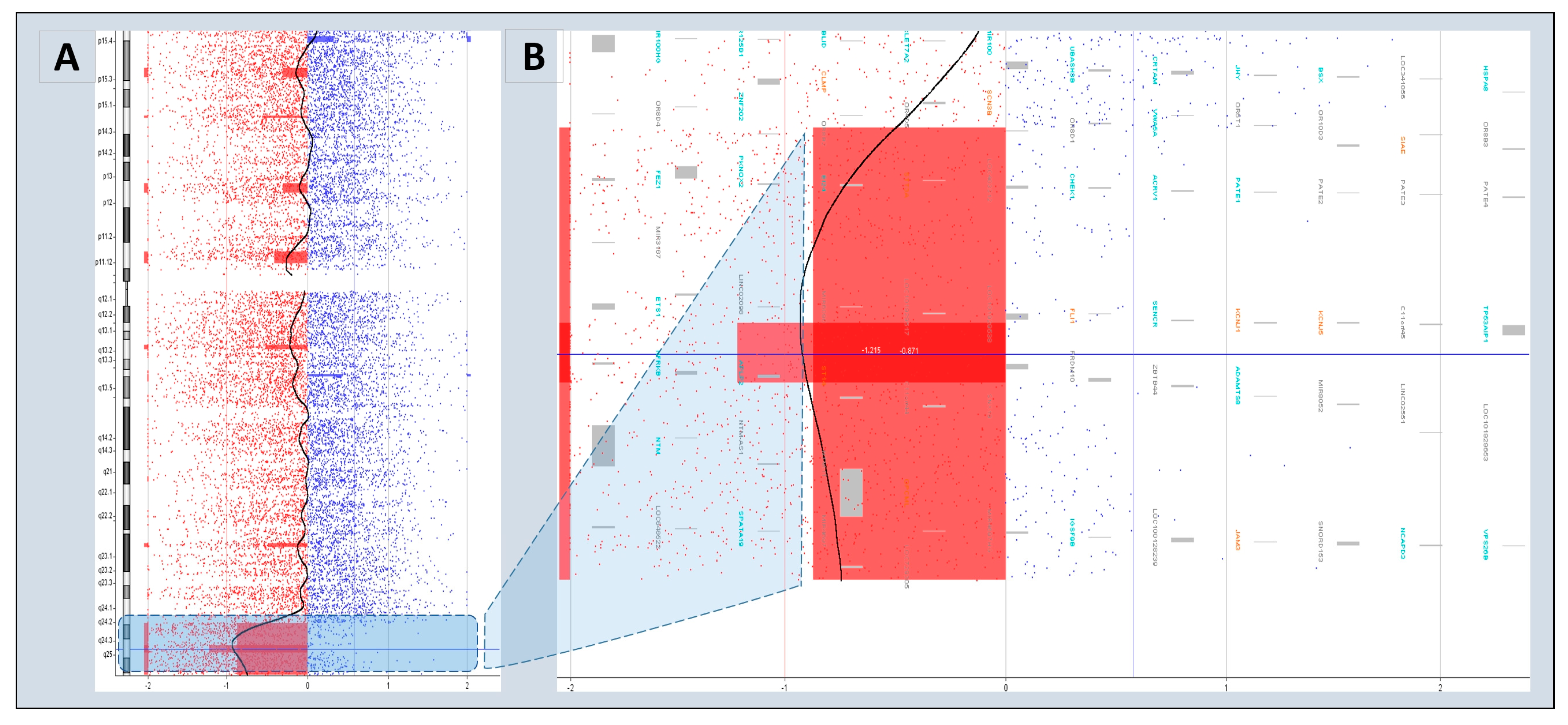
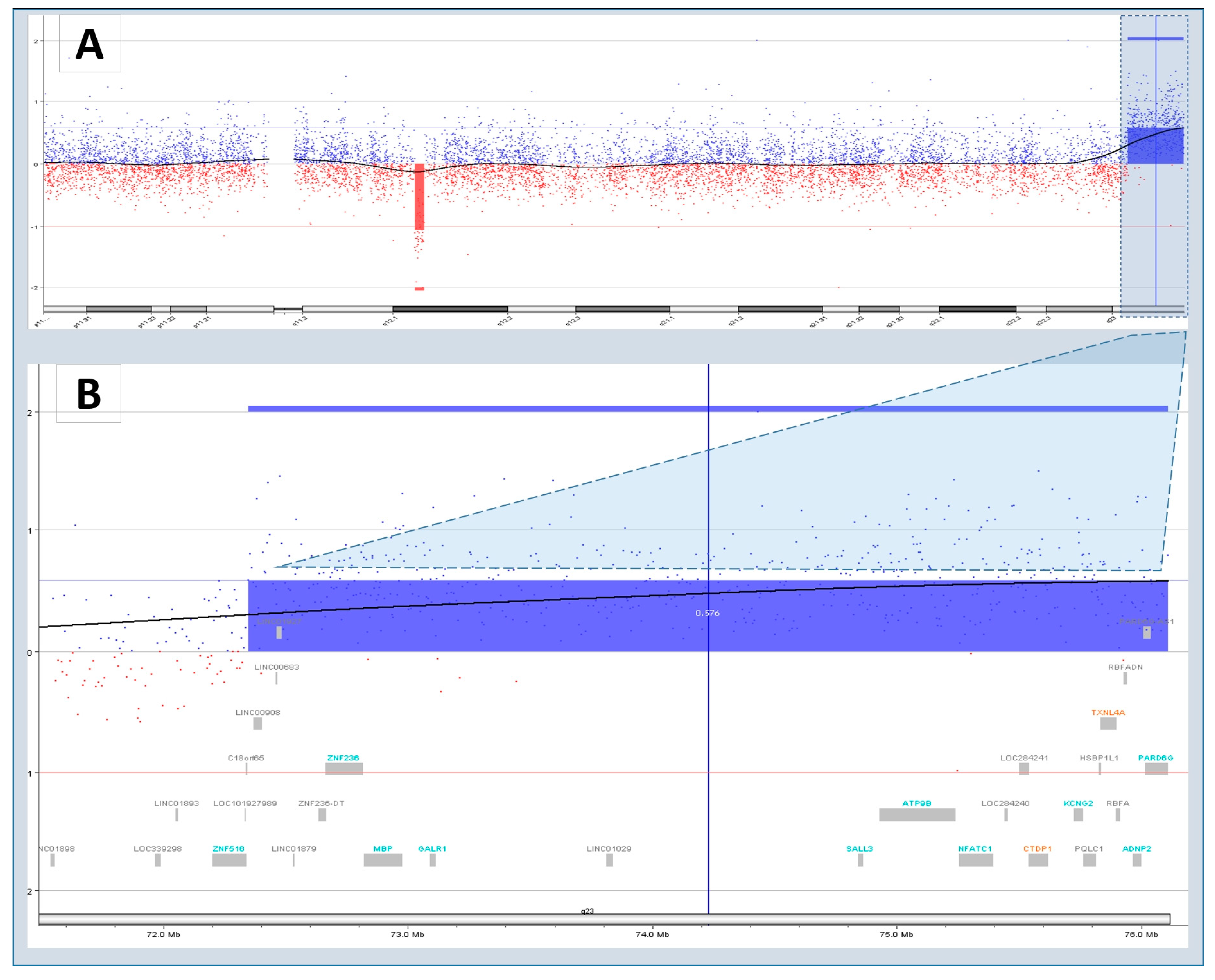
3.4. Characteristics of Disease-Associated CNVs
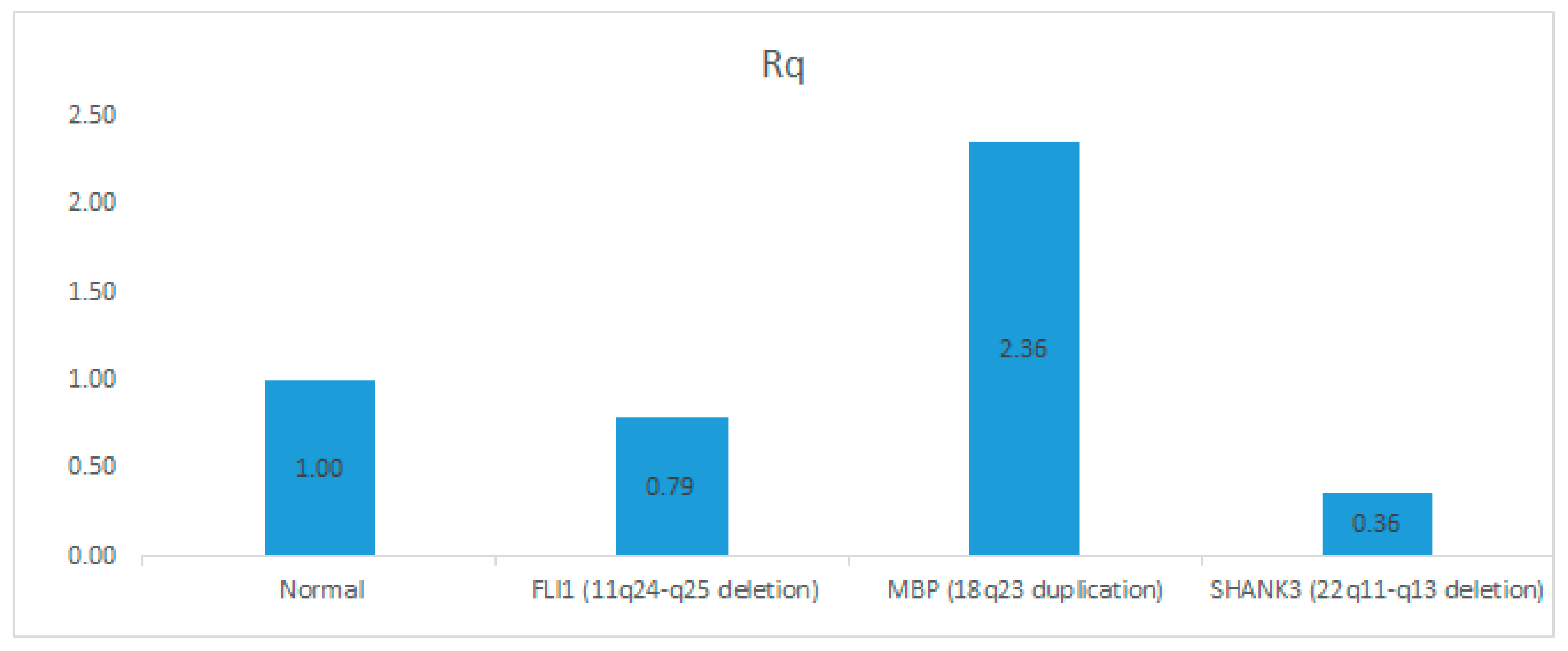
3.5. Confirmation of CNVs by Quantitative Real-Time PCR (qPCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mithyantha, R.; Kneen, R.; McCann, E.; Gladstone, M. Current evidence-based recommendations on investigating children with global developmental delay. Arch. Dis. Child. 2017, 102, 1071–1076. [Google Scholar] [CrossRef]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef]
- Jones, K.L.; Adam, M.P. Evaluation and diagnosis of the dysmorphic infant. Clin. Perinatol. 2015, 42, 243–261. [Google Scholar] [CrossRef]
- Patel, D.R.; Cabral, M.D.; Ho, A.; Merrick, J. A clinical primer on intellectual disability. Transl. Pediatr. 2020, 9, S23–S35. [Google Scholar] [CrossRef]
- Fida, N.M.; Al-Aama, J.; Nichols, W.; Nichols, W.; Alqahtani, M. A prospective study of congenital malformations among live born neonates at a University Hospital in Western Saudi Arabia. Saudi Med. J. 2007, 28, 1367–1373. [Google Scholar]
- Hussein, I.R.; Bader, R.S.; Chaudhary, A.G.; Bassiouni, R.; Alquaiti, M.; Ashgan, F.; Schulten, H.-J.; Al Qahtani, M.H. Identification of De Novo and Rare Inherited Copy Number Variants in Children with Syndromic Congenital Heart Defects. Pediatr. Cardiol. 2018, 39, 924–940. [Google Scholar] [CrossRef]
- Lee, C.L.; Lee, C.H.; Chuang, C.K.; Chiu, H.C.; Chen, Y.J.; Chou, C.L.; Wu, P.S.; Chen, C.P.; Lin, H.Y.; Lin, S.P. Array-CGH increased the diagnostic rate of developmental delay or intellectual disability in Taiwan. Pediatr. Neonatol. 2019, 60, 453–460. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Buysse, K.; Delle Chiaie, B.; Van Coster, R.; Loeys, B.; De Paepe, A.; Mortier, G.; Speleman, F.; Menten, B. Challenges for CNV interpretation in clinical molecular karyotyping: Lessons learned from a 1001 sample experience. Eur. J. Med. Genet. 2009, 52, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Zhang, J.-G.; Deng, H.-W.; Wang, Y.-P. Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS ONE 2013, 8, e59128. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Moreno-Cabrera, J.M.; Del Valle, J.; Castellanos, E.; Feliubadaló, L.; Pineda, M.; Brunet, J.; Serra, E.; Capellà, G.; Lázaro, C.; Gel, B. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur. J. Hum. Genet. EJHG 2020, 28, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bai, W.; Yuan, N.; Du, Z. Comprehensively benchmarking applications for detecting copy number variation. PLoS Comput. Biol. 2019, 15, e1007069. [Google Scholar] [CrossRef]
- Bi, W.; Borgan, C.; Pursley, A.N.; Hixson, P.; Shaw, C.A.; Bacino, C.A.; Lalani, S.R.; Patel, A.; Stankiewicz, P.; Lupski, J.R.; et al. Comparison of chromosome analysis and chromosomal microarray analysis: What is the value of chromosome analysis in today’s genomic array era? Genet. Med. 2013, 15, 450–457. [Google Scholar] [CrossRef]
- Ellison, J.W.; Ravnan, J.B.; Rosenfeld, J.A.; Morton, S.A.; Neill, N.J.; Williams, M.S.; Lewis, J.; Torchia, B.S.; Walker, C.; Traylor, R.N.; et al. Clinical utility of chromosomal microarray analysis. Pediatrics 2012, 130, e1085–e1095. [Google Scholar] [CrossRef]
- Riggs, E.R.; Wain, K.E.; Riethmaier, D.; Smith-Packard, B.; Faucett, W.A.; Hoppman, N.; Thorland, E.C.; Patel, V.C.; Miller, D.T. Chromosomal microarray impacts clinical management. Clin. Genet. 2014, 85, 147–153. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Syrmou, A.; Tzetis, M.; Fryssira, H.; Kosma, K.; Oikonomakis, V.; Giannikou, K.; Makrythanasis, P.; Kitsiou-Tzeli, S.; Kanavakis, E. Array comparative genomic hybridization as a clinical diagnostic tool in syndromic and nonsyndromic congenital heart disease. Pediatr. Res. 2013, 73, 772–776. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Moeschler, J.B.; Shevell, M. Comprehensive Evaluation of the Child With Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [PubMed]
- Serra-Juhé, C.; Rodríguez-Santiago, B.; Cuscó, I.; Vendrell, T.; Camats, N.; Torán, N.; Pérez-Jurado, L.A. Contribution of Rare Copy Number Variants to Isolated Human Malformations. PLoS ONE 2012, 7, e45530. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Jamal, H.S.; Rouzi, A.; Ardawi, M.S.M.; Schulten, H.J.; Mirza, Z.; Alansari, N.A.; Al-Quaiti, M.M.; Abusamra, H.; Naseer, M.I.; et al. Genomic answers for recurrent spontaneous abortion in Saudi Arabia: An array comparative genomic hybridization approach. Reprod. Biol. 2017, 17, 133–143. [Google Scholar] [CrossRef]
- Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Sogaty, S.; Karim, S.; Schulten, H.J.; Bibi, F.; Pushparaj, P.N.; Algahtani, H.A.; Al-Qahtani, M.H. Chromosomal Micro-aberration in a Saudi Family with Juvenile Myoclonic Epilepsy. CNS Neurol. Disord. Drug Targets 2017, 16, 1010–1017. [Google Scholar] [CrossRef]
- Ibrahim, S.M.; Karim, S.; Abusamra, H.; Pushparaj, P.N.; Khan, J.A.; Abuzenadah, A.M.; Gari, M.A.; Bakhashab, S.; Ahmed, F.; Al-Qahtani, M.H. Genomic amplification of chromosome 7 in the Doxorubicin resistant K562 cell line. Bioinformation 2018, 14, 587–593. [Google Scholar] [CrossRef]
- Rasool, M.; Pushparaj, P.N.; Mirza, Z.; Imran Naseer, M.; Abusamra, H.; Alquaiti, M.; Shaabad, M.; Sibiany, A.M.S.; Gauthaman, K.; Al-Qahtani, M.H.; et al. Array comparative genomic hybridization based identification of key genetic alterations at 2p21-p16.3 (MSH2, MSH6, EPCAM), 3p23-p14.2 (MLH1), 7p22.1 (PMS2) and 1p34.1-p33 (MUTYH) regions in hereditary non polyposis colorectal cancer (Lynch syndrome) in the Kingdom of Saudi Arabia. Saudi J. Biol. Sci. 2020, 27, 157–162. [Google Scholar] [CrossRef]
- Bibi, F.; Ali, I.; Naseer, M.I.; Ali Mohamoud, H.S.; Yasir, M.; Alvi, S.A.; Jiman-Fatani, A.A.; Sawan, A.; Azhar, E.I. Detection of genetic alterations in gastric cancer patients from Saudi Arabia using comparative genomic hybridization (CGH). PLoS ONE 2018, 13, e0202576. [Google Scholar] [CrossRef]
- Hussein, I.R.; Magbooli, A.; Huwait, E.; Chaudhary, A.; Bader, R.; Gari, M.; Ashgan, F.; Alquaiti, M.; Abuzenadah, A.; AlQahtani, M. Genome wide array-CGH and qPCR analysis for the identification of genome defects in Williams’ syndrome patients in Saudi Arabia. Mol. Cytogenet. 2016, 9, 65. [Google Scholar] [CrossRef]
- Bahamat, A.A.; Assidi, M.; Lary, S.A.; Almughamsi, M.M.; Peer Zada, A.A.; Chaudhary, A.; Abuzenadah, A.; Abu-Elmagd, M.; Al-Qahtani, M. Use of Array Comparative Genomic Hybridization for the Diagnosis of DiGeorge Syndrome in Saudi Arabian Population. Cytogenet. Genome Res. 2018, 154, 20–29. [Google Scholar] [CrossRef]
- Albesher, N.; Massadeh, S.; Hassan, S.M.; Alaamery, M. Consanguinity and Congenital Heart Disease Susceptibility: Insights into Rare Genetic Variations in Saudi Arabia. Genes 2022, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- Deans, Z.C.; Ahn, J.W.; Carreira, I.M.; Dequeker, E.; Henderson, M.; Lovrecic, L.; Õunap, K.; Tabiner, M.; Treacy, R.; van Asperen, C.J. Recommendations for reporting results of diagnostic genomic testing. Eur. J. Hum. Genet. 2022, 30, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- ISCN. An International System for Human Cytogenomic Nomenclature (2020); McGowan-Jordan, J.O., Hastings, R.J.O., Moore, S.A., Eds.; Karger: Basel, Switzerland, 2020. [Google Scholar] [CrossRef]
- Park, S.J.; Jung, E.H.; Ryu, R.S.; Kang, H.W.; Chung, H.D.; Kang, H.Y. The clinical application of array CGH for the detection of chromosomal defects in 20,126 unselected newborns. Mol. Cytogenet. 2013, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.; He, M.; Berglund, E.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Kvastad, L.; Andersson, A.; Bergenstråhle, L.; Bergenstråhle, J.; et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 2022, 608, 360–367. [Google Scholar] [CrossRef]
- Hovhannisyan, G.; Harutyunyan, T.; Aroutiounian, R.; Liehr, T. DNA Copy Number Variations as Markers of Mutagenic Impact. Int. J. Mol. Sci. 2019, 20, 4723. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, M.L.; Yang, W.; Mercer-Rosa, L.; Parnell, A.S.; Oster, M.E.; Levenbrown, Y.; Tanel, R.E.; Goldmuntz, E. 22q11.2 Deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J. Thorac. Cardiovasc. Surg. 2014, 148, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Verheij, J.B.; de Munnik, S.A.; Dijkhuizen, T.; de Leeuw, N.; Olde Weghuis, D.; van den Hoek, G.J.; Rijlaarsdam, R.S.; Thomasse, Y.E.; Dikkers, F.G.; Marcelis, C.L.; et al. An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur. J. Med. Genet. 2009, 52, 353–357. [Google Scholar] [CrossRef]
- Guilherme, R.S.; Meloni, V.A.; Perez, A.B.A.; Pilla, A.L.; de Ramos, M.A.P.; Dantas, A.G.; Takeno, S.S.; Kulikowski, L.D.; Melaragno, M.I. Duplication 9p and their implication to phenotype. BMC Med. Genet. 2014, 15, 142. [Google Scholar] [CrossRef]
- Vera-Carbonell, A.; López-González, V.; Bafalliu, J.A.; Ballesta-Martínez, M.J.; Fernández, A.; Guillén-Navarro, E.; López-Expósito, I. Clinical comparison of 10q26 overlapping deletions: Delineating the critical region for urogenital anomalies. Am. J. Med. Genet. Part A 2015, 167, 786–790. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Knight, M.A.; Hernandez, D.; Diede, S.J.; Dauwerse, H.G.; Rafferty, I.; van de Leemput, J.; Forrest, S.M.; Gardner, R.J.; Storey, E.; van Ommen, G.J.; et al. A duplication at chromosome 11q12.2-11q12.3 is associated with spinocerebellar ataxia type 20. Hum. Mol. Genet. 2008, 17, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Bernaciak, J.; Szczałuba, K.; Derwińska, K.; Wiśniowiecka-Kowalnik, B.; Bocian, E.; Sasiadek, M.M.; Makowska, I.; Stankiewicz, P.; Smigiel, R. Clinical and molecular-cytogenetic evaluation of a family with partial Jacobsen syndrome without thrombocytopenia caused by an approximately 5 Mb deletion del(11)(q24.3). Am. J. Med. Genet. Part A 2008, 146, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Favier, R.; Akshoomoff, N.; Mattson, S.; Grossfeld, P. Jacobsen syndrome: Advances in our knowledge of phenotype and genotype. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 239–250. [Google Scholar] [CrossRef]
- Walenkamp, M.J.; de Muinck Keizer-Schrama, S.M.; de Mos, M.; Kalf, M.E.; van Duyvenvoorde, H.A.; Boot, A.M.; Kant, S.G.; White, S.J.; Losekoot, M.; Den Dunnen, J.T.; et al. Successful long-term growth hormone therapy in a girl with haploinsufficiency of the insulin-like growth factor-I receptor due to a terminal 15q26.2->qter deletion detected by multiplex ligation probe amplification. J. Clin. Endocrinol. Metab. 2008, 93, 2421–2425. [Google Scholar] [CrossRef]
- Uddin, M.; Unda, B.K.; Kwan, V.; Holzapfel, N.T.; White, S.H.; Chalil, L.; Woodbury-Smith, M.; Ho, K.S.; Harward, E.; Murtaza, N.; et al. OTUD7A Regulates Neurodevelopmental Phenotypes in the 15q13.3 Microdeletion Syndrome. Am. J. Hum. Genet. 2018, 102, 278–295. [Google Scholar] [CrossRef]
- Moreno-De-Luca, A.; Helmers, S.L.; Mao, H.; Burns, T.G.; Melton, A.M.A.; Schmidt, K.R.; Fernhoff, P.M.; Ledbetter, D.H.; Martin, C.L. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet. 2011, 48, 141–144. [Google Scholar] [CrossRef]
- Burgess, T.; Brown, N.J.; Stark, Z.; Bruno, D.L.; Oertel, R.; Chong, B.; Calabro, V.; Kornberg, A.; Sanderson, C.; Kelly, J.; et al. Characterization of core clinical phenotypes associated with recurrent proximal 15q25.2 microdeletions. Am. J. Med. Genet. Part A 2014, 164, 77–86. [Google Scholar] [CrossRef]
- Shinawi, M.; Liu, P.; Kang, S.H.; Shen, J.; Belmont, J.W.; Scott, D.A.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010, 47, 332–341. [Google Scholar] [CrossRef]
- Goto, T.; Aramaki, M.; Yoshihashi, H.; Nishimura, G.; Hasegawa, Y.; Takahashi, T.; Ishii, T.; Fukushima, Y.; Kosaki, K. Large fontanelles are a shared feature of haploinsufficiency of RUNX2 and its co-activator CBFB. Congenit. Anom. 2004, 44, 225–229. [Google Scholar] [CrossRef]
- Khan, A.; Hyde, R.K.; Dutra, A.; Mohide, P.; Liu, P. Core binding factor beta (CBFB) haploinsufficiency due to an interstitial deletion at 16q21q22 resulting in delayed cranial ossification, cleft palate, congenital heart anomalies, and feeding difficulties but favorable outcome. Am. J. Med. Genet. Part A 2006, 140, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.E.; Liu, G.; Gilboa, S.M.; Ethen, M.K.; Aylsworth, A.S.; Powell, C.M.; Flood, T.J.; Mai, C.T.; Wang, Y.; Canfield, M.A.; et al. Survival of children with trisomy 13 and trisomy 18: A multi-state population-based study. Am. J. Med. Genet. Part A 2016, 170, 825–837. [Google Scholar] [CrossRef]
- Wieczorek, D.; Newman, W.G.; Wieland, T.; Berulava, T.; Kaffe, M.; Falkenstein, D.; Beetz, C.; Graf, E.; Schwarzmayr, T.; Douzgou, S.; et al. Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet. 2014, 95, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Jana, B.; Khanfar, A.; Ninan, M. Durable hematological and major cytogenetic response in a patient with isolated 20q deletion myelodysplastic syndrome treated with lenalidomide. Case Rep. Oncol. Med. 2014, 2014, 949515. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Bennett, T.M.; Knopf, H.L.; Yamada, K.; Yoshiura, K.; Niikawa, N.; Shim, S.; Hanson, P.I. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am. J. Hum. Genet. 2007, 81, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Iolascon, A.; Verissimo, F.; Trede, N.S.; Horsley, W.; Chen, W.; Paw, B.H.; Hopfner, K.P.; Holzmann, K.; Russo, R.; et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009, 41, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet (Lond. Engl.) 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol. Rev. 2019, 287, 186–201. [Google Scholar] [CrossRef]
- Rauch, R.; Hofbeck, M.; Zweier, C.; Koch, A.; Zink, S.; Trautmann, U.; Hoyer, J.; Kaulitz, R.; Singer, H.; Rauch, A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J. Med. Genet. 2010, 47, 321–331. [Google Scholar] [CrossRef]
- Kharbanda, M.; Pilz, D.T.; Tomkins, S.; Chandler, K.; Saggar, A.; Fryer, A.; McKay, V.; Louro, P.; Smith, J.C.; Burn, J.; et al. Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur. J. Med. Genet. 2017, 60, 130–135. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Biobank Code | G-Banding Result | Array CGH Results |
|---|---|---|---|
| 1 | BL-080-12 | 46,XY,del(11)(q24) | arr[hg18] 11q24.2q25(123615752_134432324)x1 |
| 2 | BL-181-13 | 47,XX,+del(9)(q22) | arr[hg18] 9p24.3p13.1(194193_38745183)x3 |
| 3 | BL-210-12 | 46,XX,dup(3)p36-p33 | arr[hg18]1p36.33p33(746956_48281628)x3 |
| 4 | BL-401-13 | 46,XX,t(13;14)(q10;q10) + 18 * | arr[hg18] 18p11.32p11.21 (75432_14092527)x3 arr[hg18] 18q11.1q23(16910049_76111023)x3 |
| 5 | Bl-628-12 | 45,X | arr[hg18]Xp22.33p11.1(2711273_58499110)x1 arr[hg18]Xq11.1q28(61848414_154582526)x1 |
| 6 | BL-664-11 | 47,XXY | arr[hg18]Xp22.33p11.1(2711273_58499110)x4 arr[hg18]Xq11.1q28(61848414_154570236)x4 |
| 7 | BL-664-13 | 45,X | arr[hg18] Xp22.33p11.1(2711273_58499110)x1 arr[hg18] Xq11.1q28(61848414_154582526)x1 |
| 8 | BL-793-13 | 46,XY,del(22)(q11.2q11.2) | arr[hg18] 22q11q21(17269039_19794119)x1 |
| 9 | BL-902-10 | 47,XY,+mar or 47,XY,+i(18)(p10) | arr[hg18] 18p11.32p11.21(114641_15062794)x4 |
| 10 | BL-1288-13 | 47,XXY | arr[hg18] Xp22.33p11.1(2711273_58499110)x4 arr[hg18] Xq11.1q28(61848414_154570236)x4 |
| S. No | Biobank Code | CNVs | CNV Classification | OMIM Genes | Associated Syndromes |
|---|---|---|---|---|---|
| 1 | BL-080-12 | arr[hg18]11q24.2q25(123615752_134432324)x1 | Pathogenic | FLI-1, KCNJ1, KCNJ5, JAM3 | Jacobsen syndrome |
| 2 | BL-161-14 | arr[hg18]16q21q23.1(65021970_74573408)x1 | VUS | CDH11, CBFB, TK2, GOT2, BEAN, | 16q22 deletion syndrome |
| 3 | BL-181-13 | arr[hg18]9p24.3p13.1(194193_38745183)x3 | Pathogenic | SMARCA2, VLDLR | Nicolaides-Baraitser syndrome |
| 4 | BL-210-12 | arr[hg18]11q12.2- q13.5(60243525_ 75206053)x3 | VUS | DAGLA, FADD, FGF3 | Spinocerebellar ataxia 20 |
| arr[hg18]16p13.3 p11.2(28087_ 31446768)x3 | Pathogenic | CREBBP, DNASE, TRAP1, TELO2 | 16p13.3 duplication syndrome | ||
| arr[hg18]22q11.1q13.33(15834835_ 49525130)x3 | VUS | LARGE, SHANK3, TBX1, HPS4 | Duplication of 22q12/13 | ||
| 5 | BL-363-12 | arr[hg18]3p23p14.2(31581603_58552982)x1 | VUS | WNT5A, HESX1, FLNB, CCK, GLYCTK | Robinow syndrome 1 |
| arr[hg18]15q13.1q26.1(27196809_89833172)x1 | Pathogenic | CHRNA7 and OTUD7 | 15q13.3 microdeletion syndrome | ||
| arr[hg18]16p13.3p11.2(1103363_33495560)x1 | VUS | CREBBP, DNASE, TRAP1, TELO2 | 16p13.3 deletion syndrome | ||
| arr[hg18]20q11.1q13.2(28252372_50274994)x1 | VUS | GDF5, EPB41L1, SAMHD1 | 20q deletion | ||
| arr[hg18]22q11.1q13.31(14513474_44350783)x1 | VUS | PEX26, SHANK3, TBX1, HPS4 | Deletion of 22q12/13 | ||
| 6 | BL-457-12 | arr[hg18]16q21q23.1(65021970_74573408)x1 | VUS | CDH11, CBFB, TK2, GOT2, BEAN, KARS1 | 16q22 deletion syndrome |
| 7 | BL-461-12 | arr[hg18]13q21.1- q32.1(57009987_95317681)x1 | VUS | DIAPH3, PIBF1, TBC1D1, FBXL3, EDNRB, PCDH17 | DD, ID, skeletal and other abnormalities |
| arr[hg18]16q21-q23.1(65109284_74852983)x3 | VUS | CDH11, CBFB, TK2, GOT2, BEAN, KARS1 | 16q22 duplication syndrome | ||
| 8 | BL-464-12 | arr[hg18]10q26.13q26.3(127272748_135284168)x3 | Pathogenic | DOCK1, C10ORF90 | 10q26 duplication syndrome |
| arr[hg18]18q23(7234653976111023)x1 | VUS | CTDP1, GALR1 | 18q23 deletion syndrome | ||
| 9 | BL-518-13 | arr[hg18] 15q25.1q26.3(78427008_100298411)x3 | Pathogenic | AP3B2, HOMER2, SH3GL3, CHD2 | Levy-Shanske syndrome |
| 10 | BL-597-12 | arr[hg18]18p11.32p11.21(121700_14112521)x4 | Pathogenic | TGIF1, NDUFV2, PIEZO2, GNAL | Partial trisomy 18p |
| arr[hg18]22q11.1q13.33(15681796_49571118)x3 | VUS | LARGE, SHANK3, TBX1, HPS4 | Duplication of 22q12/13 | ||
| 11 | BL-793-13 | arr[hg18]22q11.21(17269039_19794119)x1 | Pathogenic | PRODH, SLC25A1, SNAP29, TBX1 | DiGeorge Syndrome |
| 12 | BL-902-12 | arr[hg18]18p11.32p11.21(114641_15062794)x4 | Pathogenic | TGIF1, NDUFV2, PIEZO2, GNAL | Partial trisomy 18p |
| 13 | BL-1086-11 | arr[hg18]8q24.3(142081172_146142017)x1 | Pathogenic | PUF60, GRINA | Verheij syndrome |
| arr[hg18]11p15.5p15.4(234177_2917590)x1 | Syndromic | CDKN1C, IGF2, KCNQ1OT1 | Beckwith-Wiedemann syndrome |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karim, S.; Hussein, I.R.; Schulten, H.-J.; Alsaedi, S.; Mirza, Z.; Al-Qahtani, M.; Chaudhary, A. Identification of Extremely Rare Pathogenic CNVs by Array CGH in Saudi Children with Developmental Delay, Congenital Malformations, and Intellectual Disability. Children 2023, 10, 662. https://doi.org/10.3390/children10040662
Karim S, Hussein IR, Schulten H-J, Alsaedi S, Mirza Z, Al-Qahtani M, Chaudhary A. Identification of Extremely Rare Pathogenic CNVs by Array CGH in Saudi Children with Developmental Delay, Congenital Malformations, and Intellectual Disability. Children. 2023; 10(4):662. https://doi.org/10.3390/children10040662
Chicago/Turabian StyleKarim, Sajjad, Ibtessam Ramzi Hussein, Hans-Juergen Schulten, Saad Alsaedi, Zeenat Mirza, Mohammed Al-Qahtani, and Adeel Chaudhary. 2023. "Identification of Extremely Rare Pathogenic CNVs by Array CGH in Saudi Children with Developmental Delay, Congenital Malformations, and Intellectual Disability" Children 10, no. 4: 662. https://doi.org/10.3390/children10040662
APA StyleKarim, S., Hussein, I. R., Schulten, H.-J., Alsaedi, S., Mirza, Z., Al-Qahtani, M., & Chaudhary, A. (2023). Identification of Extremely Rare Pathogenic CNVs by Array CGH in Saudi Children with Developmental Delay, Congenital Malformations, and Intellectual Disability. Children, 10(4), 662. https://doi.org/10.3390/children10040662