
Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Objective
2.2. Study Design
2.3. Intervention
2.4. Outcome Measure
2.5. Literature Review
3. Results
3.1. Patients
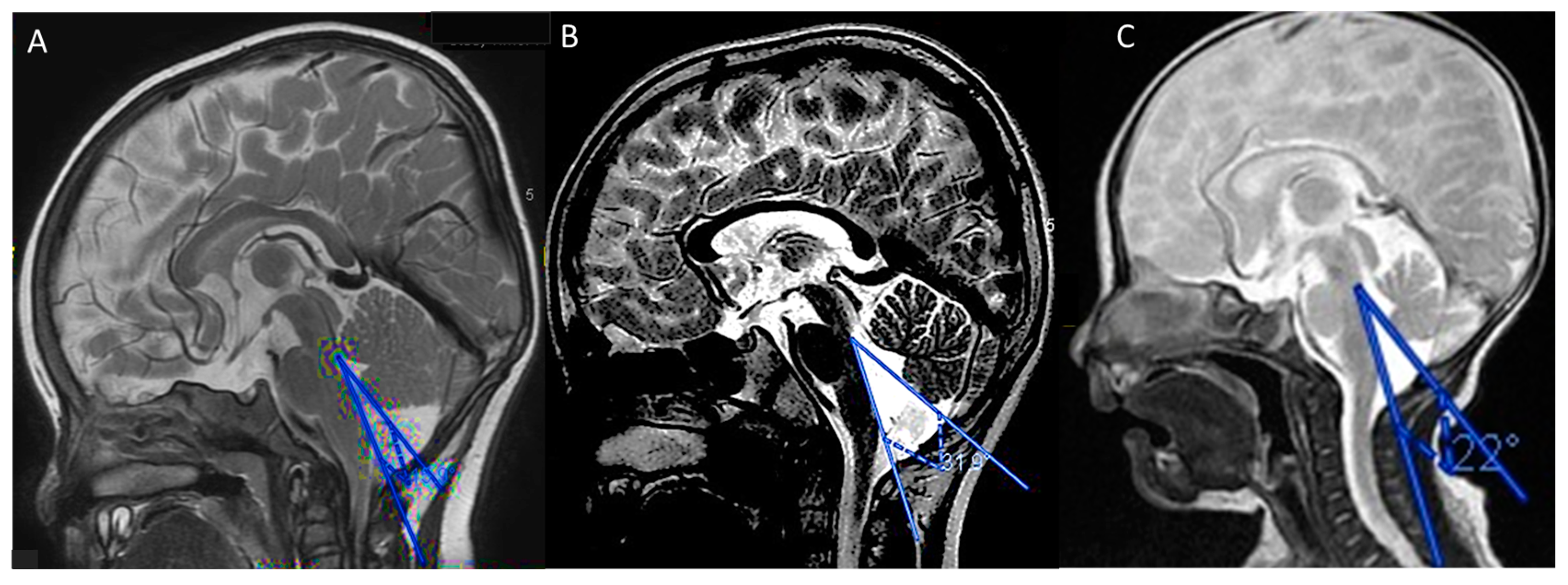
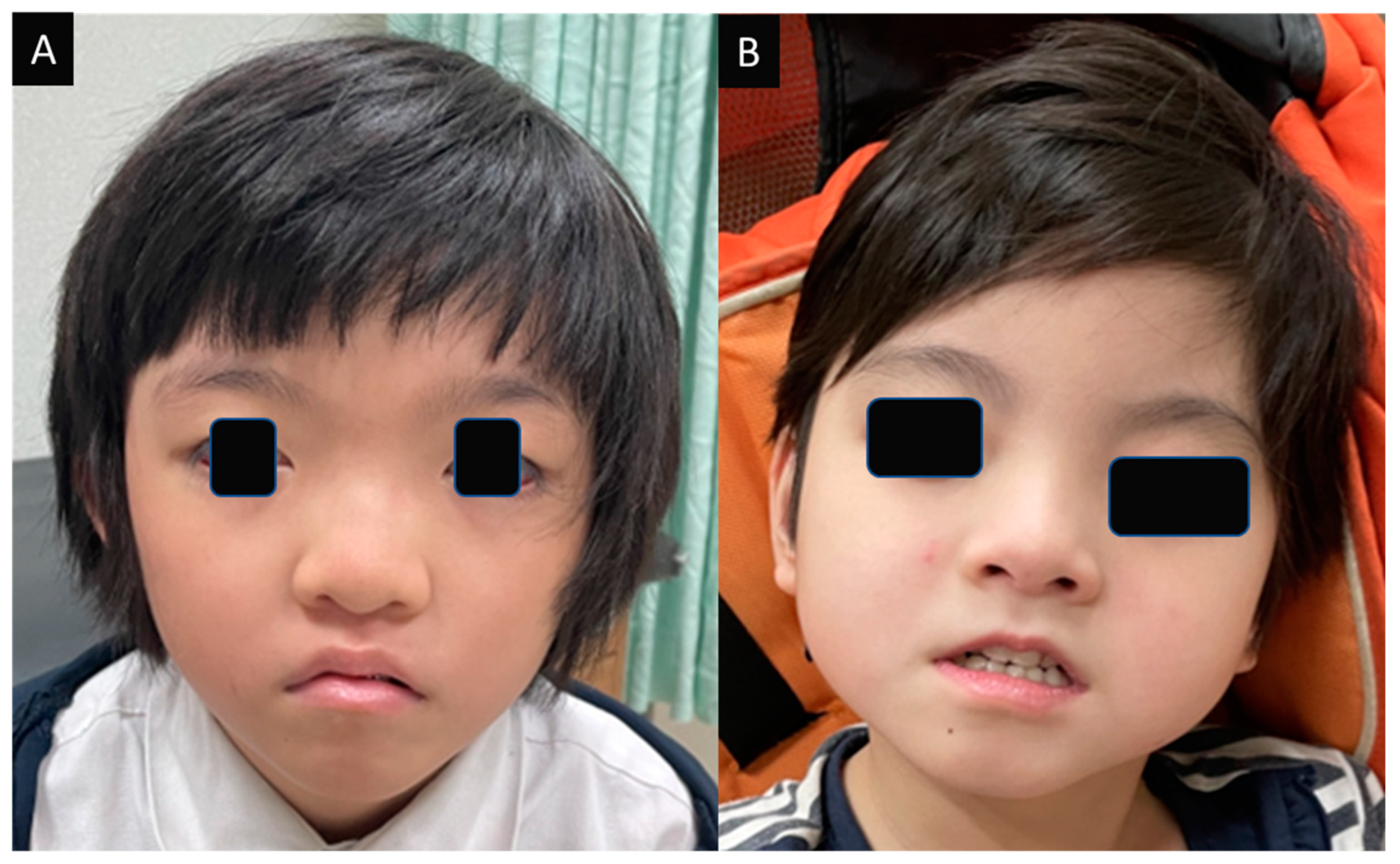
3.1.1. Case 1
3.1.2. Case 2
3.1.3. Case 3
3.2. Literature Review
3.2.1. Demographics of 32 Cases
3.2.2. Seizure Patterns, Intervention, and Outcomes
3.2.3. Dysmorphism
3.2.4. Neuroimaging Features
3.2.5. Clinical Neurodevelopmental Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuberi, S.M.; Wirrell, E.; Yozawitz, E.; Wilmshurst, J.M.; Specchio, N.; Riney, K.; Pressler, R.; Auvin, S.; Samia, P.; Hirsch, E.; et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1349–1397. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Elliott, K.S.; Shetty, J.; Armstrong, M.; Brunklaus, A.; Cutcutache, I.A.; Diver, L.; Dorris, L.; Gardiner, S.; Jollands, A.; et al. Early childhood epilepsies: Epidemiology, classification, aetiology, and socio-economic determinants. Brain 2021, 144, 2879–2891. [Google Scholar] [CrossRef]
- Chou, I.-J.; Chung, T.-T.; Liu, Y.-H.; Hung, P.-C.; Lin, J.-J.; Chiou, M.-J.; See, L.-C.; Lin, K.-L.; Wang, H.-S. Secular Trends in the Incidence, Prevalence, and Medications for Epilepsy from 2007 to 2015 in Taiwan: A Nationwide Population-Based Study. Neuroepidemiology 2021, 55, 484–494. [Google Scholar] [CrossRef]
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A key regulator of mitochondria-associated membranes (MAMs). Pharmacol. Res. 2020, 160, 105080. [Google Scholar] [CrossRef] [PubMed]
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homoeostasis and disease: Roles of the PACS proteins in membrane traffic and apoptosis. Biochem. J. 2009, 421, 1–15. [Google Scholar] [CrossRef]
- Olson, H.E.; Olson, H.E.; Jean-Marçais, N.; Yang, E.; Heron, D.; Tatton-Brown, K.; van der Zwaag, P.A.; Bijlsma, E.K.; Krock, B.L.; Backer, E.; et al. A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epi-leptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis. Am. J. Hum. Genet. 2018, 102, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, M.; Benzing, T.; Simmen, T.; Tauber, R.; Buchholz, B.; Feliciangeli, S.; Huber, T.B.; Schermer, B.; Kramer-Zucker, A.; Höpker, K.; et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 2005, 24, 705–716. [Google Scholar] [CrossRef]
- Hotka, M.; Kubista, H. The paroxysmal depolarization shift in epilepsy research. Int. J. Biochem. Cell Biol. 2019, 107, 77–81. [Google Scholar] [CrossRef]
- Pathak, D. Paroxysmal Depolarization Shift in Leech Retzius Nerve Cells Revisited. MOJ Anat. Physiol. 2017, 3, 7–9. [Google Scholar] [CrossRef]
- Feng, Y.C.; Howrigan, D.P.; Abbott, L.E.; Tashman, K.; Cerrato, F.; Singh, T.; Heyne, H.; Byrnes, A.; Churchhouse, C.; Watts, N.; et al. Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am. J. Hum. Genet. 2019, 105, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, D.; Luo, T.; Wang, Y.; Liu, Z. Phenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy. Epileptic Disord. 2022, 24, 343–352. [Google Scholar] [CrossRef]
- Mizuno, T.; Miyata, R.; Hojo, A.; Tamura, Y.; Nakashima, M.; Mizuguchi, T.; Matsumoto, N.; Kato, M. Clinical variations of epileptic syndrome associated with PACS2 variant. Brain Dev. 2020, 43, 343–347. [Google Scholar] [CrossRef]
- Wu, M.J.; Hu, C.H.; Ma, J.H.; Hu, J.S.; Liu, Z.S.; Sun, D. Early infantile epileptic encephalopathy caused by PACS2 gene variation: Three cases report and literature review. Zhonghua Er Ke Za Zhi. 2021, 59, 594–599. [Google Scholar]
- Dentici, M.L.; Barresi, S.; Niceta, M.; Ciolfi, A.; Trivisano, M.; Bartuli, A.; Digilio, M.C.; Specchio, N.; Dallapiccola, B.; Tartaglia, M. Expanding the clinical spectrum associated with PACS2 mutations. Clin. Genet. 2019, 95, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Marchese, F.; Vari, M.S.; Severino, M.; Madia, F.; Amadori, E.; Del Giudice, E.; Romano, A.; Gennaro, E.; Zara, F.; et al. A further contribution to the delineation of epileptic phenotype in PACS2-related syndrome. Seizure 2020, 79, 53–55. [Google Scholar] [CrossRef]
- Cesaroni, E.; Matricardi, S.; Cappanera, S.; Marini, C. First reported case of an inherited PACS2 pathogenic variant with variable expression. Epileptic Disord. 2022, 24, 572–576. [Google Scholar] [CrossRef]
- Perulli, M.; Picilli, M.; Contaldo, I.; Amenta, S.; Gambardella, M.L.; Quintiliani, M.; Musto, E.; Turrini, I.; Veredice, C.; Zollino, M.; et al. Pyridoxine supplementation in PACS2-related encephalopathy: A case report of possible precision therapy. Seizure 2023, 105, 14–16. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yoshihashi, H.; Uehara, T.; Miyama, S.; Kosaki, K.; Takenouchi, T. Coloboma may be a shared feature in a spectrum of disorders caused by mutations in the WDR37-PACS1-PACS2 axis. Am. J. Med. Genet. Part A 2020, 185, 884–888. [Google Scholar] [CrossRef]
- Stanojević, M.; Lopicic, S.; Jovanovic, Z.; Pathak, D.; Pavlovic, D.V.; Spasic, S.; Nedeljkov, V.; Prostran, M.; Marija, S.; Srdjan, L.; et al. Magnesium Effects on Nonsynaptic Epileptiform Activity in Leech Retzius Neurons. Folia Biol. 2015, 63, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Pathak, D.; Foehring, R.C. Functional roles of Kv1-mediated currents in genetically identified subtypes of py-ramidal neurons in layer 5 of mouse somatosensory cortex. J. Neurophysiol. 2018, 120, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Lopicic, S.; Stanojevic, M.; Nedeljkov, A.; Pavlovic, D.; Cemerikic, D.; Nedeljkov, V. Ethanol and magnesium suppress nickel-induced bursting activity in leech Retzius nerve cells. Gen. Physiol. Biophys. 2009, 28, 9–17. [Google Scholar]
- Nan, Y.; Lin, J.; Cui, Y.; Yao, J.; Yang, Y.; Li, Q. Protective role of vitamin B6 against mitochondria damage in Drosophila models of SCA3. Neurochem. Int. 2021, 144, 104979. [Google Scholar] [CrossRef]
- Zang, R.X.; Mumby, M.J.; Dikeakos, J.D. The Phosphofurin Acidic Cluster Sorting Protein 2 (PACS-2) E209K Mutation Responsible for PACS-2 Syndrome Increases Susceptibility to Apoptosis. ACS Omega 2022, 7, 34378–34388. [Google Scholar] [CrossRef]
- Kollias, S.S.; Ball, W.S., Jr.; Prenger, E.C. Cystic malformations of the posterior fossa: Differential diagnosis clarified through embryologic analysis. Radiographics 1993, 13, 1211–1231. [Google Scholar] [CrossRef] [PubMed]
- Sereno, M.I.; Diedrichsen, J.; Tachrount, M.; Testa-Silva, G.; D’Arceuil, H.; De Zeeuw, C. The human cerebellum has almost 80% of the surface area of the neocortex. Proc. Natl. Acad. Sci. USA 2020, 117, 19538–19543. [Google Scholar] [CrossRef] [PubMed]
- Volpe, P.; Contro, E.; De Musso, F.; Ghi, T.; Farina, A.; Tempesta, A.; Volpe, G.; Rizzo, N.; Pilu, G. Brainstem-vermis and brainstem-tentorium angles allow accurate categorization of fetal upward rotation of cerebellar vermis. Ultrasound Obstet. Gynecol. 2012, 39, 632–635. [Google Scholar] [CrossRef]
- Robinson, A.J.; Ederies, M.A. Diagnostic imaging of posterior fossa anomalies in the fetus. Semin. Fetal Neonatal Med. 2016, 21, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Tarui, T.; Limperopoulos, C.; Sullivan, N.R.; Robertson, R.; Du Plessis, A.J. Long-term developmental outcome of children with a fetal diagnosis of isolated inferior vermian hypoplasia. Arch. Dis. Child. Fetal Neonatal Ed. 2013, 99, F54–F58. [Google Scholar] [CrossRef] [PubMed]
- Amore, G.; Spoto, G.; Ieni, A.; Vetri, L.; Quatrosi, G.; Di Rosa, G.; Nicotera, A.G. A Focus on the Cerebellum: From Embryogenesis to an Age-Related Clinical Perspective. Front. Syst. Neurosci. 2021, 15, 646052. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Goldstein, R. The cisterna magna septa: Vestigial remnants of Blake’s pouch and a potential new marker for normal development of the rhombencephalon. J. Ultrasound Med. 2007, 26, 83–95. [Google Scholar] [CrossRef]
- Zimmer, E.Z.; Lowenstein, L.; Bronshtein, M.; Goldsher, D.; Aharon-Peretz, J. Clinical significance of isolated mega cisterna magna. Arch. Gynecol. Obstet. 2007, 276, 487–490. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Case | Sex | Age of Onset | Seizure Types | MRI | WES (PACS2, Heterozygous Missense Variant) | Inheritance | Age at Last Follow Up | AEDs at Last Follow-Up | Seizure Frequency | Development Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 2 w | GTCs, focal (upper limb) | MCM | c.625G>A (p.Glu209Lys) | de novo | 5 y | B6, LEV | controlled | hypotonia, speech delay | see Case 1 |
| 2 | F | 1 m | tonic, autonomic dysfunction | VH, MCM | c.625G>A (p.Glu209Lys) | de novo | 10 y | B6, VPA | controlled | ADHD | see Case 2 |
| 3 | F | 2 w | tonic, autonomic dysfunction, focal (head-turning, upper limbs, left leg, cycling), GTCs, vocalizations | MCM; Other: microcephaly | c.625G>A (p.Glu209Lys) | de novo | 6 y | BP, VGB, CLN | uncontrolled | severe developmental delay, severe MR, hypotonia | see Case 3 |
| 4 | M | 10 d | focal, tonic | negative | c.625G>A (p.Glu209Lys) | de novo | 5 y | B6, LEV, VPA | controlled | moderate developmental delay | Hu, C. et al. [11] |
| 5 | F | 1 m | focal, tonic | microcephaly | c.625G>A (p.Glu209Lys) | de novo | 2 y | B6, LEV, TPM, BP, VPA, KD | controlled | severe developmental delay | Hu, C. et al. [11] |
| 6 | F | 1 m | focal, tonic | negative | c.625G>A (p.Glu209Lys) | de novo | 2 y | B6, LEV, VPA | controlled | moderate developmental delay | Hu, C. et al. [11] |
| 7 | F | 6 d | focal | CD, MCM | c.625G>A (p.Glu209Lys) | de novo | 16 y | CBZ | Olson, H.E. et al. [6] | ||
| 8 | F | 4 d | GTCs | CD, VH, MCM | c.625G>A (p.Glu209Lys) | de novo | 4 y | PB, VPA | controlled | wide-based gait, sleeping, and behavioral disturbances | Olson, H.E. et al. [6] |
| 9 | M | 4 d | Other: increased subarachnoid spaces | c.625G>A (p.Glu209Lys) | de novo | 15 y | CBZ | Olson, H.E. et al. [6] | |||
| 10 | F | 7 d | GTCs | CD, VH, MCM | c.625G>A (p.Glu209Lys) | de novo | 8 y | B6, P5P, VPA, PB | controlled | ASD | Olson, H.E. et al. [6] |
| 11 | M | 2 d | clonic and GTCs | CD, VH, MCM | c.625G>A (p.Glu209Lys) | de novo | 1 y 7 m | LEV, PB, CBZ | controlled | Olson, H.E. et al. [6] | |
| 12 | M | 2 d | status epilepticus | negative | c.625G>A (p.Glu209Lys) | de novo | 8 y | TPM | controlled | OCD | Olson, H.E. et al. [6] |
| 13 | M | 2 d | focal, tonic, autonomic dysfunction | VH, other: retrocerebellar cyst | c.625G>A (p.Glu209Lys) | de novo | 1 y 4 m | PB | Olson, H.E. et al. [6] | ||
| 14 | F | 2 w | focal, tonic | negative | c.625G>A (p.Glu209Lys) | de novo | 5 y | AEDs discontinued | AEDs discontinued at 3.5 y | atypical social and behavioral features | Olson, H.E. et al. [6] |
| 15 | F | 2 d | focal, tonic, myoclonic, GTCs | CD, MCM | c.625G>A (p.Glu209Lys) | de novo | 3 y | LEV, PB | controlled | atypical social and behavioral features | Olson, H.E. et al. [6] |
| 16 | M | 1–2 m | clonic, GTCs | MCM | c.625G>A (p.Glu209Lys) | de novo | 7 y | LEV | controlled | ASD | Olson, H.E. et al. [6] |
| 17 | M | 1 d | focal, GTCs | VH | c.625G>A (p.Glu209Lys) | de novo | 12.5 y | VPA | ASD | Olson, H.E. et al. [6] | |
| 18 | F | 3 d | focal, tonic, GTCs, status epilepticus | CD, MCM | c.625G>A (p.Glu209Lys) | de novo | 9 m | LEV, PB, OCZ | Olson, H.E. et al. [6] | ||
| 19 | F | 2 w | focal, GTCs | CD | c.625G>A (p.Glu209Lys) | de novo | 3.5 y | B6, P5P, LEV, VGB, LMT, VPA, CLB | Olson, H.E. et al. [6] | ||
| 20 | F | 3 d | tonic, GTCs | CD, MCM; Other: SAH | c.625G>A (p.Glu209Lys) | de novo | 5.5 y | B6, PB, LEV, LCS | wide-based gait, selective mutism | Olson, H.E. et al. [6] | |
| 21 | F | 2 w | tonic, head-turning | other: signs of perinatal injury | c.625G>A (p.Glu209Lys) | de novo | 2 y | CBZ, CLB | controlled | hypotonia, mild developmental delay | Mizuno, T. et al. [12] |
| 22 | F | 3 d | focal, GTCs | Normal | c.625G>A (p.Glu209Lys) | de novo | 12 y | LMT, VPA, CLN | 4 y controlled; 9 y Lennox–Gastaut syndrome; 12 y almost controlled | ASD, severe MR, hypotonia, walking alone | Mizuno, T. et al. [12] |
| 23 | F | 3 d | tonic | Other: right venous sinus thrombosis | c.625G>A (p.Glu209Lys) | de novo | 3 y | AEDs discontinued | AEDs discontinued | normal psychomotor development | Mizuno, T. et al. [12] |
| 24 | F | 7 d | focal, GTC | c.625G>A (p.Glu209Lys) | de novo | 2 y 2 m | VPA | uncontrolled (poor compliance) | Wu, M.J. et al. [13] | ||
| 25 | F | 5 d | focal | CD | c.625G>A (p.Glu209Lys) | de novo | 5 m | LEV, VPA | controlled | Wu, M.J. et al. [13] | |
| 26 | F | 3 d | tonic | c.625G>A (p.Glu209Lys) | de novo | 5 m | LEV, VPA | controlled | Wu, M.J. et al. [13] | ||
| 27 | M | 3 d | focal (upper limb) | CD | c.631G>A (p.Glu211Lys) | de novo | 7 y | AEDs discontinued | AEDs discontinued | moderate MR (IQ = 47) | Dentici, M.L. et al. [14] |
| 28 | F | 2 d | tonic (upper limbs), autonomic dysfunction | CD | c.625G>A (p.Glu209Lys) | de novo | 2 y 2 m | CBZ | controlled | Terrone, G. et al. [15] | |
| 29 | M | 7 d | focal | VH, MCM | c.625G>A (p.Glu209Lys) | Maternal Inheritance | 1 y 7 m | CBZ | uncontrolled | hypotonia, moderate to severe developmental retardation | Cesaroni, E. et al. [16] |
| 30 | F | a few weeks | tonic | CT: negative | c.625G>A (p.Glu209Lys) | 37 y | seizure free | AEDs discontinued at several years old | learning disability | Cesaroni, E. et al. [16] | |
| 31 | M | 8 d | tonic | MCM | c.625G>A (p.Glu209Lys) | de novo | 11 y | seizure free | AEDs discontinued; B6, P5P, folinic acid | mild MR (IQ = 62), ASD, motor and speech delay | Perulli, M. et al. [17] |
| 32 | M | 2 m | tonic | CD, other: cerebellar progressive atrophy | c.625G>A (p.Glu209Lys) | de novo | 23 y | seizure free | AEDs discontinued at 9 y | low average IQ (IQ = 85), hypotonia, wide-based gait | Sakaguchi, Y. et al. [18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, I.-J.; Hou, J.-Y.; Fan, W.-L.; Tsai, M.-H.; Lin, K.-L. Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review. Children 2023, 10, 621. https://doi.org/10.3390/children10040621
Chou I-J, Hou J-Y, Fan W-L, Tsai M-H, Lin K-L. Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review. Children. 2023; 10(4):621. https://doi.org/10.3390/children10040621
Chicago/Turabian StyleChou, I-Jun, Ju-Yin Hou, Wen-Lang Fan, Meng-Han Tsai, and Kuang-Lin Lin. 2023. "Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review" Children 10, no. 4: 621. https://doi.org/10.3390/children10040621
APA StyleChou, I.-J., Hou, J.-Y., Fan, W.-L., Tsai, M.-H., & Lin, K.-L. (2023). Long-Term Outcome of Neonatal Seizure with PACS2 Mutation: Case Series and Literature Review. Children, 10(4), 621. https://doi.org/10.3390/children10040621