
Modelling Nonalcoholic Steatohepatitis In Vivo—A Close Transcriptomic Similarity Supports the Guinea Pig Disease Model

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
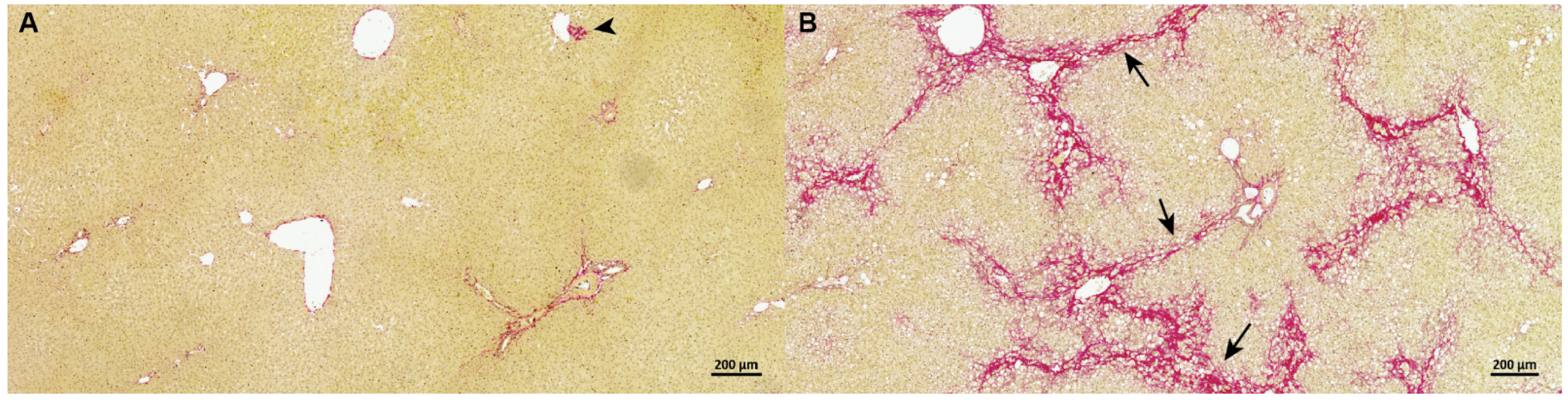
2.2. Liver Samples and Histology
2.3. Guinea Pig Hepatic RNA Sequencing
2.4. Transcriptome Analyses
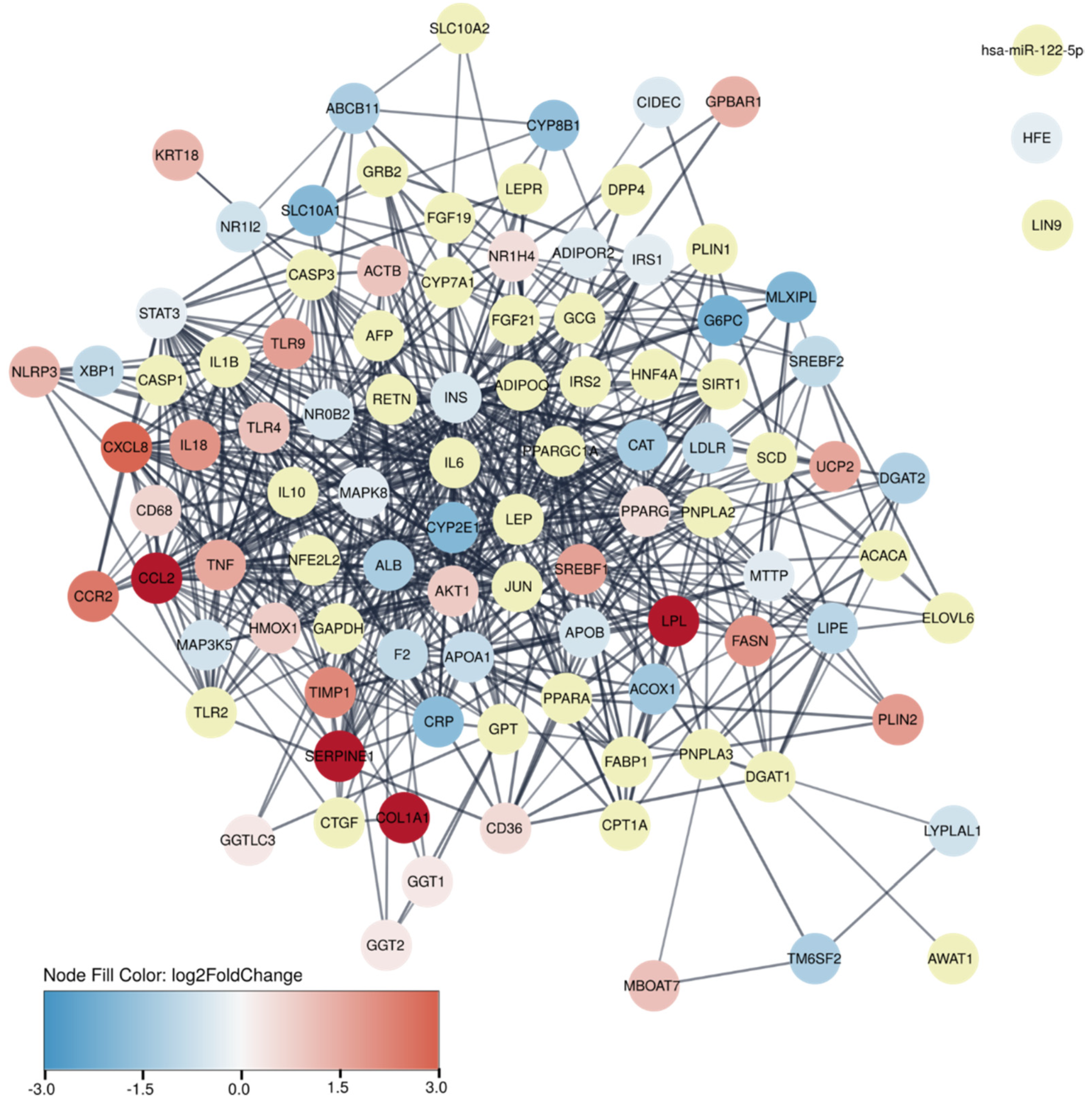
2.5. Protein–Protein Association Network
2.6. Translational Aspects of the Guinea Pig Model
2.7. Correlation Analysis of Gene Expression and Fibrosis Quantification
2.8. Statistical Analysis
3. Results
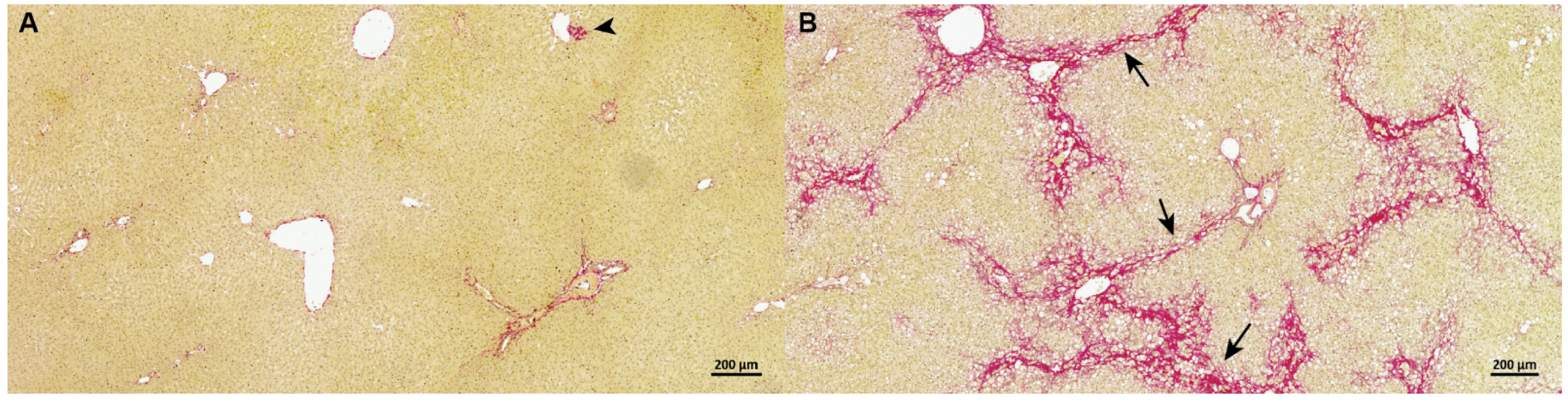
3.1. Guinea Pig NASH Development and Disease Stage
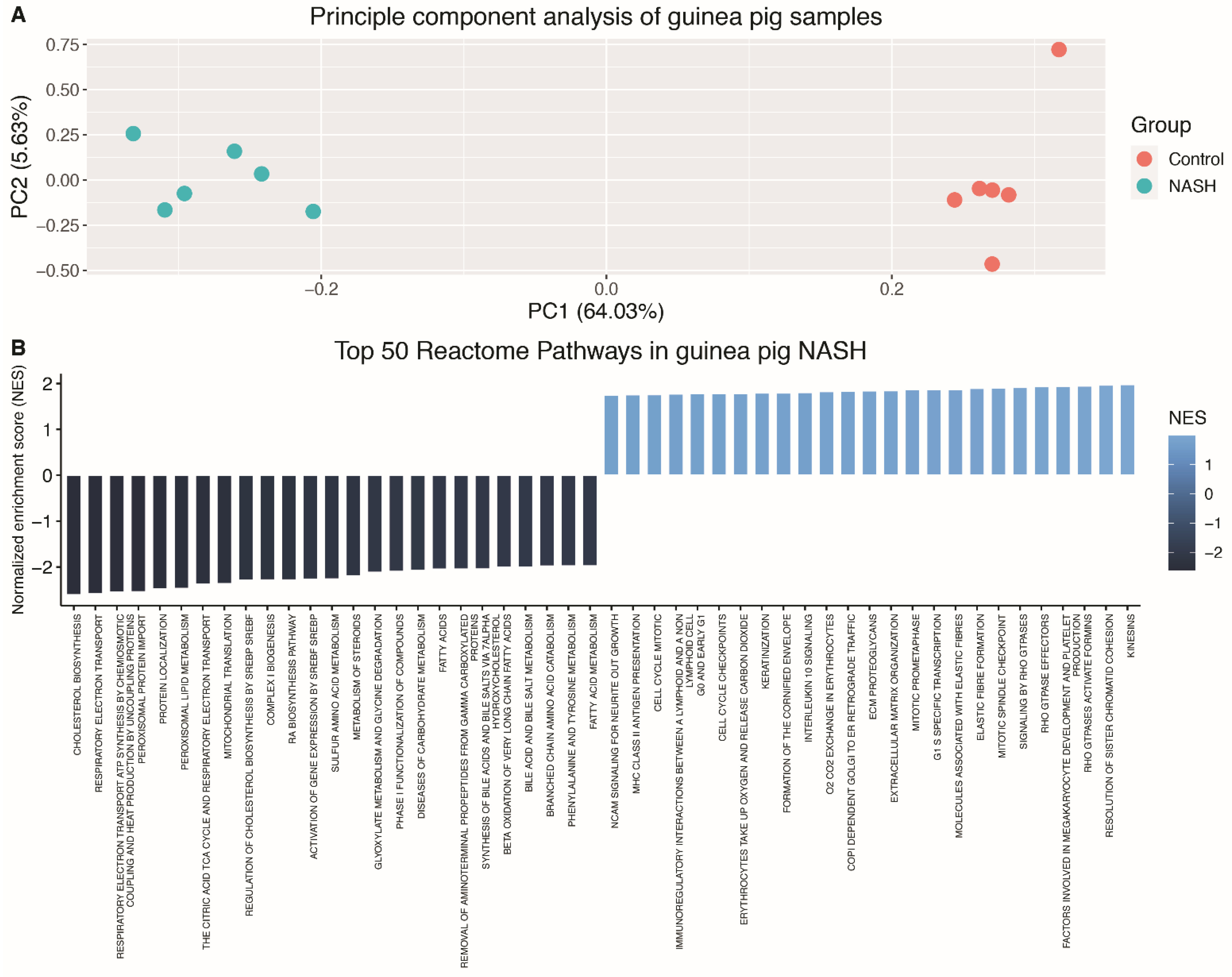
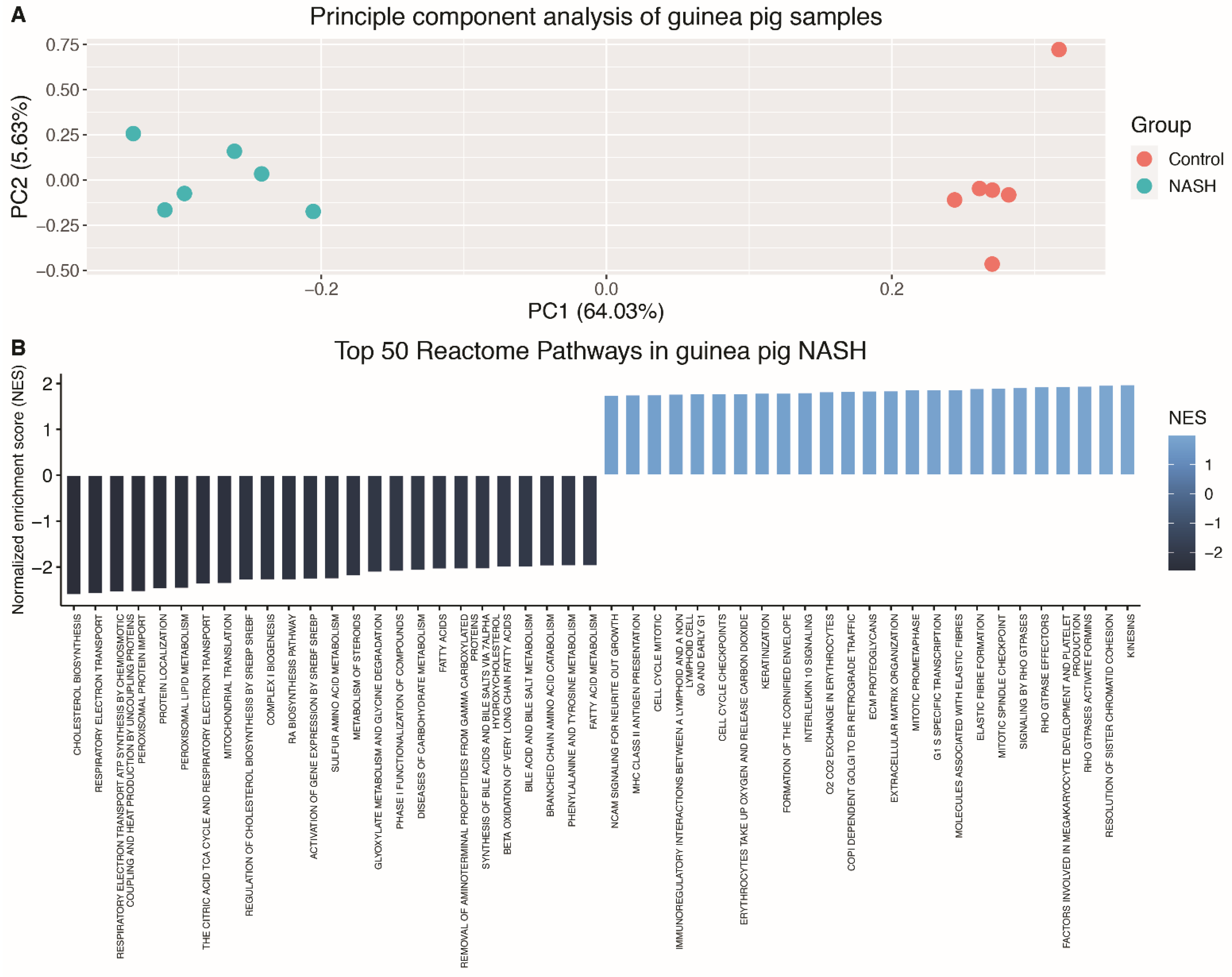
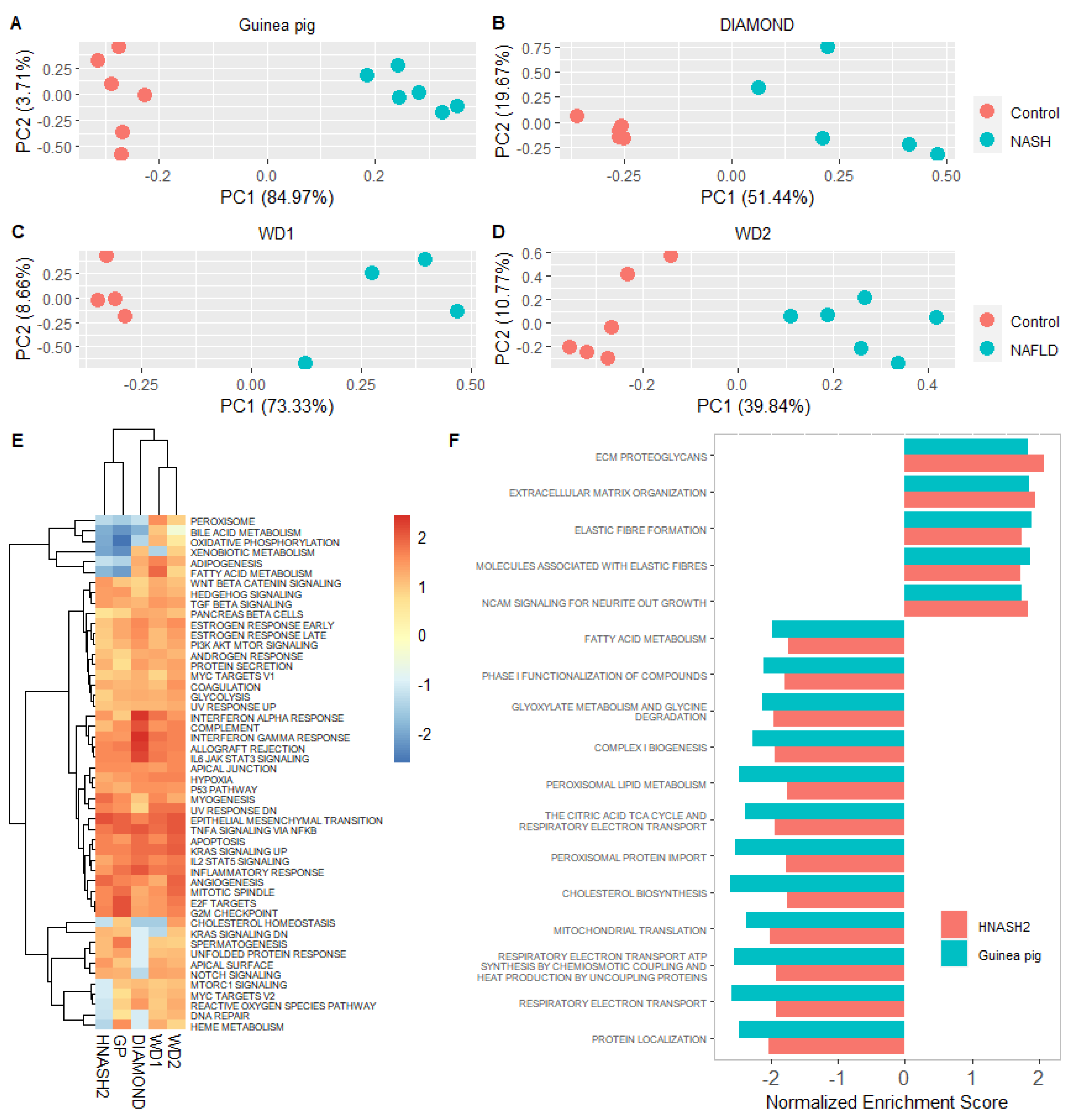
3.2. The Hepatic Transcriptome Clearly Distinguishes Guinea Pigs with NASH from Controls
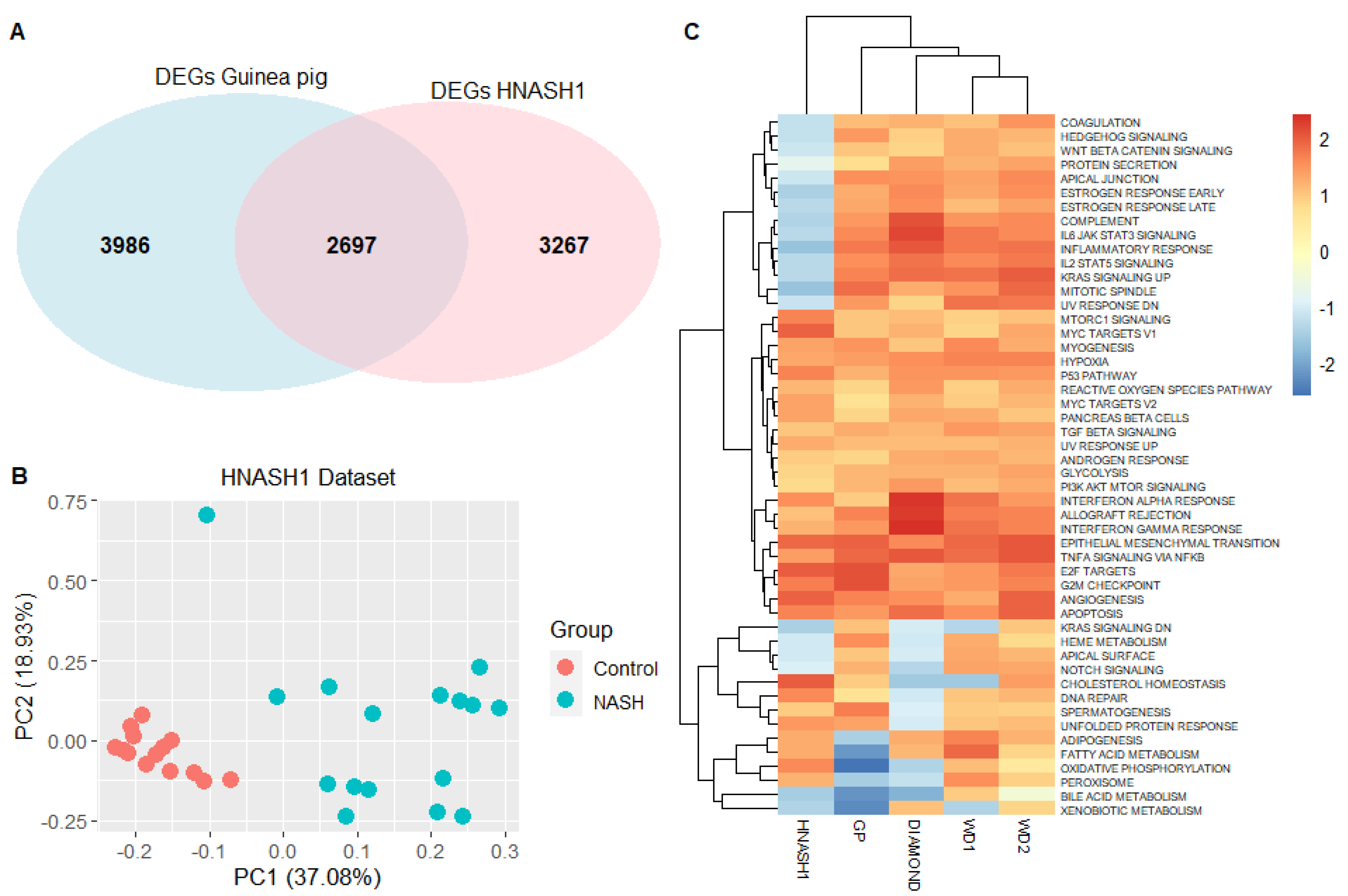
3.3. The Guinea Pig NASH Transcriptome Is Similar to That of Humans with Early-Stage NASH
3.4. Guinea Pig NASH Transcriptome Resembles Human Advanced NASH
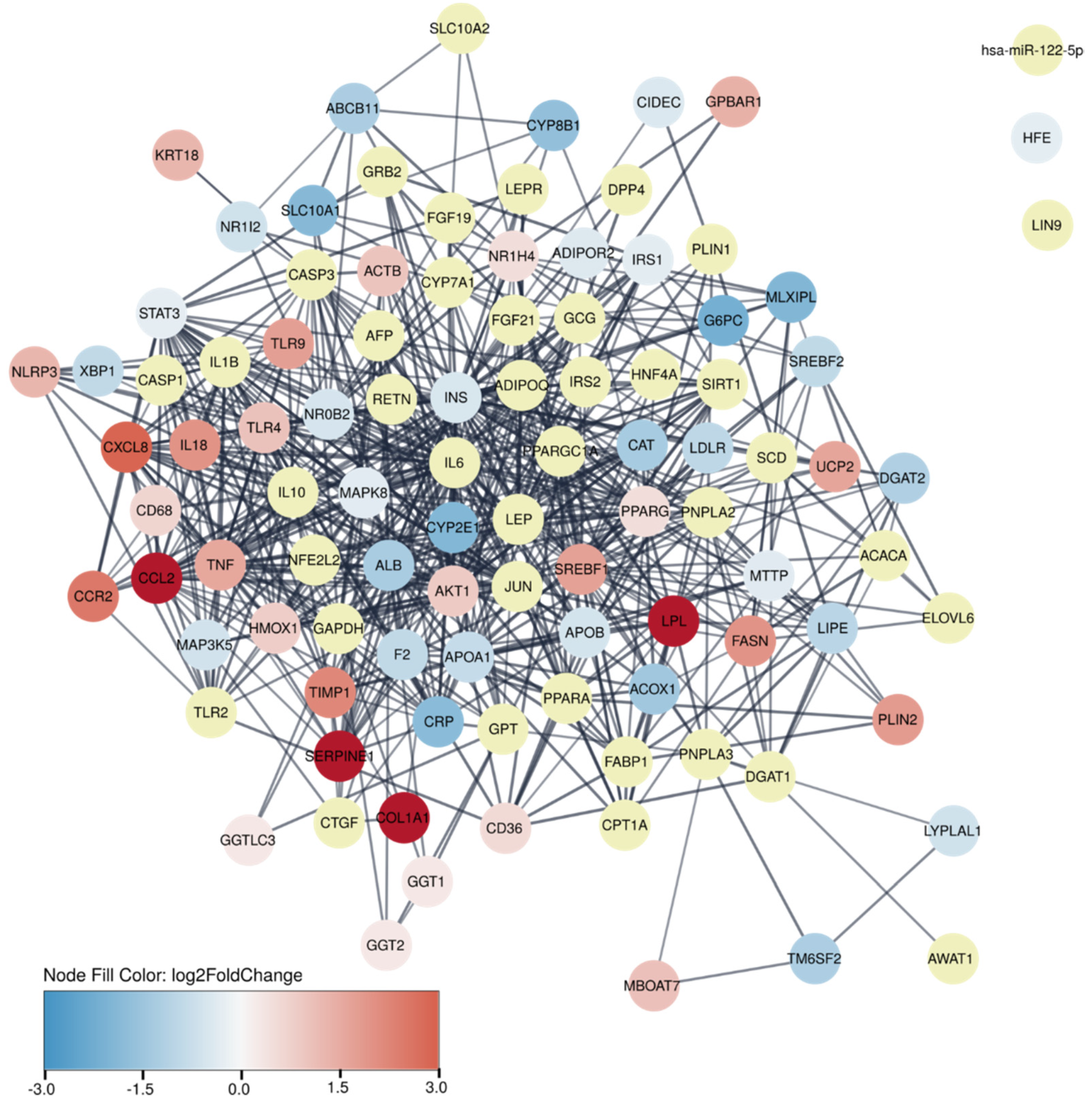
3.5. Identification of Potential New Biomarkers of Fibrosis Deposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagstrom, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wong, V.W.-S.; Peleg, N.; et al. Association btween fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis. Gastroenterology 2020, 158, 1611–1625.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease? Adv. Nutr. 2020, 11, 1696–1711. [Google Scholar] [CrossRef] [PubMed]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525.e0. [Google Scholar] [CrossRef] [PubMed]
- deOgburn, R.; Leite, J.O.; Ratliff, J.; Volek, J.S.; McGrane, M.M.; Fernandez, M.L. Effects of increased dietary cholesterol with carbohydrate restriction on hepatic lipid metabolism in Guinea pigs. Comp. Med. 2012, 62, 109–115. [Google Scholar]
- Ipsen, D.H.; Tveden-Nyborg, P.; Rolin, B.; Rakipovski, G.; Beck, M.; Mortensen, L.W. High-fat but not sucrose intake is essential for induction of dyslipidemia and non-alcoholic steatohepatitis in guinea pigs. Nutr. Metab. 2016, 13, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tveden-Nyborg, P.; Birck, M.M.; Ipsen, D.H.; Thiessen, T.; Feldmann, L.B.; Lindblad, M.M.; Jensen, H.E.; Lykkesfeldt, J. Diet-induced dyslipidemia leads to nonalcoholic fatty liver disease and oxidative stress in guinea pigs. Transl. Res. 2016, 168, 146–160. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Skat-Rordam, J.; Tsamouri, M.M.; Latta, M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular drivers of non-alcoholic steatohepatitis are sustained in mild-to-late fibrosis progression in a guinea pig model. Mol. Genet. Genom. 2019, 6, 309–313. [Google Scholar] [CrossRef]
- deOgburn, R.; Murillo, G.; Luz Fernandez, M. Guinea pigs as models for investigating non-alcoholic fatty liver disease. Integr. Food Nutr. Metab. 2016, 3, 309–313. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- De Rudder, M.; Bouzin, C.; Nachit, M.; Louvegny, H.; Vande Velde, G.; Jule, Y.; Leclercq, I.A. Automated computerized image analysis for the user-independent evaluation of disease severity in preclinical models of NAFLD/NASH. Lab. Invest. 2020, 100, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Cabibi, D.; Bronte, F.; Porcasi, R.; Ingrao, S.; Giannone, A.G.; Maida, M.; Bavetta, M.G.; Petta, S.; Di Marco, V.; Calvaruso, V. Comparison of Histochemical Stainings in Evaluation of Liver Fibrosis and Correlation with Transient Elastography in Chronic Hepatitis. Anal. Cell. Pathol. 2015, 2015, 431750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TimeTree: A Resource for Timelines, Timetrees and Divergence Times. Available online: http://www.timetree.org/ (accessed on 12 December 2019).
- Padilla-Carlin, D.J.; McMurray, D.N.; Hickey, A.J. The guinea pig as a model of infectious diseases. Comp. Med. 2008, 58, 324–340. [Google Scholar]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef]
- Diseases: Text Mining and Data Integration of Disease–Gene Associations. Available online: https://diseases.jensenlab.org/Entity?order=textmining,knowledge,experiments&textmining=10&knowledge=10&experiments=10&type1=-26&type2=9606&id1=DOID:0080547 (accessed on 1 July 2020).
- String: Functional Protein Association Networks. Available online: https://string-db.org/ (accessed on 1 July 2020).
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M. The string database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Gene Expression Omnibus. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 12 April 2020).
- Bedossa, P.; Poitou, C.; Veyrie, N.; Bouillot, J.L.; Basdevant, A.; Paradis, V. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012, 56, 1751–1759. [Google Scholar] [CrossRef]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heeboll, S.; Eriksen, P.L.; Demant, M. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Engelmann, J.C.; Saugspier, M.; Koch, A.; Hartmann, A.; Muller, M. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab. Invest. 2014, 94, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Oike, H. Chronic dietary intake of quercetin alleviates hepatic fat accumulation associated with consumption of a Western-style diet in C57/BL6J mice. Mol. Nutr. Food Res. 2011, 55, 530–540. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Biorxiv 2019, 12, 1554. [Google Scholar]
- MSigDB: Molecular Signatures Database. Available online: https://www.gsea-msigdb.org/gsea/msigdb/index.jsp (accessed on 7 April 2020).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueno, A.L.; Castano, G.O.; Pirola, C.J. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis 2011, 218, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Novak, P.; Lake, A.D.; Shipkova, P.; Aranibar, N.; Robertson, D. Characterization of hepatocellular carcinoma related genes and metabolites in human nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014, 59, 365–374. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hwang, S.; Cai, Y.; Kim, S.J.; Xu, M.; Yang, D. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology 2019, 70, 1150–1167. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z. Von Willebrand Factor Is Associated With Increased Collagen Deposition in Patients With Nonalcoholic Steatohepatitis (NASH): 2084. Am. J. Gastroenterol. 2015, 110, pS874. [Google Scholar] [CrossRef]
- Maeso-Diaz, R.; Boyer-Diaz, Z.; Lozano, J.J.; Ortega-Ribera, M.; Peralta, C.; Bosch, J. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease. Cells 2019, 8, 1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information: Gene. Available online: https://www.ncbi.nlm.nih.gov/gene (accessed on 15 May 2020).
- Aizarani, N.; Saviano, A.; Sagar Mailly, L.; Durand, S.; Herman, J.S. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- A Human Liver Cell Atlas reveals Heterogeneity and Epithelial Progenitors. Available online: http://human-liver-cell-atlas.ie-freiburg.mpg.de/ (accessed on 15 May 2020).
- Juarez-Hernandez, E.; Chavez-Tapia, N.C.; Uribe, M.; Barbero-Becerra, V.J. Role of bioactive fatty acids in nonalcoholic fatty liver disease. Nutr. J. 2016, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.T.; Tseng, P.H.; Tseng, F.Y.; Chi, Y.C.; Han, D.S.; Yang, W.S. The serum level of a novel lipogenic protein Spot 14 was reduced in metabolic syndrome. PLoS ONE 2019, 14, e0212341. [Google Scholar] [CrossRef] [Green Version]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Jin, Q.; Chen, H.; Wood, G.C.; Petrick, A.; Strodel, W. CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J. Transl. Med. 2018, 16, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, G.S.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; DiStefano, J.K. Transcriptomic Profiling of Obesity-Related Nonalcoholic Steatohepatitis Reveals a Core Set of Fibrosis-Specific Genes. J. Endocr. Soc. 2018, 2, 710–726.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patouraux, S.; Rousseau, D.; Bonnafous, S.; Lebeaupin, C.; Luci, C.; Canivet, C.M. CD44 is a key player in non-alcoholic steatohepatitis. J. Hepatol. 2017, 67, 328–338. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Kubes, P. Interactions between CD44 and Hyaluronan in Leukocyte Trafficking. Front. Immunol. 2015, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Egan, C.E.; Daugherity, E.K.; Rogers, A.B.; Abi Abdallah, D.S.; Denkers, E.Y.; Maurer, K.J. CCR2 and CD44 promote inflammatory cell recruitment during fatty liver formation in a lithogenic diet fed mouse model. PLoS ONE 2013, 8, e65247. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Agerskov, R.H.; Klaebel, J.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. The development of nonalcoholic steatohepatitis is subjected to breeder dependent variation in guinea pigs. Sci. Rep. 2021, 11, 2955. [Google Scholar] [CrossRef]
- Ferlitsch, M.; Reiberger, T.; Hoke, M.; Salzl, P.; Schwengerer, B.; Ulbrich, G. Von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012, 56, 1439–1447. [Google Scholar] [CrossRef]
- Schwarzer, R.; Reiberger, T.; Mandorfer, M.; Kivaranovic, D.; Hametner, S.; Hametner, S. The von Willebrand Factor antigen to platelet ratio (VITRO) score predicts hepatic decompensation and mortality in cirrhosis. J. Gastroenterol. 2020, 55, 533–542. [Google Scholar] [CrossRef] [PubMed]
- van Mourik, J.A.; Boertjes, R.; Huisveld, I.A.; Fijnvandraat, K.; Pajkrt, D.; van Genderen, P.J. von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood 1999, 94, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Maieron, A.; Salzl, P.; Peck-Radosavljevic, M.; Trauner, M.; Hametner, S.; Schofl, R. Von Willebrand Factor as a new marker for non-invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2014, 39, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Chen, E.; Chen, J.; Zhang, J.; Zhang, N.; Chen, Z. Overexpression of proteinase inhibitor 9 is associated with poor prognosis in human hepatocellular carcinoma and with proliferation and apoptosis in HepG2 cells in vitro. Int. J. Clin. Exp. Pathol. 2019, 12, 3719–3727. [Google Scholar]
- Unutmaz, D.; Willberg, C.B.; Ward, S.M.; Clayton, R.F.; Naoumov, N.V.; McCormick, C. Protection of Hepatocytes from Cytotoxic T Cell Mediated Killing by Interferon-Alpha. PLoS ONE 2007, 2, 8. [Google Scholar]
- Rowshani, A.T.; Strik, M.C.M.; Molenaar, R.; Yong, S.-L.; Wolbink, A.M.; Bemelman, F.J. The Granzyme B Inhibitor SERPINB9 (Protease Inhibitor 9) Circulates in Blood and Increases on Primary Cytomegalovirus Infection after Renal Transplantation. J. Infect. Dis. 2005, 192, 1908–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Groth, E.; Pruessmeyer, J.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Dreymueller, D. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim. Biophys. Acta 2016, 1863, 2795–2808. [Google Scholar] [CrossRef]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Model | Sex | Species: Strain | Weeks on Diet | Histological Phenotype |
|---|---|---|---|---|
| Guinea pig | Female | Guinea pig: Dunkin-Hartley | 25 | NASH with fibrosis. Histological scoring: NASH CRN [10] |
| DIAMOND [32] | Male | Mouse: B6/129 (isogenic cross between C57BL/6J and 129S1/SvImJ) | 52 | NASH with fibrosis Histological scoring: NASH CRN [10] |
| WD1 [30] | Male | Mouse: C57BL/6N | 12 | NAFLD No histological scoring. Positive α-sma liver stain and picrosirius red indicative of activated hepatic stellate cells and fibrosis, respectively |
| WD2 [31] | Male | Mouse: C57BL/6J | 20 | NAFLD. No histological scoring |
| Group | Control (n = 6) | NASH (n = 6) |
|---|---|---|
| Steatosis 1 | 0 | 3 ** |
| Ballooning 1 | 0 | 2 (1–2) * |
| Inflammation 1 | 0 (0–1) | 3 ** |
| Fibrosis 1 | 0 | 3 ** |
| Relative fibrosis area 2 | 1.39 ± 0.24 | 7.48 ± 1.81 *** |
| Gene Name (Full Name) | Log2 Fold Change | Adjusted p-Value | Function |
|---|---|---|---|
| ADAMDEC1 (ADAM like decysin 1) | 8.01 | 4.94 ×10−60 | Secreted protein invovled in dendritic cell maturation. |
| ADGRG3 (adhesion G protein-coupled receptor 3) | 7.66 | 3.79 × 10−40 | GPCR possibly invovled tumor angiogenesis. |
| KRT23 (keratin 23) | 10.97 | 1.53 × 10−35 | Member of keratin family of intermediate filaments responsible for structural integrity of epithelial cells. |
| ATP6V0A4 (ATPase H + transporting V0 subunit a4) | 9.02 | 3.01 × 10−32 | Vacuolar ATPase mediating acidification of intracellular compartments necessary for protein sorting, zymogen activation, receptor-mediated endocytosis and synaptic vesicle protein gradient generation. |
| TMEM213 (transmembrane protein 213) | 10.12 | 1.28 × 10−29 | No listed function. |
| CIB4 (calcium and integrin binding family member 4) | −9.70 | 1.82 × 10−26 | No listed function. |
| PAK6 (p21 (RAC1) activated kinase 6) | 9.38 | 5.69 × 10−26 | p21 stimulated serine/threonine kinase involved in cytoskeleton rearrangement, apoptosis and MAP kinase signalling pathway. |
| TMC1 (transmembrane channel like 1) | 9.27 | 1.82 × 10−25 | No listed function. |
| CCL7 (C–C motif chemokine ligand 7) | 9.09 | 2.30 × 10−25 | Encodes MCP3-a secreted chemokine recruiting macrophages during inflammation, and also a substrate of MMP2. |
| PTPRN (protein tyrosine phosphatase receptor type N) | 9.23 | 2.58 × 10−24 | Signalling molecule regulating processes such as cell growth, differentiation, mitotic cycle, and oncogenic transformation. |
| VSIG1 (V-set and immunoglobulin domain containing 1) | 8.26 | 1.06 × 10−22 | Encodes a member of the junctional adhesion molecule (JAM) family. |
| SLC34A2 (solute carrier family 34 member 2) | 8.49 | 1.13 × 10−20 | pH-sensitive sodium-dependent phosphate transporter |
| DSG4 (desmoglein 4) | 8.15 | 2.78 × 10−19 | Desmosomal cadherin possibly playing a role in cell–cell adhesion in epithelial cells. |
| TNFSF18 (TNF super family member 18) | 7.74 | 1.92 × 10−17 | Cytokine belonging to the TNF ligand family that plays a role in T-lymphocyte survival and the interaction between endothelial cells and T lymphocytes. |
| MTHFD2 (methylenetetrahydrofolate dehydrogenase (NADP + dependent) 2, methenyltetrahydrofolate cyclohydrolase) | 7.63 | 6.53 × 10−17 | Nuclear encoded mitochondrial bifunctional enzyme with methylenetetrahydrofolate dehydrogenase and methenyltetrahydrofolate cyclohydrolase activities. |
| SPOCK1 (SPARC (osteonectin), cwcv and kazal-like domains proteoglycan 1) | 7.72 | 8.38 × 10−17 | Seminal plasma proteoglycan containing chondroitin and heperan sulfate chains. |
| ECT2 (epithelial cell transforming 2) | 8.43 | 5.94 × 10−16 | Guanine nucleotide exchange factor, expressed at high levels in mitotic cells in the regenerating liver. |
| SPTA1 (spectrin alpha, erythrocytic 1) | 7.99 | 7.35 × 10−15 | Molecular scaffold protein that links the plasma membrane to the actin cytoskeleton and determines the cell shape. |
| STAR (steroidogenic acute regulatory protein) | 8.36 | 2.48 × 10−14 | Involved in the acute regulation of steroid hormone synthesis by enhancing the conversion of cholesterol into pregnolone. |
| KEL (Kell metalloendopeptidase (Kell blood group)) | 7.66 | 1.34 × 10−12 | Encodes a type II transmembrane glycoprotein of the Kell blood group antigen. |
| Gene | Pearson’s ρ | p Value | GPLog2FC | HLog2FC | Function 1 | Secreted | Role in NASH | Cell-Specific Expression 2 |
|---|---|---|---|---|---|---|---|---|
| ACKR3 | All: 0.91 NASH: 0.87 | All: 4.54 × 10−5 NASH: 0.025 | 1.36 | HNASH1: ND HNASH2: 0.69 | GPCR, orphan receptor | NO | ? | Endothelial cells |
| BIRC3 | All: 0.88 NASH: 0.82 | All: 1.77 × 10−4 NASH: 0.045 | 1.1 | HNASH1: 1.9 HNASH2: 0.7 | Inhibits apoptosis | NO | YES (hypoxia induced) [39] | Immune cells, cholangiocytes, endothelial cells, and hepatocytes |
| CHST11 | All: 0.95 NASH: 0.87 | All: 1.42 × 10−6 NASH: 0.023 | 1.16 | HNASH1: −0.35 HNASH2: 0.19 | Promotes synthesis of chondroitin (ECM) | NO | ? | Immune cells |
| EMP3 | All: 0.93 NASH: 0.81 | All: 9.41 × 10−6 NASH: 0.049 | 1.5 | HNASH1: 0.98 HNASH2: 0.19 | Membrane protein, cell proliferation | NO | ? | Immune cells |
| FZD7 | All: 0.87 NASH: 0.83 | All: 2.22 × 10−4 NASH: 0.041 | 1.2 | HNASH1: ND HNASH2: 0.6 | Wnt signalling | NO | YES in HCC [40] | Cholangiocytes, HSC |
| RGS14 | All: −0.83 NASH: −0.80 | All: 8.26 × 10−4 NASH: 0.053 | −1.58 | HNASH1: −0.3 HNASH2: −0.2 | Regulates GPCR (increases microtubule assembly) | NO | ? | Immune cells |
| RHBDF1 | All: 0.96 NASH: 0.92 | All: 4.11 × 10−7 NASH: 0.010 | 1.17 | HNASH1: 0.6 HNASH2: 0.03 | Regulates ADAM17 and release of TNF-α | NO | ? | Cholangiocytes |
| SERPINB9 | All: 0.9 NASH: 0.84 | All: 6.19 × 10−5 NASH: 0.037 | 1.4 | HNASH1: −0.7 HNASH2: 0.5 | Inhibits activity of granzyme B | YES | YES [41] | Immune cells, endothelial cells, stellate cells, and myofibroblasts, macrovascular endothelial cells |
| VWF | All: 0.97 NASH: 0.86 | All: 7.52 × 10−8 NASH: 0.027 | 1.46 | HNASH1: −0.03 HNASH2: 0.5 | Platelet aggregation | YES | YES [42,43] | Macrovascular endothelial cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skat-Rørdam, J.; Ipsen, D.H.; Seemann, S.E.; Latta, M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Modelling Nonalcoholic Steatohepatitis In Vivo—A Close Transcriptomic Similarity Supports the Guinea Pig Disease Model. Biomedicines 2021, 9, 1198. https://doi.org/10.3390/biomedicines9091198
Skat-Rørdam J, Ipsen DH, Seemann SE, Latta M, Lykkesfeldt J, Tveden-Nyborg P. Modelling Nonalcoholic Steatohepatitis In Vivo—A Close Transcriptomic Similarity Supports the Guinea Pig Disease Model. Biomedicines. 2021; 9(9):1198. https://doi.org/10.3390/biomedicines9091198
Chicago/Turabian StyleSkat-Rørdam, Josephine, David H. Ipsen, Stefan E. Seemann, Markus Latta, Jens Lykkesfeldt, and Pernille Tveden-Nyborg. 2021. "Modelling Nonalcoholic Steatohepatitis In Vivo—A Close Transcriptomic Similarity Supports the Guinea Pig Disease Model" Biomedicines 9, no. 9: 1198. https://doi.org/10.3390/biomedicines9091198
APA StyleSkat-Rørdam, J., Ipsen, D. H., Seemann, S. E., Latta, M., Lykkesfeldt, J., & Tveden-Nyborg, P. (2021). Modelling Nonalcoholic Steatohepatitis In Vivo—A Close Transcriptomic Similarity Supports the Guinea Pig Disease Model. Biomedicines, 9(9), 1198. https://doi.org/10.3390/biomedicines9091198