
The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease



Abstract
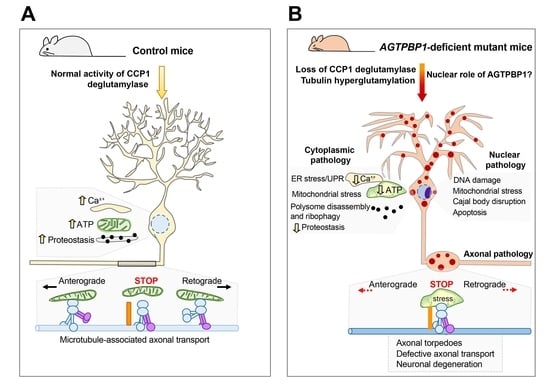
1. Introduction
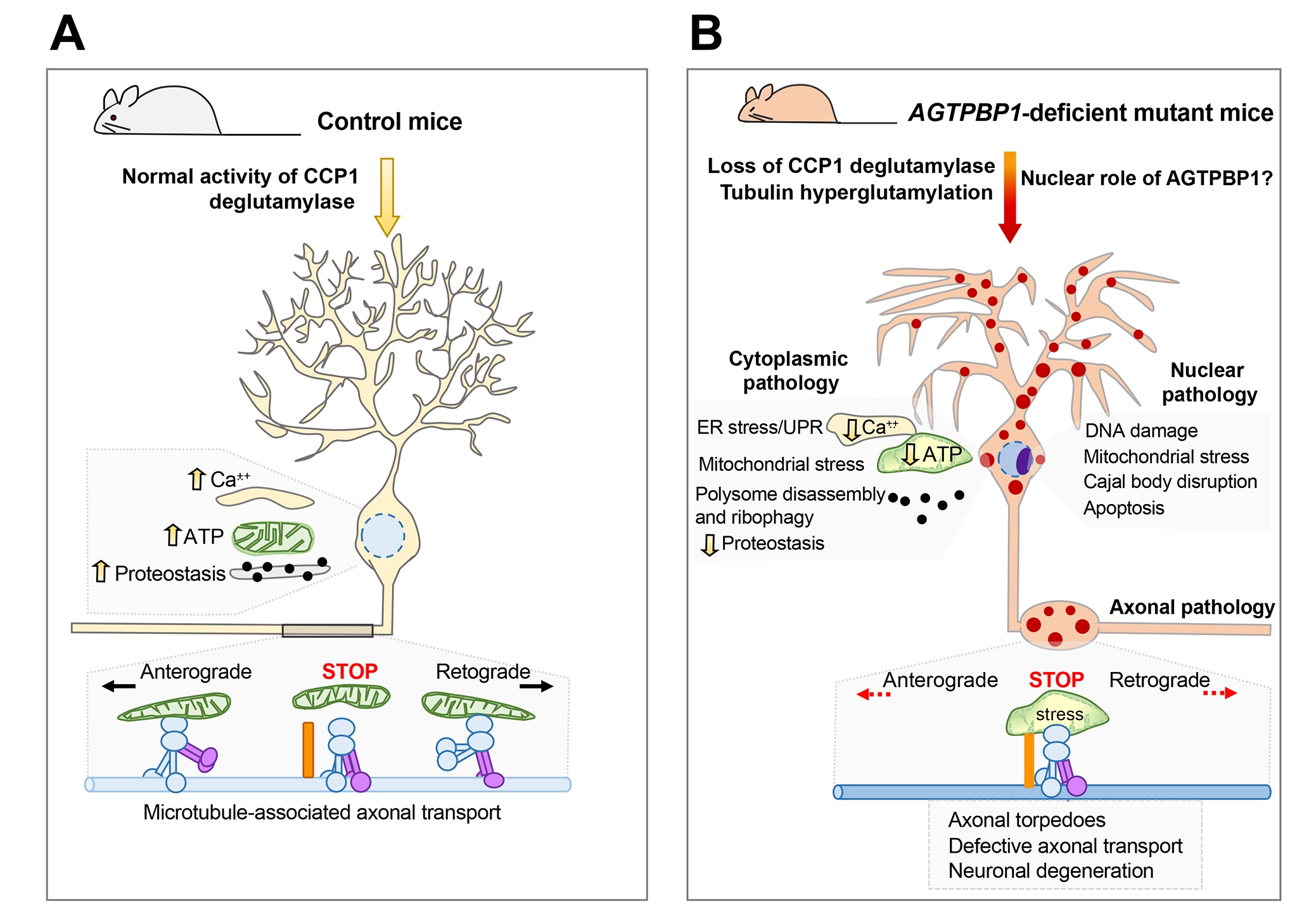
2. The AGTPBP1 Gene
2.1. Genomic Structure and Organisation
2.2. Expression Pattern
3. The AGTPBP1 Protein
3.1. Modular Domain Structure
3.2. Expression Pattern
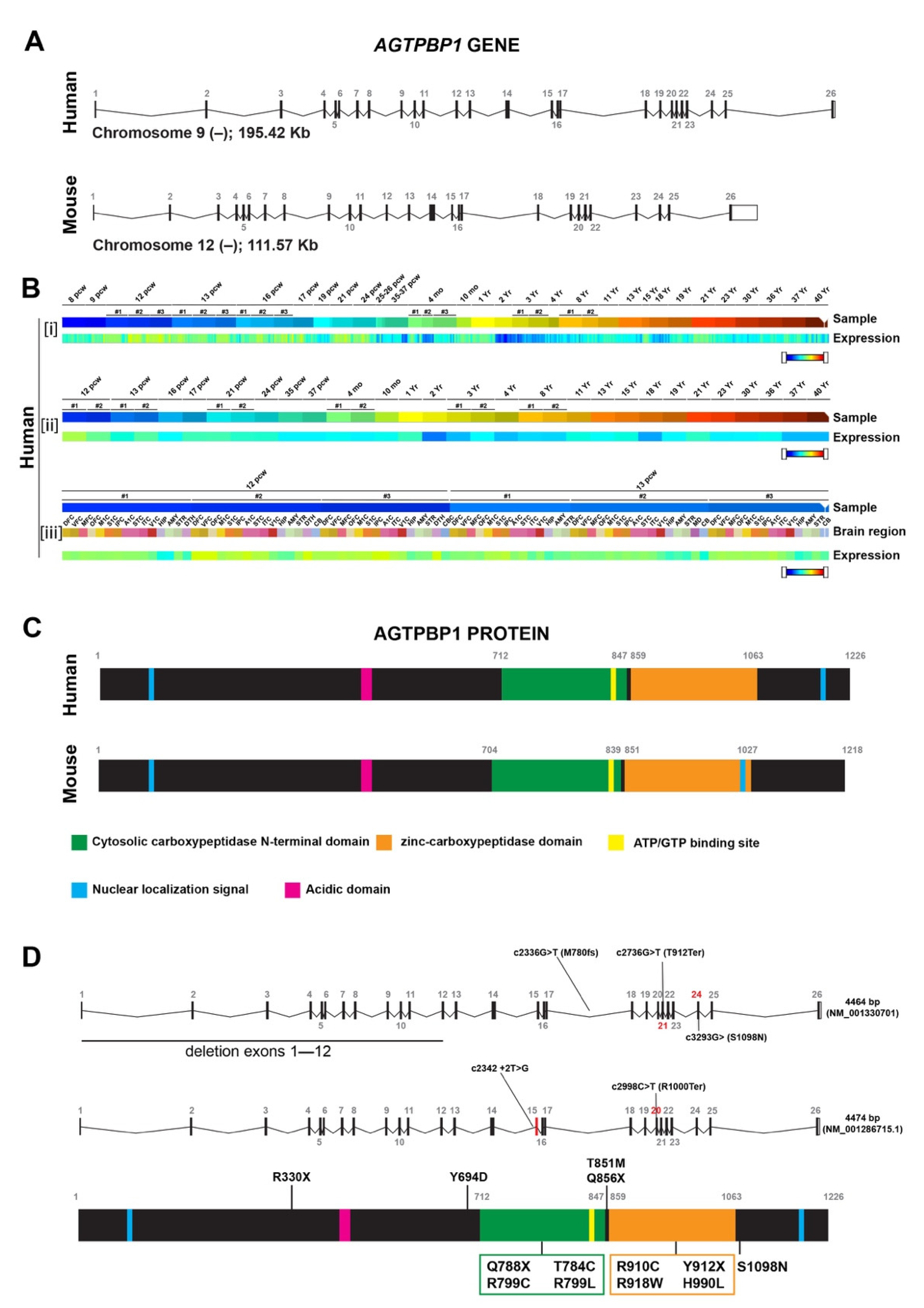
3.3. Role of the AGTPBP1 Protein
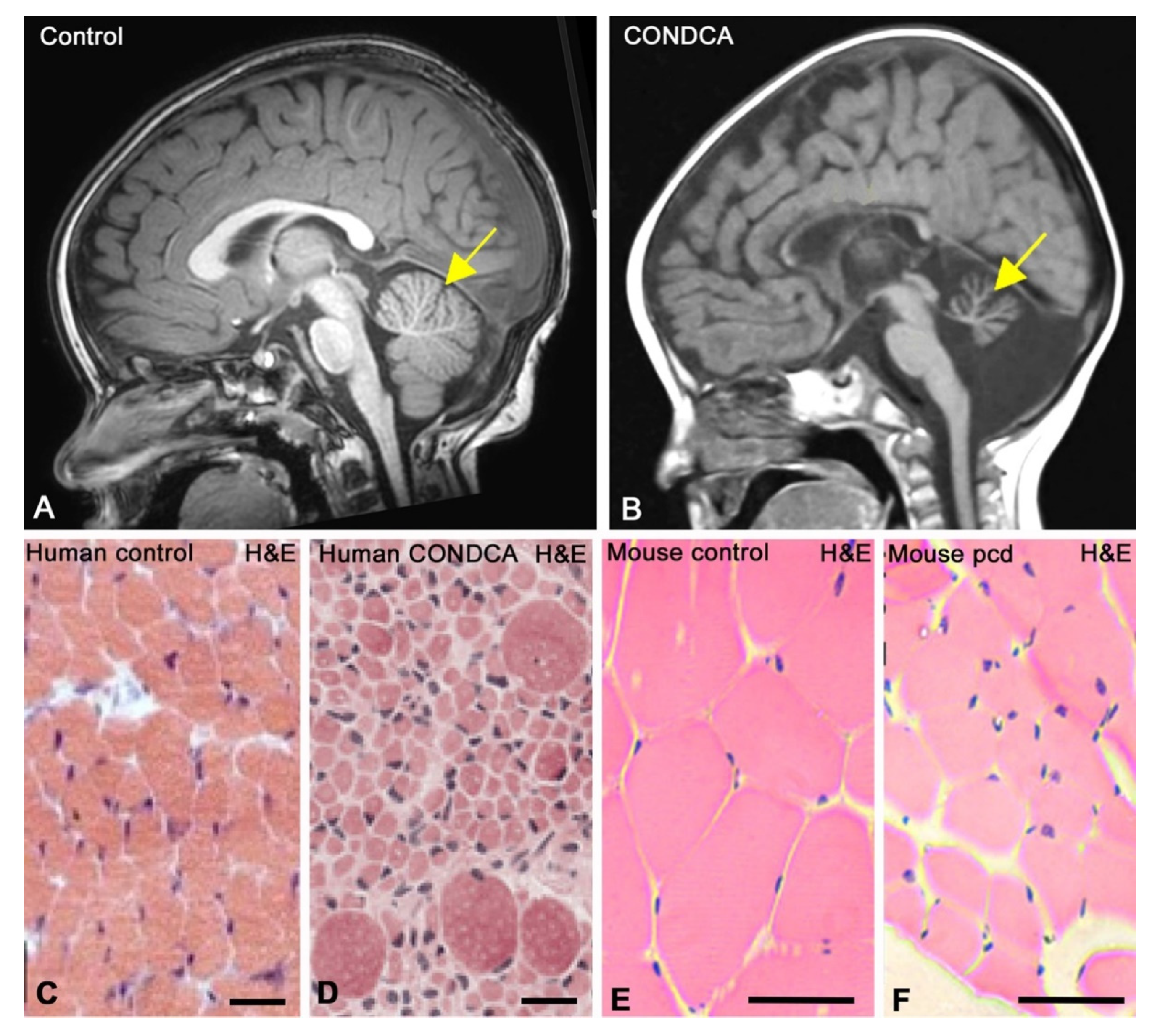
4. AGTPBP1 Mutation-Related Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA)
5. The pcd Mouse as an Animal Model for Studying AGTPBP1 Mutation-Related CONDCA
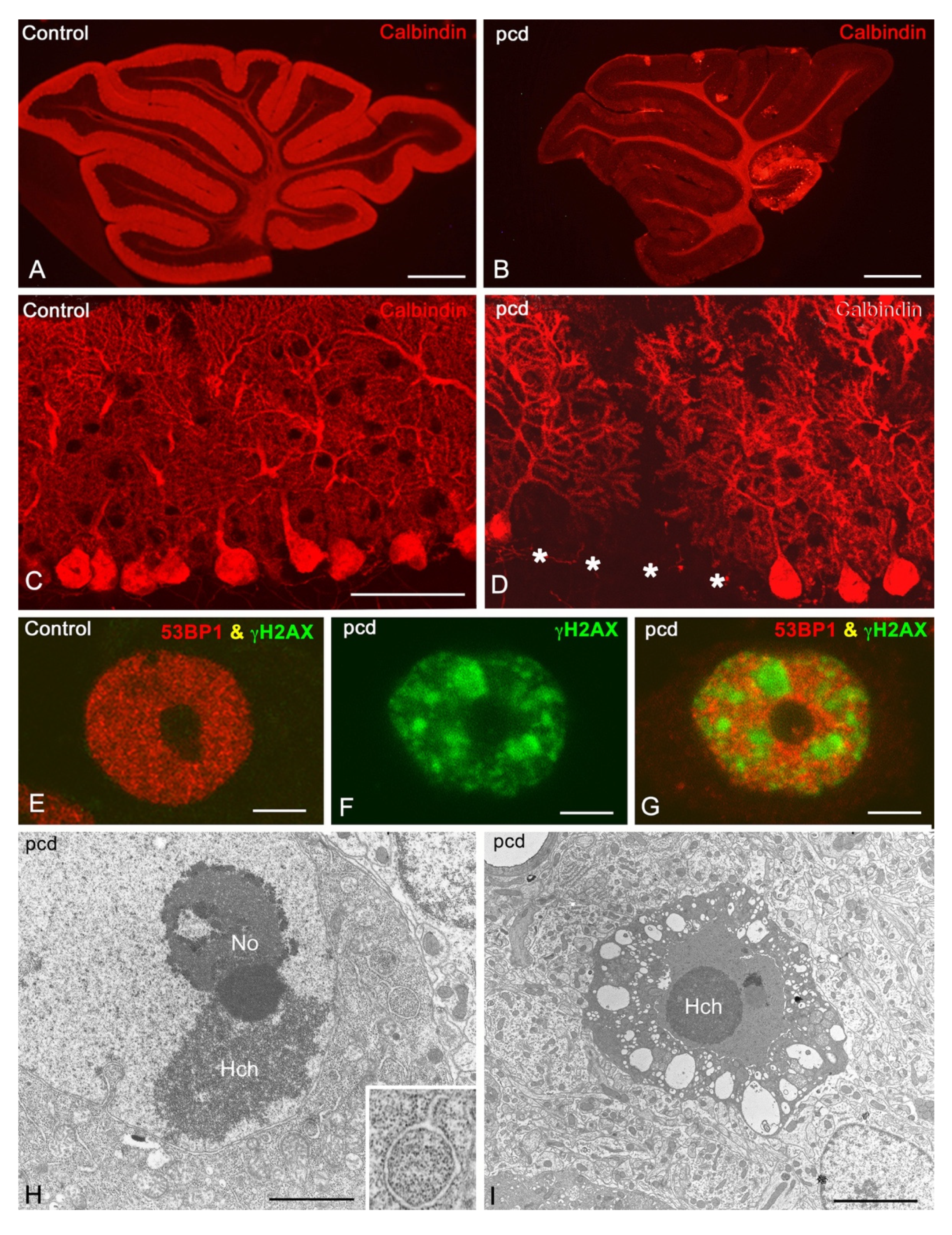
5.1. Degeneration in the Cerebellum
5.1.1. Degeneration of Purkinje Cells
5.1.2. Alterations in Other Neuronal Types in the Cerebella of pcd Mice
5.1.3. Reorganisation of Cerebellar Circuitry in pcd Mice after Purkinje Cell Loss
5.1.4. Alterations in Cerebellar-Dependent Tasks
5.2. Degeneration in the Olfactory Bulb
5.2.1. Degeneration of Mitral Cells
5.2.2. Reorganisation of Synaptic Circuitry after Mitral Cell Loss
5.2.3. Neural Plasticity in the Olfactory Bulb after Mitral Cell Loss
5.2.4. Alterations in Olfactory Task Performance after Mitral Cell Loss
5.3. Degeneration in the Thalamus
5.4. Degeneration in the Retina
5.5. Degeneration of Other Neuronal Types
5.6. Therapeutic Strategies
5.6.1. Stem Cell-Based Transplantation
5.6.2. Preservation of Degenerating Neurons in pcd Mice
5.6.3. Genetically Mediated Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Shashi, V.; Magiera, M.M.; Klein, D.; Zaki, M.; Schoch, K.; Rudnik-Schöneborn, S.; Norman, A.; Lopes Abath Neto, O.; Dusl, M.; Yuan, X.; et al. Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 2018, 37, e100540. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Gur, M.; Brooks, R.; Salah, S.; Daana, M.; Fraenkel, N.; Eisenstein, E.; Rabie, M.; Nevo, Y.; Jalas, C.; et al. Biallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy. Eur. J. Hum. Genet. 2019, 27, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Paketci, C.; Altmueller, J.; Thiele, H.; Hoelker, I.; Yis, U.; Wirth, B. Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am. J. Med. Genet. A 2019, 179, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2019, 21, 736–742. [Google Scholar] [CrossRef]
- Mullen, R.J.; Eicher, E.M.; Sidman, R.L. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl. Acad. Sci. USA 1976, 73, 208–212. [Google Scholar] [CrossRef]
- Blanks, J.C.; Mullen, R.J.; Lavail, M.M. Retinal degeneration in thepcd cerebellar mutant mouse. II. Electron microscopic analysis. J. Comp. Neurol. 1982, 212, 231–246. [Google Scholar] [CrossRef]
- Harris, A.; Morgan, J.I.; Pecot, M.; Soumare, A.; Osborne, A.; Soares, H.D. Regenerating motor neurons express Nna1, a novel ATP/GTP-binding protein related to zinc carboxypeptidases. Mol. Cell. Neurosci. 2000, 16, 578–596. [Google Scholar] [CrossRef]
- Greer, C.A.; Shepherd, G.M. Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD). Brain Res. 1982, 235, 156–161. [Google Scholar] [CrossRef]
- Wang, T.; Morgan, J.I. The Purkinje cell degeneration (pcd) mouse: An unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007, 1140, 26–40. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; La Spada, A.R.; Treadaway, J.; Higdon, J.C.; Harris, B.S.; Sidman, R.L.; Morgan, J.I.; Zuo, J. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 2002, 295, 1904–1906. [Google Scholar] [CrossRef]
- Zhao, X.; Onteru, S.K.; Dittmer, K.E.; Parton, K.; Blair, H.T.; Rothschild, M.F.; Garrick, D.J. A missense mutation in AGTPBP1 was identified in sheep with a lower motor neuron disease. Heredity 2012, 109, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.; Biswas, R.; Berezniuk, I.; Hermoso, A.; Aviles, F.X.; Fricker, L.D. A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J. 2007, 21, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.R.; Sevilla, R.G.; Hermoso, A.; Lorenzo, J.; Tanco, S.; Diez, A.; Fricker, L.D.; Bautista, J.M.; Aviles, F.X. Nnal-like proteins are active metallocarboxypeptidases of a new and diverse M14 subfamily. FASEB J. 2007, 21, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Parris, J.; Li, L.; Morgan, J.I. The carboxypeptidase-like substrate-binding site in Nna1 is essential for the rescue of the Purkinje cell degeneration (pcd) phenotype. Mol. Cell. Neurosci. 2006, 33, 200–213. [Google Scholar] [CrossRef]
- Rogowski, K.; van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.-C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef]
- Berezniuk, I.; Vu, H.T.; Lyons, P.J.; Sironi, J.J.; Xiao, H.; Burd, B.; Setou, M.; Angeletti, R.H.; Ikegami, K.; Fricker, L.D. Cytosolic carboxypeptidase 1 is involved in processing α- and β-tubulin. J. Biol. Chem. 2012, 287, 6503–6517. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Casafont, I.; Lafarga, V.; Weruaga, E.; Alonso, J.R.; Berciano, M.T.; Lafarga, M. Purkinje Cell Degeneration in pcd Mice Reveals Large Scale Chromatin Reorganization and Gene Silencing Linked to Defective DNA Repair. J. Biol. Chem. 2011, 286, 28287–28302. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Zahra, R.; Jackson, S.M.; Kazemi-Esfarjani, P.; Sopher, B.L.; Mason, A.G.; Toneff, T.; Ryu, S.; Shaffer, S.; Kansy, J.W.; et al. Mitochondrial Dysfunction in NnaD Mutant Flies and Purkinje Cell Degeneration Mice Reveals a Role for Nna Proteins in Neuronal Bioenergetics. Neuron 2010, 66, 835–847. [Google Scholar] [CrossRef]
- Li, J.; Gu, X.; Ma, Y.; Calicchio, M.L.; Kong, D.; Teng, Y.D.; Yu, L.; Crain, A.M.; Vartanian, T.K.; Pasqualini, R.; et al. Nna1 mediates purkinje cell dendritic development via lysyl oxidase propeptide and NF-κB signaling. Neuron 2010, 68, 45–60. [Google Scholar] [CrossRef]
- Valero, J.; Berciano, M.T.; Weruaga, E.; Lafarga, M.; Alonso, J.R. Pre-neurodegeneration of mitral cells in the pcd mutant mouse is associated with DNA damage, transcriptional repression, and reorganization of nuclear speckles and Cajal bodies. Mol. Cell. Neurosci. 2006, 33, 283–295. [Google Scholar] [CrossRef]
- O’Gorman, S.; Sidman, R.L. Degeneration of thalamic neurons in? Purkinje cell degeneration? mutant mice. I. Distribution of neuron loss. J. Comp. Neurol. 1985, 234, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Magiera, M.M.; Bodakuntla, S.; Žiak, J.; Lacomme, S.; Marques Sousa, P.; Leboucher, S.; Hausrat, T.J.; Bosc, C.; Andrieux, A.; Kneussel, M.; et al. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 2018, 37, e100440. [Google Scholar] [CrossRef]
- Strzyz, P. Neurodegenerative polyglutamylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Bodakuntla, S.; Schnitzler, A.; Villablanca, C.; Gonzalez-Billault, C.; Bieche, I.; Janke, C.; Magiera, M.M. Tubulin polyglutamylation is a general traffic control mechanism in hippocampal neurons. J. Cell Sci. 2020, 133, jcs241802. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, R.; Díaz, D.; Peris, L.; Andrieux, A.; Bosc, C.; Muñoz-Castañeda, J.M.; Janke, C.; Alonso, J.R.; Moutin, M.-J.; Weruaga, E. Cytoskeleton stability is essential for the integrity of the cerebellum and its motor- and affective-related behaviors. Sci. Rep. 2018, 8, 3072. [Google Scholar] [CrossRef]
- De La Vega Otazo, M.R.; Lorenzo, J.; Tort, O.; Avilés, F.X.; Bautista, J.M. Functional segregation and emerging role of cilia-related cytosolic carboxypeptidases (CCPs). FASEB J. 2013, 27, 424–431. [Google Scholar] [CrossRef]
- Lyons, P.J.; Sapio, M.R.; Fricker, L.D. Zebrafish cytosolic carboxypeptidases 1 and 5 are essential for embryonic development. J. Biol. Chem. 2013, 288, 30454–30462. [Google Scholar] [CrossRef]
- Kimura, Y.; Kurabe, N.; Ikegami, K.; Tsutsumi, K.; Konishi, Y.; Kaplan, O.I.; Kunitomo, H.; Iino, Y.; Blacque, O.E.; Setou, M. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 2010, 285, 22936–22941. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Wang, T.; Li, L.; Correia, K.; Morgan, J.I. A structural and functional analysis of Nna1 in Purkinje cell degeneration (pcd) mice. FASEB J. 2012, 26, 4468–4480. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Eng, J.; Martinez, R.A.; Jackson, S.; Huang, J.; Possin, D.E.; Sopher, B.L.; La Spada, A.R. The zinc-binding domain of Nna1 is required to prevent retinal photoreceptor loss and cerebellar ataxia in Purkinje cell degeneration (pcd) mice. Vis. Res. 2008, 48, 1999–2005. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Bulinski, J.C. Microtubules and Neurodegeneration: The Tubulin Code Sets the Rules of the Road. Curr. Biol. 2019, 29, R28–R30. [Google Scholar] [CrossRef] [PubMed]
- Janke, C. The tubulin code: Molecular components, readout mechanisms, functions. J. Cell Biol. 2014, 206, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Tanco, S.; Tort, O.; Demol, H.; Aviles, F.X.; Gevaert, K.; Van Damme, P.; Lorenzo, J. C-terminomics Screen for Natural Substrates of Cytosolic Carboxypeptidase 1 Reveals Processing of Acidic Protein C termini. Mol. Cell. Proteom. 2015, 14, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Gilmore-Hall, S.; Kuo, J.; Ward, J.M.; Zahra, R.; Morrison, R.S.; Perkins, G.; La Spada, A.R. CCP1 promotes mitochondrial fusion and motility to prevent Purkinje cell neuron loss in pcd mice. J. Cell Biol. 2018, 218, 206–219. [Google Scholar] [CrossRef]
- Türay, S.; Eröz, R.; Başak, A.N. A novel pathogenic variant in the 3′ end of the AGTPBP1 gene gives rise to neurodegeneration without cerebellar atrophy: An expansion of the disease phenotype? Neurogenetics 2021, 22, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Díaz, D.; Piquer-Gil, M.; Recio, J.S.; Martínez-Losa, M.M.; Alonso, J.R.; Weruaga, E.; Álvarez-Dolado, M. Bone marrow transplantation improves motor activity in a mouse model of ataxia. J. Tissue Eng. Regen. Med. 2018, 12, e1950–e1961. [Google Scholar] [CrossRef]
- Akhmanova, A.; Hoogenraad, C.C. More is not always better: Hyperglutamylation leads to neurodegeneration. EMBO J. 2018, 37, e101023. [Google Scholar] [CrossRef]
- Kitano, S.; Kino, Y.; Yamamoto, Y.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y.; Arima, K.; Satoh, J.-I. Bioinformatics Data Mining Approach Suggests Coexpression of AGTPBP1 with an ALS-linked Gene C9orf72. J. Cent. Nerv. Syst. Dis. 2015, 7, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Hossain, M.I.; Yamazaki, M.; Abe, M.; Natsume, R.; Konno, K.; Kageyama, S.; Komatsu, M.; Watanabe, M.; Sakimura, K.; et al. Deletion of exons encoding carboxypeptidase domain of Nna1 results in Purkinje cell degeneration (pcd) phenotype. J. Neurochem. 2018, 147, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Marchena, M.; Lara, J.; Aijón, J.; Germain, F.; de la Villa, P.; Velasco, A. The retina of the PCD/PCD mouse as a model of photoreceptor degeneration. A structural and functional study. Exp. Eye Res. 2011, 93, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ghetti, B.; Lee, W.H. Decreased IGF-I gene expression during the apoptosis of Purkinje cells in pcd mice. Dev. Brain Res. 1997, 98, 164–176. [Google Scholar] [CrossRef]
- Ghetti, B.; Norton, J.; Triarhou, L.C. Nerve cell atrophy and loss in the inferior olivary complex of “Purkinje cell degeneration” mutant mice. J. Comp. Neurol. 1987, 260, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Triarhou, L.C.; Norton, J.; Ghetti, B. Anterograde transsynaptic degeneration in the deep cerebellar nuclei of Purkinje cell degeneration (pcd) mutant mice. Exp. Brain Res. 1987, 66, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Ford, G.D.; Ford, B.D.; Steele, E.C.; Gates, A.; Hood, D.; Matthews, M.A.B.; Mirza, S.; MacLeish, P.R. Analysis of transcriptional profiles and functional clustering of global cerebellar gene expression in PCD3J mice. Biochem. Biophys. Res. Commun. 2008, 377, 556–561. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Berciano, M.T.; Valero, J.; Gómez, C.; Díaz, D.; Alonso, J.R.; Lafarga, M.; Weruaga, E. Differential glial activation during the degeneration of Purkinje cells and mitral cells in the PCD mutant mice. Glia 2013, 61, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Landis, S.C.; Mullen, R.J. The development and degeneration of Purkinje cells in pcd mutant mice. J. Comp. Neurol. 1978, 177, 125–143. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Casafont, I.; Weruaga, E.; Alonso, J.R.; Berciano, M.T.; Lafarga, M. Nucleolar Disruption and Cajal Body Disassembly are Nuclear Hallmarks of DNA Damage-Induced Neurodegeneration in Purkinje Cells. Brain Pathol. 2011, 21, 374–388. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Berciano, M.T.; Tapia, O.; Narcis, J.O.; Lafarga, V.; Díaz, D.; Weruaga, E.; Santos, E.; Lafarga, M. Nucleolin reorganization and nucleolar stress in Purkinje cells of mutant PCD mice. Neurobiol. Dis. 2019, 127, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Díaz, D.; Recio, J.S.; Weruaga, E.; Alonso, J.R. Mild cerebellar neurodegeneration of aged heterozygous PCD mice increases cell fusion of Purkinje and bone marrow-derived cells. Cell Transplant. 2012, 21, 1595–1602. [Google Scholar] [CrossRef]
- Li, J.; Snyder, E.Y.; Tang, F.H.F.; Pasqualini, R.; Arap, W.; Sidman, R.L. Nna1 gene deficiency triggers Purkinje neuron death by tubulin hyperglutamylation and ER dysfunction. JCI Insight 2020, 5, e136078. [Google Scholar] [CrossRef]
- Kyuhou, S.I.; Kato, N.; Gemba, H. Emergence of endoplasmic reticulum stress and activated microglia in Purkinje cell degeneration mice. Neurosci. Lett. 2006, 396, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial complex 1 activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- Date, H.; Onodera, O.; Tanaka, H.; Iwabuchi, K.; Uekawa, K.; Igarashi, S.; Koike, R.; Hiroi, T.; Yuasa, T.; Awaya, Y.; et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001, 29, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef]
- Suraweera, A.; Becherel, O.J.; Chen, P.; Rundle, N.; Woods, R.; Nakamura, J.; Gatei, M.; Criscuolo, C.; Filla, A.; Chessa, L.; et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007, 177, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.M.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA Repair and Neurodegenerative Disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef]
- Shackelford, D.A. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, M.; Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T. Cajal’s contribution to the knowledge of the neuronal cell nucleus. Chromosoma 2009, 118, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Rieker, C.; Engblom, D.; Kreiner, G.; Domanskyi, A.; Schober, A.; Stotz, S.; Neumann, M.; Yuan, X.; Grummt, I.; Schütz, G.; et al. Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J. Neurosci. 2011, 31, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Pietrzak, M. Emerging roles of the neuronal nucleolus. Trends Neurosci. 2012, 35, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Parlato, R.; Kreiner, G. Nucleolar activity in neurodegenerative diseases: A missing piece of the puzzle? J. Mol. Med. 2013, 91, 541–547. [Google Scholar] [CrossRef]
- Garcia-Esparcia, P.; Sideris-Lampretsas, G.; Hernandez-Ortega, K.; Grau-Rivera, O.; Sklaviadis, T.; Gelpi, E.; Ferrer, I. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am. J. Neurodegener. Dis. 2017, 6, 15–25. [Google Scholar]
- Hernández-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Tapia, O.; Narcís, J.O.; Riancho, J.; Tarabal, O.; Piedrafita, L.; Calderó, J.; Berciano, M.T.; Lafarga, M. Cellular bases of the RNA metabolism dysfunction in motor neurons of a murine model of spinal muscular atrophy: Role of Cajal bodies and the nucleolus. Neurobiol. Dis. 2017, 108, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Berezniuk, I.; Fricker, L.D. A defect in cytosolic carboxypeptidase 1 (Nna1) causes autophagy in Purkinje cell degeneration mouse brain. Autophagy 2010, 6, 558–559. [Google Scholar] [CrossRef][Green Version]
- El-Bazzal, L.; Rihan, K.; Bernard-Marissal, N.; Castro, C.; Chouery-Khoury, E.; Desvignes, J.P.; Atkinson, A.; Bertaux, K.; Koussa, S.; Lévy, N.; et al. Loss of Cajal bodies in motor neurons from patients with novel mutations in VRK1. Hum. Mol. Genet. 2019, 28, 2378–2394. [Google Scholar] [CrossRef]
- Pessina, F.; Gioia, U.; Brandi, O.; Farina, S.; Ceccon, M.; Francia, S.; d’Adda di Fagagna, F. DNA Damage Triggers a New Phase in Neurodegeneration. Trends Genet. 2020, 337–354. [Google Scholar]
- Zhou, L.; Araki, A.; Nakano, A.; Sezer, C.; Harada, T. Different types of neural cell death in the cerebellum of the ataxia and male sterility (AMS) mutant mouse. Pathol. Int. 2006, 56, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Blosa, M.; Bursch, C.; Weigel, S.; Holzer, M.; Jäger, C.; Janke, C.; Matthews, R.T.; Arendt, T.; Morawski, M. Reorganization of synaptic connections and perineuronal nets in the deep cerebellar nuclei of purkinje cell degeneration mutant mice. Neural Plast. 2016, 2016, 2828536. [Google Scholar] [CrossRef] [PubMed]
- Triarhou, L.C.; Ghetti, B. Stabilisation of neurone number in the inferior olivary complex of aged “Purkinje cell degeneration” mutant mice. Acta Neuropathol. 1991, 81, 597–602. [Google Scholar] [CrossRef]
- Grüsser-Cornehls, U.; Bäurle, J. Mutant mice as a model for cerebellar ataxia. Prog. Neurobiol. 2001, 63, 489–540. [Google Scholar] [CrossRef]
- Ghetti, B.; Perry, K.W.; Fuller, R.W. Serotonin concentration and turnover in cerebelum and other brain regions of pcd mutant mice. Brain Res. 1988, 458, 367–371. [Google Scholar] [CrossRef]
- Triarhou, L.C.; Ghetti, B. Serotonin-immunoreactivity in the cerebellum of two neurological mutant mice and the corresponding wild-type genetic stocks. J. Chem. Neuroanat. 1991, 4, 421–428. [Google Scholar] [CrossRef]
- Le Marec, N.; Hébert, C.; Amdiss, F.; Botez, M.I.; Reader, T.A. Regional distribution of 5-HT transporters in the brain of wild type and “Purkinje cell degeneration” mutant mice: A quantitative autoradiographic study with [3H]citalopram. J. Chem. Neuroanat. 1998, 15, 155–171. [Google Scholar] [CrossRef]
- Felten, D.L.; Felten, S.Y.; Perry, K.W.; Fuller, R.W.; Nurnberger, J.I.; Ghetti, B. Noradrenergic innervation of the cerebellar cortex in normal and in Purkinje cell degeneration mutant mice: Evidence for long term survival following loss of the two major cerebellar cortical neuronal populations. Neuroscience 1986, 18, 783–793. [Google Scholar] [CrossRef]
- Roffler-Tarlov, S.; Landis, S.C.; Zigmond, M.J. Effects of Purkinje cell degeneration on the noradrenergic projection to mouse cerebellar cortex. Brain Res. 1984, 298, 303–311. [Google Scholar] [CrossRef]
- Strazielle, C.; Lalonde, R.; Hébert, C.; Reader, T.A. Regional brain distribution of noradrenaline uptake sites, and of α1-, α2- and β-adrenergic receptors in PCD mutant mice: A quantitative autoradiographic study. Neuroscience 1999, 94, 287–304. [Google Scholar] [CrossRef]
- Delis, F.; Mitsacos, A.; Giompres, P. Dopamine receptor and transporter levels are altered in the brain of Purkinje Cell Degeneration mutant mice. Neuroscience 2004, 125, 255–268. [Google Scholar] [CrossRef]
- Le Marec, N.; Lalonde, R. Sensorimotor learning and retention during equilibrium tests in Purkinje cell degeneration mutant mice. Brain Res. 1997, 768, 310–316. [Google Scholar] [CrossRef]
- Wu, H.Y.; Rong, Y.; Correia, K.; Min, J.; Morgan, J.I. Comparison of the Enzymatic and Functional Properties of Three Cytosolic Carboxypeptidase Family Members. J. Biol. Chem. 2015, 290, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.S.; Darmohray, D.M.; Fayad, J.; Marques, H.G.; Carey, M.R. A quantitative framework for whole-body coordination reveals specific deficits in freely walking ataxic mice. Elife 2015, 4, e07892. [Google Scholar] [CrossRef]
- Chen, L.; Bao, S.; Lockard, J.M.; Kim, J.J.; Thompson, R.F. Impaired classical eyeblink conditioning in cerebellar-lesioned and Purkinje cell degeneration (pcd) mutant mice. J. Neurosci. 1996, 16, 2829–2838. [Google Scholar] [CrossRef]
- Brown, K.L.; Agelan, A.; Woodruff-Pak, D.S. Unimpaired trace classical eyeblink conditioning in Purkinje cell degeneration (pcd) mutant mice. Neurobiol. Learn. Mem. 2010, 93, 303–311. [Google Scholar] [CrossRef][Green Version]
- Goodlett, C.R.; Hamre, K.M.; West, J.R. Dissociation of spatial navigation and visual guidance performance in Purkinje cell degeneration (pcd) mutant mice. Behav. Brain Res. 1992, 47, 129–141. [Google Scholar] [CrossRef]
- Lalonde, R.; Strazielle, C. The effects of cerebellar damage on maze learning in animals. Cerebellum 2003, 2, 300–309. [Google Scholar]
- Tuma, J.; Kolinko, Y.; Vozeh, F.; Oendelint, J. Mutation-related differences in exploratory, spatial, and depressive-like behavior in pcd and Lurcher cerebellar mutant mice. Front. Behav. Neurosci. 2015, 9, 116. [Google Scholar] [CrossRef]
- Bartolomei, J.C.; Greer, C.A. The organization of piriform cortex and the lateral olfactory tract following the loss of mitral cells in PCD mice. Exp. Neurol. 1998, 154, 537–550. [Google Scholar] [CrossRef]
- Greer, C.A. Golgi analyses of dendritic organization among denervated olfactory bulb granule cells. J. Comp. Neurol. 1987, 257, 442–452. [Google Scholar] [CrossRef]
- Recio, J.S.; Weruaga, E.; Gomez, C.; Valero, J.; Briñón, J.G.; Alonso, J.R. Changes in the connections of the main olfactory bulb after mitral cell selective neurodegeneration. J. Neurosci. Res. 2007, 85, 2407–2421. [Google Scholar] [CrossRef]
- Baker, H.; Greer, C.A. Region-specific consequences of PCD gene expression in the olfactory system. J. Comp. Neurol. 1990, 293, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.; Curto, G.G.; Baltanás, F.C.; Valero, J.; O’Shea, E.; Colado, M.I.; Díaz, D.; Weruaga, E.; Alonso, J.R. Changes in the serotonergic system and in brain-derived neurotrophic factor distribution in the main olfactory bulb of pcd mice before and after mitral cell loss. Neuroscience 2012, 201, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Airado, C.; Gómez, C.; Recio, J.S.; Baltanás, F.C.; Weruaga, E.; Alonso, J.R. Zincergic innervation from the anterior olfactory nucleus to the olfactory bulb displays plastic responses after mitral cell loss. J. Chem. Neuroanat. 2008, 36, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Valero, J.; Weruaga, E.; Murias, A.R.; Recio, J.S.; Curto, G.G.; Gómez, C.; Alonso, J.R. Changes in cell migration and survival in the olfactory bulb of the pcd/pcd mouse. Dev. Neurobiol. 2007, 67, 839–859. [Google Scholar] [CrossRef]
- Chu, M.W.; Li, W.L.; Komiyama, T. Balancing the Robustness and Efficiency of Odor Representations during Learning. Neuron 2016, 92, 174–186. [Google Scholar] [CrossRef]
- Díaz, D.; Lepousez, G.; Gheusi, G.; Alonso, J.R.; Lledo, P.M.; Weruaga, E. Bone marrow cell transplantation restores olfaction in the degenerated olfactory bulb. J. Neurosci. 2012, 32, 9053–9058. [Google Scholar] [CrossRef]
- Díaz, D.; Gómez, C.; Muñoz-Castañeda, R.; Baltanás, F.; Alonso, J.R.; Weruaga, E. The olfactory system as a puzzle: Playing with its pieces. Anat. Rec. 2013, 296, 1383–1400. [Google Scholar] [CrossRef]
- O’Gorman, S. Degeneration of thalamic neurons in “Purkinje cell degeneration” mutant mice. II. Cytology of neuron loss. J. Comp. Neurol. 1985, 234, 298–316. [Google Scholar] [CrossRef] [PubMed]
- Kyuhou, S.-i.; Gemba, H. Fast cortical oscillation after thalamic degeneration: Pivotal role of NMDA receptor. Biochem. Biophys. Res. Commun. 2007, 356, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, L.; Neal, J.T.; Miles, M.; Martinez, R.A.; Smith, A.C.; Sopher, B.L.; La Spada, A.R. The Purkinje cell degeneration 5J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm. Genome 2006, 17, 103–110. [Google Scholar] [CrossRef] [PubMed]
- LaVail, M.M.; Blanks, J.C.; Mullen, R.J. Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographio analysis. J. Comp. Neurol. 1982, 212, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Grau, M.B.; Masson, C.; Gadadhar, S.; Rocha, C.; Tort, O.; Sousa, P.M.; Vacher, S.; Bieche, I.; Janke, C. Alterations in the balance of tubulin glycylation and glutamylation in photoreceptors leads to retinal degeneration. J. Cell Sci. 2017, 130, 938–949. [Google Scholar] [CrossRef]
- Okubo, A.; Sameshima, M.; Unoki, K.; Uehara, F. The ultrastructural study of ribosomes in photoreceptor inner segments of the pcd cerebellar mutant mouse. Jpn. J. Ophthalmol. 1995, 39, 152–161. [Google Scholar]
- LaVail, M.M.; Gorrin, G.M.; Yasumura, D.; Matthes, M.T. Increased susceptibility to constant light in nr and pcd mice with inherited retinal degenerations. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1020–1024. [Google Scholar]
- Gardette, R.; Alvarado-Mallart, R.M.; Crepel, F.; Sotelo, C. Electrophysiological demonstration of a synaptic integration of transplanted purkinje cells into the cerebellum of the adult purkinje cell degeneration mutant mouse. Neuroscience 1988, 24, 777–789. [Google Scholar] [CrossRef]
- Sotelo, C.; Alvarado-Mallart, R.M. Embryonic and adult neurons interact to allow Purkinje cell replacement in mutant cerebellum. Nature 1987, 327, 421–423. [Google Scholar] [CrossRef]
- Sotelo, C.; Alvarado-Mallart, R.M. Reconstruction of the defective cerebellar circuitry in adult purkinje cell degeneration mutant mice by Purkinje cell replacement through transplantation of solid embryonic implants. Neuroscience 1987, 20, 1–22. [Google Scholar] [CrossRef]
- Sotelo, C.; Alvarado-Mallart, R.M.; Gardette, R.; Crepel, F. Fate of grafted embryonic purkinje cells in the cerebellum of the adult “purkinje cell degeneration” mutant mouse. I. Development of reciprocal graft-host interactions. J. Comp. Neurol. 1990, 295, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Gardette, R.; Crepel, F.; Alvarado-Mallart, R.M.; Sotelo, C. Fate of grafted embryonic purkinje cells in the cerebellum of the adult “purkinje cell degeneration” mutant mouse. II. Development of synaptic responses: An in vitro study. J. Comp. Neurol. 1990, 295, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Triarhou, L.C.; Low, W.C.; Ghetti, B. Serotonin fiber innervation of cerebellar cell suspensions intraparenchymally grafted to the cerebellum of pcd mutant mice. Neurochem. Res. 1992, 17, 475–482. [Google Scholar] [CrossRef]
- Sotelo, C.; Alvarado-Mallart, R.M. Growth and differentiation of cerebellar suspensions transplanted into the adult cerebellum of mice with heredodegenerative ataxia. Proc. Natl. Acad. Sci. USA 1986, 83, 1135–1139. [Google Scholar] [CrossRef]
- Chang, A.C.; Ghetti, B. Embryonic cerebellar graft development during acute phase of gliosis in the cerebellum of pcd mutant mice. Chin. J. Physiol. 1993, 36, 141–149. [Google Scholar]
- Triarhou, L.C.; Zhang, W.; Lee, W.H. Graft-induced restoration of function in hereditary cerebellar ataxia. Neuroreport 1995, 6, 1827–1932. [Google Scholar] [CrossRef]
- Triarhou, L.C.; Zhang, W.; Lee, W.H. Amelioration of the behavioral phenotype in genetically ataxic mice through bilateral intracerebellar grafting of fetal Purkinje cells. Cell Transplant. 1996, 5, 269–277. [Google Scholar] [CrossRef]
- Zhang, W.; Lee, W.H.; Triarhou, L.C. Grafted cerebellar cells in a mouse model of hereditary ataxia express IGF-I system genes and partially restore behavioral function. Nat. Med. 1996, 2, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Recio, J.S.; Álvarez-Dolado, M.; Díaz, D.; Baltanás, F.C.; Piquer-Gil, M.; Alonso, J.R.; Weruaga, E. Bone marrow contributes simultaneously to different neural types in the central nervous system through different mechanisms of plasticity. Cell Transplant. 2011, 20, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Díaz, D.; del Pilar, C.; Carretero, J.; Alonso, J.R.; Weruaga, E. Daily bone marrow cell transplantations for the management of fast neurodegenerative processes. J. Tissue Eng. Regen. Med. 2019, 13, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Cendelin, J. Transplantation and Stem Cell Therapy for Cerebellar Degenerations. Cerebellum 2016, 15, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aleman, I.; Barrios, V.; Lledo, A.; Berciano, J. The insulin-like growth factor I system in cerebellar degeneration. Ann. Neurol. 1996, 39, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, C.; Torres-Aleman, I.; Lopez-Lopez, C.; Carro, E.; Espejo, L.; Torrado, S.; Torrado, J.J. Microspheres containing insulin-like growth factor I for treatment of chronic neurodegeneration. Biomaterials 2004, 25, 707–714. [Google Scholar] [CrossRef][Green Version]
- Sanz-Gallego, I.; Rodriguez-de-Rivera, F.J.; Pulido, I.; Torres-Aleman, I.; Arpa, J. IGF-1 in autosomal dominant cerebellar ataxia—Open-label trial. Cerebellum Ataxias 2014, 1, 13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pérez-Martín, E.; Muñoz-Castañeda, R.; Moutin, M.J.; Ávila-Zarza, C.A.; Muñoz-Castañeda, J.M.; Del Pilar, C.; Alonso, J.R.; Andrieux, A.; Díaz, D.; Weruaga, E. Oleoylethanolamide Delays the Dysfunction and Death of Purkinje Cells and Ameliorates Behavioral Defects in a Mouse Model of Cerebellar Neurodegeneration. Neurotherapeutics 2021. [Google Scholar] [CrossRef]
- Henry, R.J.; Ritzel, R.M.; Barrett, J.P.; Doran, S.J.; Jiao, Y.; Leach, J.B.; Szeto, G.L.; Wu, J.; Stoica, B.A.; Faden, A.I.; et al. Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J. Neurosci. 2020, 40, 2960–2974. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (Age; Sex; Consanguinity) | Allelic Variant | Consequence | Region Affected | Reference |
|---|---|---|---|---|
| 2-year-old; F; NO | NM_001330701 c.2336-1G>T | Transversion in intron 17. Results in a splice site aberration, a frameshift and premature termination (M780fs) | Cytosolic carboxypeptidase N-terminal domain | [1] |
| NM_001330701 c.2736delC | Deletion in exon 21. Results in a frameshift and premature termination (T912Ter) | Zinc-carboxypeptidase domain | [1] | |
| 12-month-old; M; YES | NM_001330701 c.2752C>T | Transition in exon 21. Results in R918W substitution | Zinc-carboxypeptidase domain | [1] |
| 7-month-old; M; YES | - | Deletion of exons 1 to 12 | Non-defined | [1] |
| Not available | NM_015239.2 c.2632C>T | Cerebellar hypoplasia and lower motor neuron degeneration. Results in R878W substitution | Zinc-carboxypeptidase domain | [4] |
| 4-year-old; M; NO | NM_001286715 c.2351A>G | Results in a T784C substitution | Non-defined | [2] |
| NM_001286715 c.2998C>T | Results in a frameshift and premature termination (R1000Ter) | Zinc-carboxypeptidase domain | ||
| 15-month-old; M; YES | NM_001286715 c.2342C>T+2T>G | Skips exon 15 (loss of 29 highly conserved aa) | Non-defined modular domain | [2] |
| 5-year-old; F; NO | NM_001330701 c.2752C>T | Transition in exon 21. Results in R918W substitution | Zinc-carboxypeptidase domain | [1] |
| NM_001330701 c.2080T>G | Transversion in exon 15. Results in a Y694D substitution | Non-defined | ||
| 16-month-old; F; YES | NM_001330701 c.2566C>T | Homozygous transition in exon 19. Results in a Q856 * | Non-defined | [1] |
| 8-year-old; M; YES | NM_001330701 c.2395C>T | Results in a R799C substitution | Cytosolic carboxypeptidase N-terminal domain | [1] |
| 8-year-old; M; YES | NM_001330701 c.2566C>T | Results in a P799C substitution | Cytosolic carboxypeptidase N-terminal domain | [1] |
| 7-month-old; M; YES | NM_001330701 c.2396G>T | Results in a P799L substitution | Cytosolic carboxypeptidase N-terminal domain | [3] |
| 2-year-old; M; YES | NM_001330701 c.2396G>T | Results in a P799L substitution | Cytosolic carboxypeptidase N-terminal domain | [3] |
| 20-month-old; F; NO | NM_001330701 c.988C>T | Results in a R330 * | Non-defined | [1] |
| Results in a Y912X substitution | Zinc-carboxypeptidase domain | |||
| 8-year-old; M; YES and 5-year-old; F; YES | NM_001330701 c.2728C>T | Results in a R910C substitution | Zinc-carboxypeptidase domain | [1] |
| 3 infant sibs; YES | NM_001330701 c.2362C>T | Transition in exon 18. Results in a Q788 * | Cytosolic carboxypeptidase N-terminal domain | [1] |
| 14-year-old; M; NO | NM_001330701 c.2552C>T | Transition in exon 19, resulting in a T851M substitution | Non-defined | [1] |
| NM_001330701 c.2969A>T | transversion in exon 22, resulting in a H990L substitution | Zinc-carboxypeptidase domain | ||
| 21-month-old; M; YES | NM_001330701 c.3293G>A | Mutation in exon 24, resulting in a S1098N substitution | 3′ end domain | [36] |
| 17-year-old; F; YES | NM_001330701 c.3293G>A | Mutation in exon 24, resulting in an S1098N substitution | 3′ end domain | [36] |
| Feature | Data from [1,2,3,4,36] |
|---|---|
| Onset | Birth to 20 months |
| Gender | 10F, 9M Not available (1) |
| Consanguinity | 14/19 Not available (1) |
| Progressive degenerative course | 19/20 |
| Microcephaly | 11/20 Not available (1) |
| Motor delay | 20/20 |
| Hypotonia | 19/20 Not available (1) |
| Muscle weakness | 16/18 Not available (2) |
| Muscle weakness pattern | Tetraparesis/plegia (8) Lower limb (2) Neck (3) Diaphragm/intercostal (4) Not specified (8) |
| Muscle atrophy | 9/18 |
| Tongue fasciculations | 7/20 Not available (13/20) |
| Tendon reflexes | Low or absent (10/20) Normal (3/20) Increased (6/29) Not available (1) |
| Ataxia | Yes (6) Not available (12) |
| Dystonia | 5/20 Not available (1) |
| Spasticity | 7/20 Not available (3/18) |
| Respiratory distress | 9/20 Not available (1) |
| Feeding difficulties | 13/20 Not available (1) |
| Eye movement abnormalities | Detected (12/20) Not detected (6/20) Not available (2/20) |
| Hearing | Impaired (1/20) Normal (5/20) Not available (14/20) |
| Cognitive delay | 17/20 Not available (3/20) |
| Brain MRI | Cerebellar atrophy (18/20) Dysplastic corpus callosum (6/20) Small pons (1/20) Enlarged CSF spaces (1/20) |
| Nerve conduction studies | Motor neuropathy (2/20) Axonal motor neuropathy (5/20) Normal (1/18) Not available (12/20) |
| Electromyography | Denervation (5/20) Neurogenic (2/20) Normal (1/20) Not available (12/20) |
| Allele Name Mutation | Mutation | Clinical Features | Genetic Mutation in AGTPBP1 |
|---|---|---|---|
| Agtpbp1pcd−1J | Spontaneous | Reduced body size; Ataxia; cerebellar atrophy; postnatal degeneration of thalamic neurons, PCs, MCs and retinal photoreceptors; male infertility; female partial fertility. | Unknown (possibly in regulatory region) |
| Agtpbp1pcd−2J | Spontaneous | Hylomorphic allele with reduced | Insertion (~7.8Kb) between exons 14–15 |
| Agtpbp1pcd−3J | Spontaneous | Reduced body size; Ataxia; Cerebellar atrophy; postnatal degeneration of thalamic neurons, PCs, MCs and photoreceptors; male infertility; female partial fertility; Reduced number of antral follicles. | Deletion (~12.2 Kb) between intron 5 and exon 8 |
| Agtpbp1pcd−4J | ENU-induced mutagenesis | Ataxia; degeneration of PCs | Unknown |
| Agtpbp1pcd−5J | Spontaneous | Ataxia; Degeneration of PCs and MCs | Insertion of an aspartic acid residue (D775) in exon 18 |
| Agtpbp1pcd−6J | ENU-induced mutagenesis | Ataxia; cerebellar and testicular atrophy; postnatal degeneration of PCs, MCs and photoreceptors; decreased skeletal muscle fiber size; male infertility. | Unknown |
| Agtpbp1pcd−7J | Spontaneous | Ataxia; postnatal degeneration of PCs; enlarged hippocampus; abnormal hearing | Unknown |
| Agtpbp1pcd−8J | Spontaneous | Affectation of behavior; low size body; Alteration of nervous system development, reproductive, and vision. | Unknown |
| Agtpbp1pcd−9J | Spontaneous | Ataxia, but has a slightly later onset than that caused by the original pcd allele. | Unknown |
| Agtpbp1pcd-Tg(Dhfr)1jwg | Transgene insertion | Ataxia; degeneration of PCs, MCs and photoreceptor cells; some male infertility, female partial fertility; degeneration of sperm | Random gene disruption |
| Agtpbp1Drunk | Mutagenesis | Degeneration of Purkinje cells and photoreceptor cells; Male infertility | Unknown |
| Agtpbp1Rio | Mutagenesis | Tremor and abnormal sperm | Unknown |
| Agtpbp1babe | ENU-induced mutagenesis | Ataxia; paraparesis | P804 arginine to a termination codon |
| Agtpbp1pcd-Btlr | ENU-induced mutagenesis | Ataxia; degeneration of PCs, MCs and photoreceptor cells; male infertility, oligozoospermia and teratozoospermia | a T-to-A transversion in the donor splice site of intron 11 |
| Agtpbp1pcd-m2Btlr | ENU-induced mutagenesis | Tremors; decreased body size; reduced activated sperm motility | an A to G transition; destroys the acceptor splice site of intron 7 of the gene |
| Agtpbp1pcd-Sid | Spontaneous | Reduced body size; Ataxia; Cerebellar atrophy. | Deletion of exon 7 |
| Agtpbp1Gt(IST13517F11)Tigm | Gene trapped allele | one ES cell; unclassified | Chr13:59477801-59478055 bp (-);Chr13:59477801-59477979 bp (-) |
| Agtpbp1Gt(OST186151)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59531904-59544452 bp (-) |
| Agtpbp1Gt(OST188387)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59531902-59533237 bp (-) |
| Agtpbp1Gt(OST252171)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59531904-59544452 bp (-) |
| Agtpbp1Gt(OST300426)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59531904-59544452 bp (-) |
| Agtpbp1Gt(OST300428)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59536248-59536374 bp (-) |
| Agtpbp1Gt(OST301743)Lex | Gene trapped allele | Lex-1 (ES Cell) | Chr13:59531913-59536374 bp (-) |
| pcdKO | Knock-out | Ataxia; cerebellar atrophy, postnatal degeneration of PCs and photoreceptors. | Deletion of exons 21 and 22 |
| Physiopathological Feature | CONDCA Patients | pcd Mice |
|---|---|---|
| Early-onset | YES | YES |
| Progressive degenerative course | YES | YES |
| Microcephaly | YES | YES |
| Motor delay | YES | YES |
| Hypotonia | YES | N.E |
| Muscle weakness | YES | YES |
| Muscle atrophy | YES | YES |
| Tongue fasciculations | Frequent | N.E |
| Alteration of tendon reflexes | Frequent | N.E |
| Ataxia | Frequent | YES |
| Dystonia | Frequent | N.E |
| Spasticity | Frequent | N.E |
| Respiratory distress | Frequent | N.E |
| Feeding difficulties | Frequent | YES |
| Eye movement abnormalities | Frequent | N.E |
| Defective hearing | Occasional | YES |
| Cognitive delay | YES | YES |
| Motor and axonal motor neuropathy | Frequent | YES |
| Denervation | Frequent | YES |
| Olfactory dysfunction | N.E | YES |
| Visual deficiency | N.E | YES |
| Defective sperm | N.E | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baltanás, F.C.; Berciano, M.T.; Santos, E.; Lafarga, M. The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease. Biomedicines 2021, 9, 1157. https://doi.org/10.3390/biomedicines9091157
Baltanás FC, Berciano MT, Santos E, Lafarga M. The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease. Biomedicines. 2021; 9(9):1157. https://doi.org/10.3390/biomedicines9091157
Chicago/Turabian StyleBaltanás, Fernando C., María T. Berciano, Eugenio Santos, and Miguel Lafarga. 2021. "The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease" Biomedicines 9, no. 9: 1157. https://doi.org/10.3390/biomedicines9091157
APA StyleBaltanás, F. C., Berciano, M. T., Santos, E., & Lafarga, M. (2021). The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease. Biomedicines, 9(9), 1157. https://doi.org/10.3390/biomedicines9091157