
Long Term Response to Circulating Angiogenic Cells, Unstimulated or Atherosclerotic Pre-Conditioned, in Critical Limb Ischemic Mice

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
2.2. AP Secretome Acquisition
2.3. Murine Model of CLI. CACs Administration
2.4. Follow-Up of Physiological Changes after CLI and Cell Administration
2.5. Tissue Extraction
2.6. Immunohistochemical Analysis
2.7. Alu-Based Quantification
2.8. Proteomic Analysis
2.9. Cytokine Expression Analysis
2.10. Statistical Analysis
3. Results
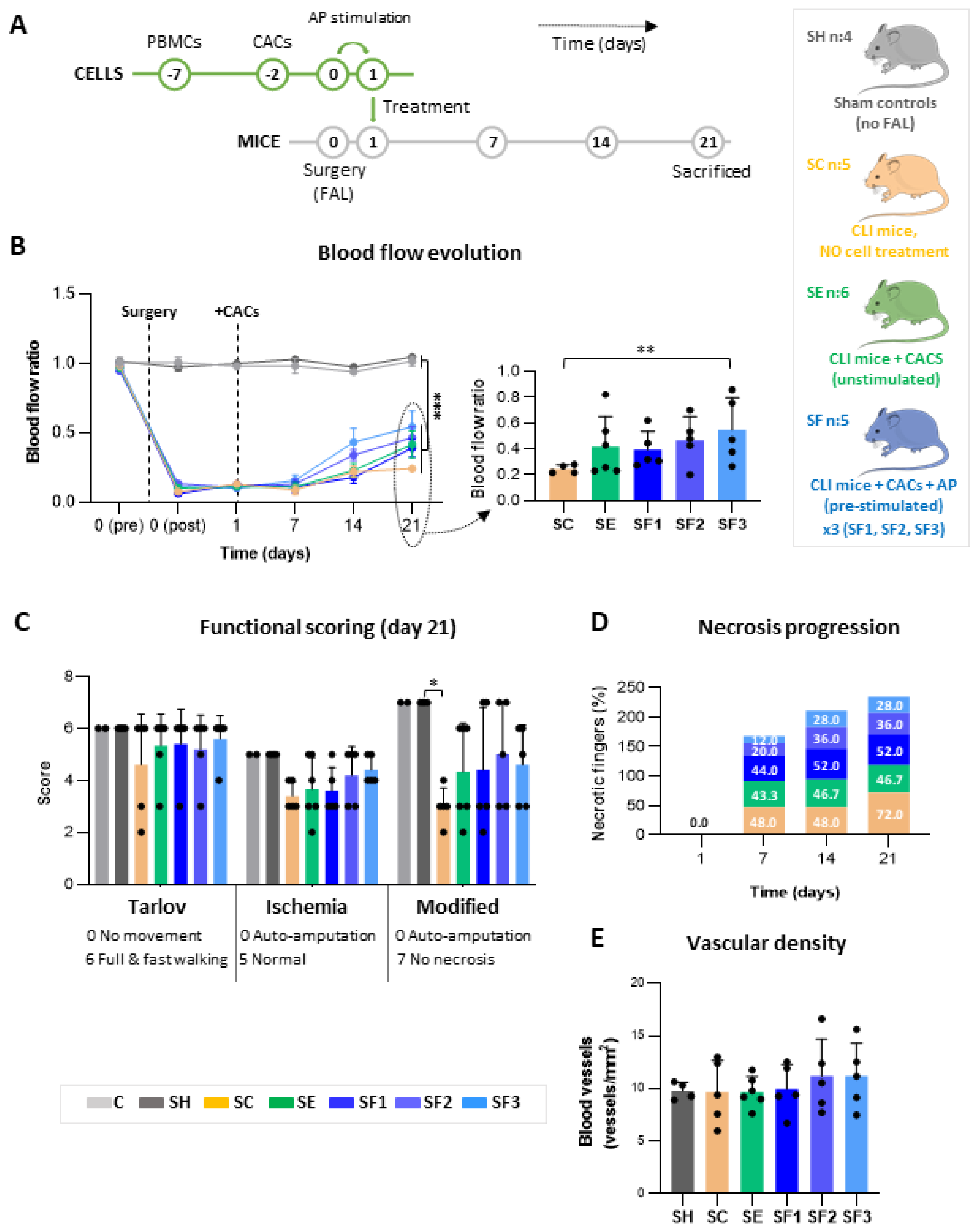
3.1. CACs Promote Blood Flow Recovery in CLI Mice
3.2. CACs Delays Ischemic Progression in CLI Mice
3.3. Vascular Density Stabilizes at Day 21
3.4. Human CACs Are Not Present in Ischemic Tissues after 21 Days Post-Transplantation
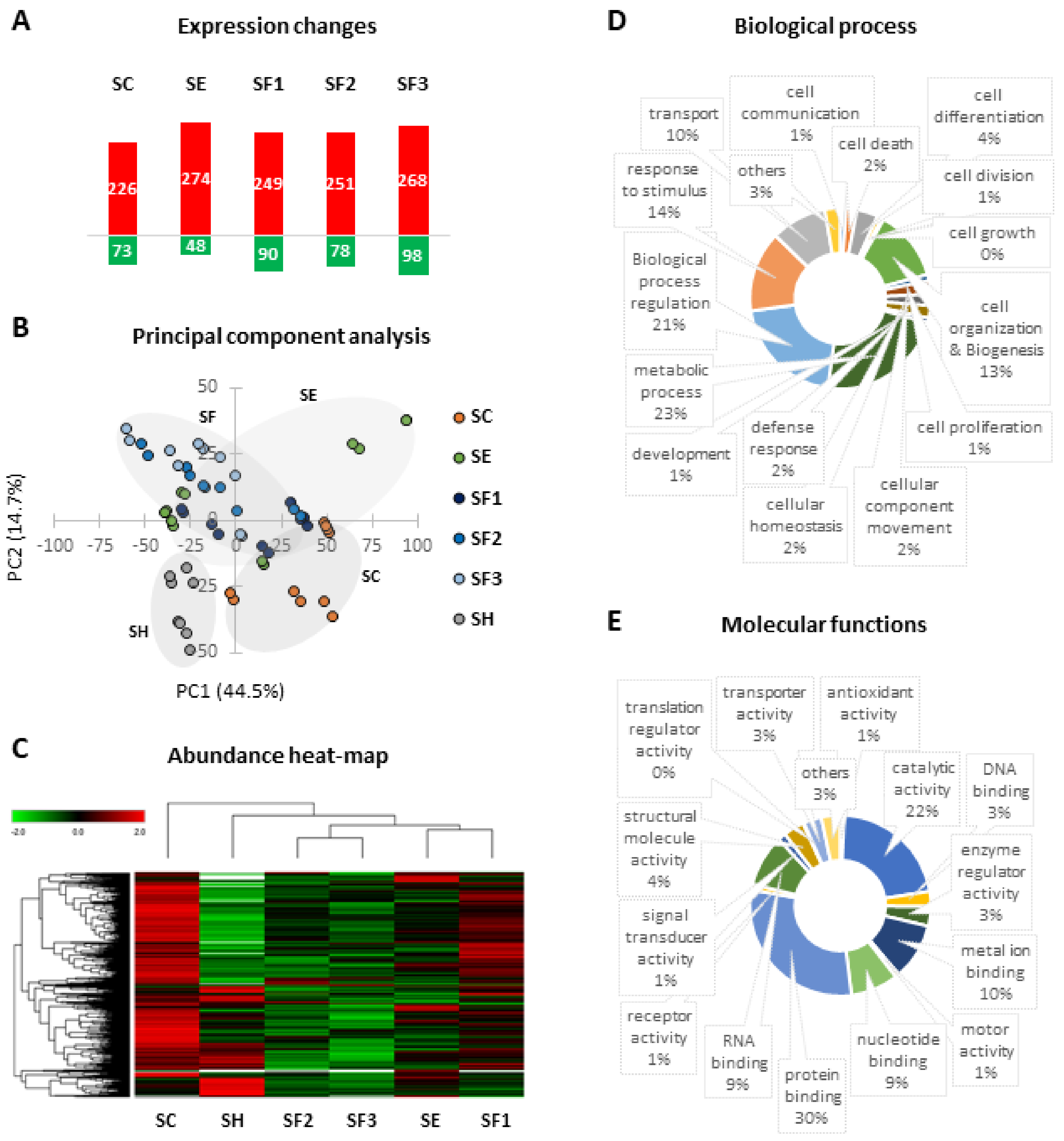
3.5. Proteomic Changes Related to Long-Term CLI and CACs Treatment
3.5.1. Long-Term Protein Changes Due to Ischemia
3.5.2. CACs Long Term Effect at the Molecular Level
3.5.3. CACs Atherosclerotic Pre-Conditioning Effect
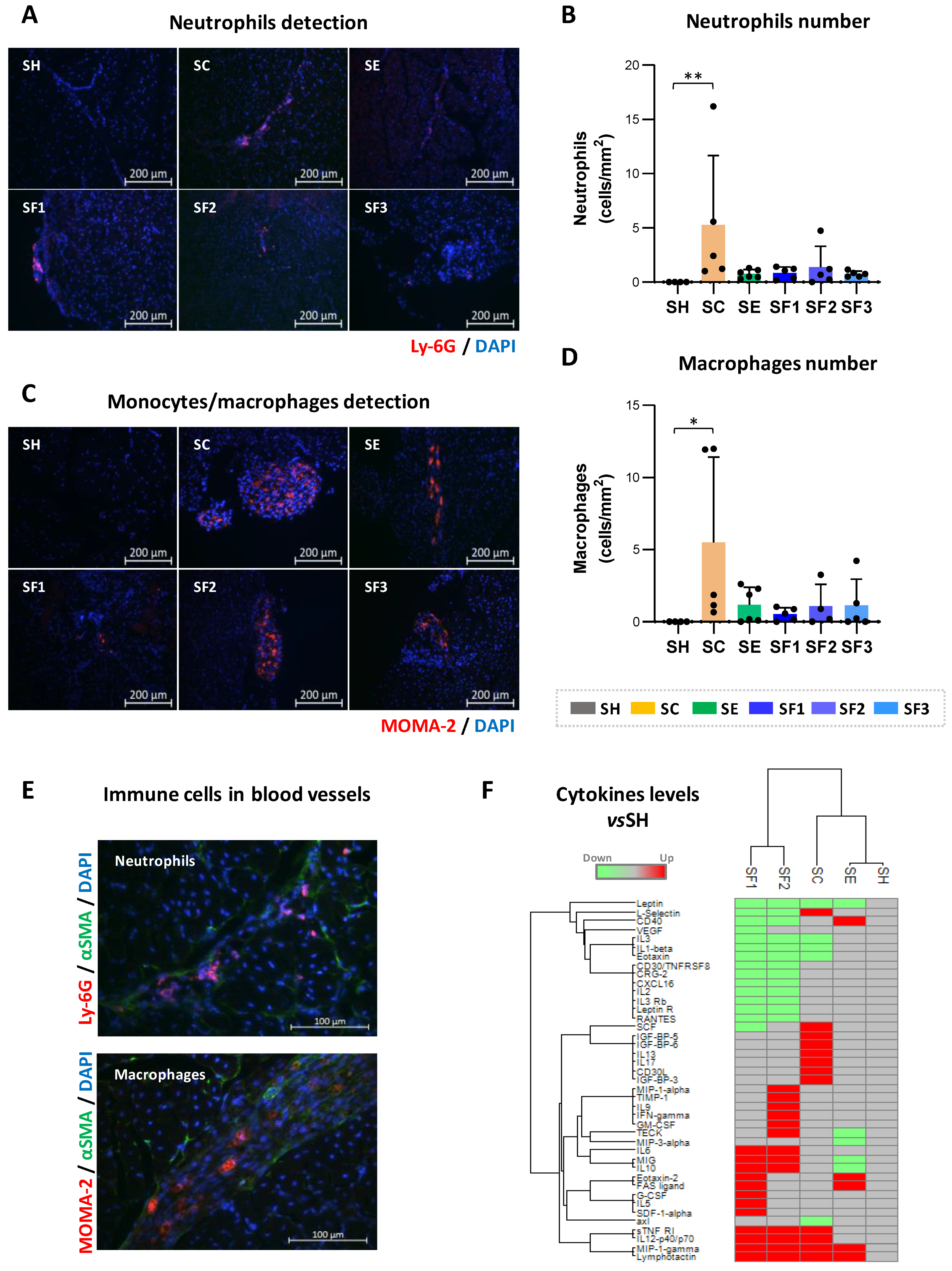
3.6. CACs Treatment Decrease Recruitment of Immune Cells
4. Discussion
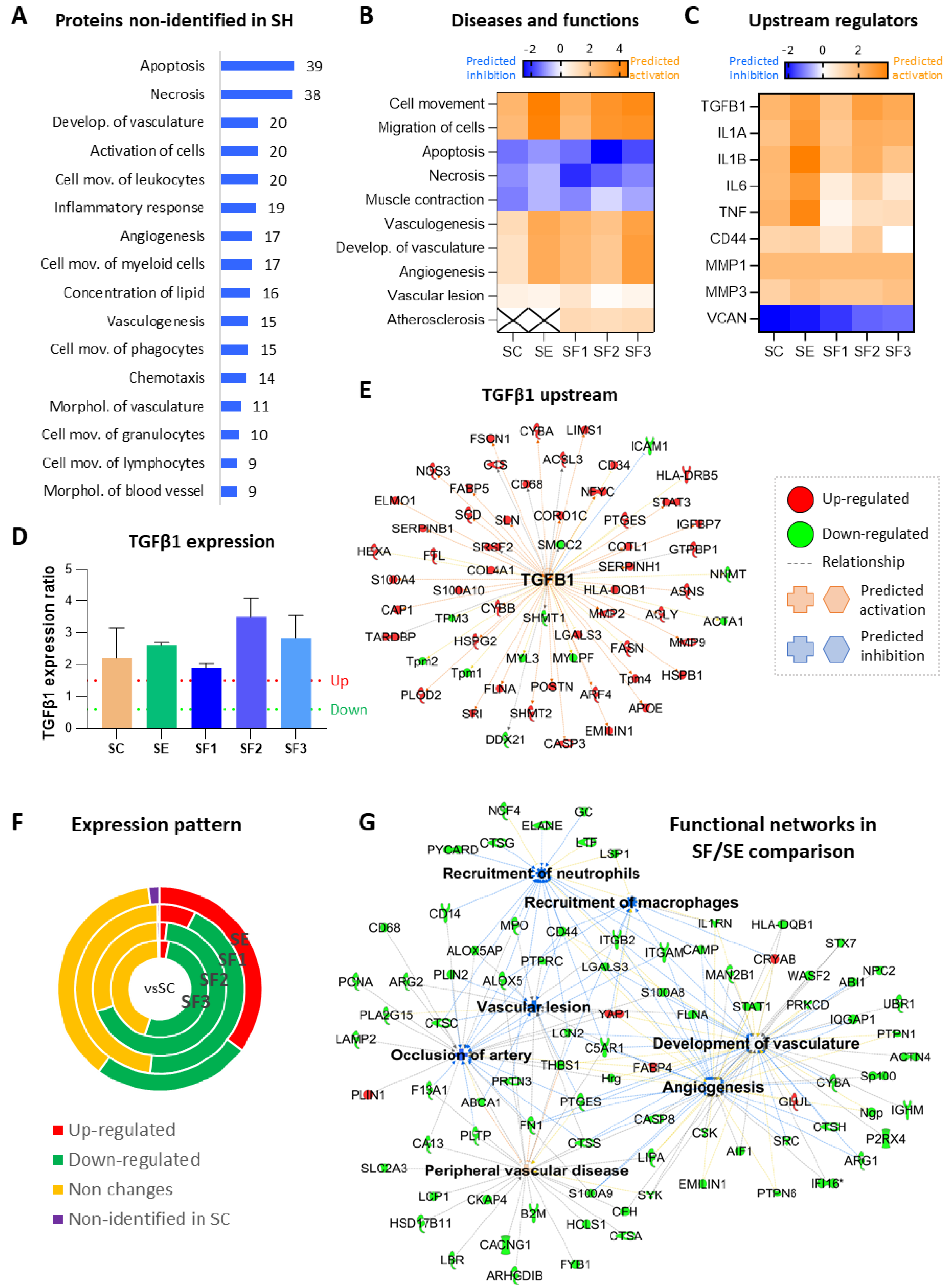
4.1. Long Term Protein Changes Related to Ischemia
4.2. Molecular Changes in Response to CACs
4.3. CACs Pre-Stimulation with Atherosclerotic Plaque Secretomes
4.4. Modulation of Immune Cell Recruitment by CACs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Weel, V.; Van Tongeren, R.B.; Van Hinsbergh, V.W.M.; Van Bockel, J.H.; Quax, P.H.A. Vascular Growth in Ischemic Limbs: A Review of Mechanisms and Possible Therapeutic Stimulation. Ann. Vasc. Surg. 2008, 22, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Krishna, S.M.; Moxon, J.V.; Golledge, J. A Review of the Pathophysiology and Potential Biomarkers for Peripheral Artery Disease. Int. J. Mol. Sci. 2015, 16, 11294–11322. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.S.; Pomposelli, F.B.; Clair, D.G.; Geraghty, P.J.; McKinsey, J.F.; Mills, J.L.; Moneta, G.L.; Murad, M.H.; Powell, R.J.; Reed, A.B.; et al. Society for Vascular Surgery practice guidelines for atherosclerotic occlusive disease of the lower extremities: Management of asymptomatic disease and claudication. J. Vasc. Surg. 2015, 61 (Suppl. 3), 2S–41S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.L.; Kearns, B.; Stevenson, M.D.; Cantrell, A.J.; Littlewood, C.; Michaels, J.A. Enhancements to angioplasty for peripheral arterial occlusive disease: Systematic review, cost-effectiveness assessment and expected value of information analysis. Health Technol. Assess. 2014, 18, 1–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.H.; Krankenberg, H.; Balzer, J.O.; Kalka, C.; Baumgartner, I.; Schlüter, M.; Tonn, T.; Seeger, F.; Dimmeler, S.; Lindhoff-Last, E.; et al. Intraarterial Administration of Bone Marrow Mononuclear Cells in Patients With Critical Limb Ischemia. Circ. Cardiovasc. Interv. 2011, 4, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Losordo, D.W. Cell Therapy for Critical Limb Ischemia. Circ. Cardiovasc. Interv. 2011, 4, 2–5. [Google Scholar] [CrossRef] [Green Version]
- MacAskill, M.G.; Saif, J.; Condie, A.; Jansen, M.; MacGillivray, T.; Tavares, A.; Fleisinger, L.; Spencer, H.L.; Besnier, M.; Martin, E.; et al. Robust Revascularization in Models of Limb Ischemia Using a Clinically Translatable Human Stem Cell-Derived Endothelial Cell Product. Mol. Ther. 2018, 26, 1669–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Prater, D.N.; Case, J.; Ingram, D.A.; Yoder, M. Working hypothesis to redefine endothelial progenitor cells. Leukemia 2007, 21, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, K.; Yoder, M.C. Tissue regeneration using endothelial colony-forming cells: Promising cells for vascular repair. Pediatr. Res. 2018, 83, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Beltran-Camacho, L.; Jimenez-Palomares, M.; Rojas-Torres, M.; Gomar, I.S.; Rosal-Vela, A.; Eslava-Alcon, S.; Perez-Segura, M.C.; Serrano, A.; Antequera-González, B.; Alonso-Piñero, J.A.; et al. Identification of the initial molecular changes in response to circulating angiogenic cells-mediated therapy in critical limb ischemia. Stem Cell Res. Ther. 2020, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Eslava-Alcon, S.; Extremera-García, M.; Sanchez-Gomar, I.; Beltrán-Camacho, L.; Rosal-Vela, A.; Muñoz, J.; Ibarz, N.; Alonso-Piñero, J.; Rojas-Torres, M.; Jiménez-Palomares, M.; et al. Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo. Int. J. Mol. Sci. 2020, 21, 5256. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Ng, W.K.; Chan, J.K.Y. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. STEM CELLS Transl. Med. 2016, 5, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Yoder, M.C. Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Yoder, M.C. Endothelial progenitor cell: A blood cell by many other names may serve similar functions. J. Mol. Med. 2013, 91, 285–295. [Google Scholar] [CrossRef]
- Annex, B.H. Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 2013, 10, 387–396. [Google Scholar] [CrossRef]
- Bayraktutan, U. Endothelial progenitor cells: Potential novel therapeutics for ischaemic stroke. Pharmacol. Res. 2019, 144, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Assmus, B.; Schächinger, V.; Teupe, C.; Britten, M.; Lehmann, R.; Döbert, N.; Grünwald, F.; Aicher, A.; Urbich, C.; Martin, H.; et al. Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE-AMI). Circulation 2002, 106, 3009–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meglio, F.; Nurzynska, D.; Castaldo, C.; Arcucci, A.; DE Santo, L.S.; De Feo, M.; Cotrufo, M.; Montagnani, S.; Giordano-Lanza, G. In vitro cultured progenitors and precursors of cardiac cell lineages from human normal and post-ischemic hearts. Eur. J. Histochem. 2007, 51, 275–282. [Google Scholar]
- Liu, P.; Zhou, B.; Gu, D.; Zhang, L.; Han, Z.C. Endothelial progenitor cell therapy in atherosclerosis: A double-edged sword? Ageing Res. Rev. 2009, 8, 83–93. [Google Scholar] [CrossRef]
- Vega, F.M.; Gautier, V.; Fernandez-Ponce, C.M.; Extremera, M.; Altelaar, A.; Millan, J.; Tellez, J.C.; Hernandez-Campos, J.A.; Conejero, R.; Bolivar, J.; et al. The atheroma plaque secretome stimulates the mobilization of endothelial progenitor cells ex vivo. J. Mol. Cell. Cardiol. 2017, 105, 12–23. [Google Scholar] [CrossRef]
- Hristov, M.; Weber, C. Ambivalence of progenitor cells in vascular repair and plaque stability. Curr. Opin. Lipidol. 2008, 19, 491–497. [Google Scholar] [CrossRef]
- Yoon, C.-H.; Hur, J.; Park, K.-W.; Kim, J.-H.; Lee, C.-S.; Oh, I.-Y.; Kim, T.-Y.; Cho, H.-J.; Kang, H.-J.; Chae, I.-H.; et al. Synergistic Neovascularization by Mixed Transplantation of Early Endothelial Progenitor Cells and Late Outgrowth Endothelial Cells. Circulation 2005, 112, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Jujo, K.; Ii, M.; Losordo, D.W. Endothelial progenitor cells in neovascularization of infarcted myocardium. J. Mol. Cell. Cardiol. 2008, 45, 530–544. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.T.; Zhang, S.; Wu, H.D.; Körbling, M.; Willerson, J.T.; Estrov, Z. Transdifferentiation of Human Peripheral Blood CD34 + -Enriched Cell Population Into Cardiomyocytes, Endothelial Cells, and Smooth Muscle Cells In Vivo. Circulation 2003, 108, 2070–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslava-Alcon, S.; Extremera-García, M.; González-Rovira, A.; Rosal-Vela, A.; Rojas-Torres, M.; Beltran-Camacho, L.; Sanchez-Gomar, I.; Jiménez-Palomares, M.; Alonso-Piñero, J.; Conejero, R.; et al. Molecular signatures of atherosclerotic plaques: An up-dated panel of protein related markers. J. Proteom. 2020, 221, 103757. [Google Scholar] [CrossRef] [PubMed]
- Tarlov, I.M. Spinal cord compression studies. III. Time limits for recovery after gradual compression in dogs. AMA Arch. Neurol. Psychiatry 1954, 71, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Marston, N.; Sandoval, Y.; Pierpont, G.; Adabag, S.; Brenes, J.; Santilli, S.; McFalls, E.O. Prognostic value of 12-lead electrocardiogram and peak troponin I level after vascular surgery. J. Vasc. Surg. 2013, 57, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobellis, G.; Silvestroni, A.; Lillo, S.; Sica, G.; Botti, C.; Maione, C.; Schiavone, V.; Rocco, S.; Brando, G.; Sica, V. Long-term effects of repeated autologous transplantation of bone marrow cells in patients affected by peripheral arterial disease. Bone Marrow Transplant. 2008, 42, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Torres, M.; Jiménez-Palomares, M.; Martín-Ramírez, J.; Beltrán-Camacho, L.; Sánchez-Gomar, I.; Eslava-Alcon, S.; Rosal-Vela, A.; Gavaldá, S.; Durán-Ruiz, M.C. REX-001, a BM-MNC Enriched Solution, Induces Revascularization of Ischemic Tissues in a Murine Model of Chronic Limb-Threatening Ischemia. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Lee, P.Y.; Wang, J.-X.; Parisini, E.; Dascher, C.C.; Nigrovic, P.A. Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 2013, 94, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Soria-Juan, B.; Escacena, N.; Capilla-González, V.; Aguilera, Y.; Llanos, L.; Tejedo, J.R.; Bedoya, F.J.; Juan, V.; De La Cuesta, A.; Ruiz-Salmerón, R.; et al. Cost-Effective, Safe, and Personalized Cell Therapy for Critical Limb Ischemia in Type 2 Diabetes Mellitus. Front. Immunol. 2019, 10, 1151. [Google Scholar] [CrossRef]
- Funakoshi, K.; Bagheri, M.; Zhou, M.; Suzuki, R.; Abe, H.; Akashi, H. Highly sensitive and specific Alu-based quantification of human cells among rodent cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Qadura, M.; Terenzi, D.C.; Verma, S.; Al-Omran, M.; Hess, D.A. Concise review: Cell therapy for critical limb ischemia: An integrated review of preclinical and clinical studies. Stem Cells 2018, 36, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrán-Camacho, L.; Rojas-Torres, M.; Durán-Ruiz, M. Current Status of Angiogenic Cell Therapy and Related Strategies Applied in Critical Limb Ischemia. Int. J. Mol. Sci. 2021, 22, 2335. [Google Scholar] [CrossRef]
- Watson, E.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell. Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Pan, Y.-Y.; Wang, X.-X.; Qiu, Y.-G.; Mao, W. Endothelial progenitor cells in age-related vascular remodeling. Cell Transplant. 2018, 27, 786–795. [Google Scholar] [CrossRef]
- Yang, Z.; Von Ballmoos, M.W.; Faessler, D.; Voelzmann, J.; Ortmann, J.; Diehm, N.; Kalka-Moll, W.; Baumgartner, I.; Di Santo, S.; Kalka, C. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis 2010, 211, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Macrophages and Secrete Angiogenic Growth Factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Aly, H.; Hamed, Z.; Mohsen, L.; Ramy, N.; Arnaoot, H.; Lotfy, A. Serum amyloid A protein and hypoxic ischemic encephalopathy in the newborn. J. Perinatol. 2010, 31, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Kawamoto, A. Stem cell-based peripheral vascular regeneration. Adv. Drug Deliv. Rev. 2017, 120, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone Marrow-Derived Cells Do Not Incorporate Into the Adult Growing Vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.E.; Lyass, A.; Courchesne, P.; Chen, G.; Liu, C.; Yin, X.; Hwang, S.J.; Massaro, J.M.; Larson, M.G.; Levy, D. Protein Biomarkers of Cardiovascular Disease and Mortality in the Community. J. Am. Heart Assoc. 2018, 7, e008108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum Amyloid A Directly Accelerates the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.M.; Pai, J.K.; Bertoia, M.L.; Gansevoort, R.T.; Bakker, S.J.L.; Cooke, J.P.; Rimm, E.B.; Mukamal, K.J. β2-Microglobulin, Cystatin C, and Creatinine and Risk of Symptomatic Peripheral Artery Disease. J. Am. Hear. Assoc. 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Liu, J.; Rong, D.; Ge, Y.; Li, H.; Jia, S.; Sun, G.; Zhang, H.; Liu, X.; Guo, W. Association between Cystatin C and Peripheral Artery Disease in the Chinese Han Population. Ann. Vasc. Surg. 2021, 73, 244–253. [Google Scholar] [CrossRef]
- Ekman, C.; Gottsäter, A.; Lindblad, B.; Dahlbäck, B. Plasma concentrations of Gas6 and soluble Axl correlate with disease and predict mortality in patients with critical limb ischemia. Clin. Biochem. 2010, 43, 873–876. [Google Scholar] [CrossRef]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Faga, T.; Michael, A.; Ielapi, N.; Grande, R.; Sapienza, P.; De Franciscis, S.; Mastroroberto, P.; et al. The Association of Matrix Metalloproteinases with Chronic Kidney Disease and Peripheral Vascular Disease: A Light at the End of the Tunnel? Biomolecules 2020, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Beaudeux, J.-L.; Giral, P.; Bruckert, E.; Bernard, M.; Foglietti, M.-J.; Chapman, M. Serum matrix metalloproteinase-3 and tissue inhibitor of metalloproteinases-1 as potential markers of carotid atherosclerosis in infraclinical hyperlipidemia. Atherosclerosis 2003, 169, 139–146. [Google Scholar] [CrossRef]
- Martínez-Aguilar, E.; Gomez-Rodriguez, V.; Orbe, J.; Rodriguez, J.; Fernández-Alonso, L.; Roncal, C.; Paramo, J.A. Matrix metalloproteinase 10 is associated with disease severity and mortality in patients with peripheral arterial disease. J. Vasc. Surg. 2015, 61, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Morishita, T.; Uzui, H.; Nakano, A.; Mitsuke, Y.; Geshi, T.; Ueda, T.; Lee, J.-D. Number of Endothelial Progenitor Cells in Peripheral Artery Disease as a Marker of Severity and Association with Pentraxin-3, Malondialdehyde-Modified Low-Density Lipoprotein and Membrane Type-1 Matrix Metalloproteinase. J. Atheroscler. Thromb. 2012, 19, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Muhs, B.E.; Plitas, G.; Delgado, Y.; Ianus, I.; Shaw, J.P.; Adelman, M.A.; Lamparello, P.; Shamamian, P.; Gagne, P. Temporal expression and activation of matrix metalloproteinases-2, -9, and membrane type 1-matrix metalloproteinase following acute hindlimb ischemia. J. Surg. Res. 2003, 111, 8–15. [Google Scholar] [CrossRef]
- Tayebjee, M.H.; Tan, K.T.; MacFadyen, R.J.; Lip, G.Y.H. Abnormal circulating levels of metalloprotease 9 and its tissue inhibitor 1 in angiographically proven peripheral arterial disease: Relationship to disease severity. J. Intern. Med. 2004, 257, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Le Nghia, T. The dual personalities of matrix metalloproteinases in inflammation. Front. Biosci. 2007, 12, 1475–1487. [Google Scholar] [CrossRef] [Green Version]
- Decock, J.; Thirkettle, S.; Wagstaff, L.; Edwards, D. Matrix metalloproteinases: Protective roles in cancer. J. Cell. Mol. Med. 2011, 15, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Kratz, G.; Haegerstrand, A.; Ståhle-Bäckdahl, M. Collagenase Expression Is Rapidly Induced in Wound-Edge Keratinocytes After Acute Injury in Human Skin, Persists During Healing, and Stops at Re-Epithelialization. J. Investig. Dermatol. 1995, 104, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauris, J.; Woodward, A.M.; Cao, Z.; Panjwani, N.; Argüeso, P. Molecular basis for MMP9 induction and disruption of epithelial cell-cell contacts by galectin-3. J. Cell Sci. 2014, 127, 3141–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivak, J.; Fini, M. MMPs in the eye: Emerging roles for matrix metalloproteinases in ocular physiology. Prog. Retin. Eye Res. 2002, 21, 1–14. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, K.H.; Lee, W.M.; Jun, J.H.; Kim, D.H. CD44 Disruption Attenuates Murine Hepatic Ischemia/Reperfusion Injury. J. Korean Med Sci. 2011, 26, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, C.; Mas, S.; Martín-Ventura, J.L.; Meilhac, O.; Michel, J.B.; Gallego-Delgado, J.; Lazaro, A.; Tuñon, J.; Egido, J.; Vivanco, F. Proteomic analysis of human vessels: Application to atherosclerotic plaques. Proteomics 2003, 3, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.; Toledo, A.H.; Lopez-Neblina, F.; Anaya-Prado, R.; Toledo-Pereyra, L.H. Nitric Oxide Mechanism of Protection in Ischemia and Reperfusion Injury. J. Investig. Surg. 2009, 22, 46–55. [Google Scholar] [CrossRef]
- Hamed, S.; Brenner, B.; Roguin, A. Nitric oxide: A key factor behind the dysfunctionality of endothelial progenitor cells in diabetes mellitus type-2. Cardiovasc. Res. 2010, 91, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Brown, D.A.; Neufer, P.D.; McClung, J.M. Mitochondrial Regulation of the Muscle Microenvironment in Critical Limb Ischemia. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, P.A.; Mccarty, J.H. TGF-β Activation and Signaling in Angiogenesis. In Physiologic and Pathologic Angiogenesis—Signaling Mechanisms and Targeted Therapy; IntechOpen: London, UK, 2017; p. 2017. [Google Scholar]
- Bei, Y.; Pan, L.-L.; Zhou, Q.; Zhao, C.; Xie, Y.; Wu, C.; Meng, X.; Gu, H.; Xu, J.; Zhou, L.; et al. Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med. 2019, 17, 1–20. [Google Scholar] [CrossRef]
- Kawamoto, A.; Katayama, M.; Handa, N.; Kinoshita, M.; Takano, H.; Horii, M.; Sadamoto, K.; Yokoyama, A.; Yamanaka, T.; Onodera, R.; et al. Intramuscular Transplantation of G-CSF-Mobilized CD34+Cells in Patients With Critical Limb Ischemia: A Phase I/IIa, Multicenter, Single-Blinded, Dose-Escalation Clinical Trial. STEM CELLS 2009, 27, 2857–2864. [Google Scholar] [CrossRef]
- Masuda, H.; Kalka, C.; Takahashi, T.; Yoshida, M.; Wada, M.; Kobori, M.; Itoh, R.; Iwaguro, H.; Eguchi, M.; Iwami, Y.; et al. Estrogen-Mediated Endothelial Progenitor Cell Biology and Kinetics For Physiological Postnatal Vasculogenesis. Circ. Res. 2007, 101, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Chang, T.-T.; Chang, C.-C.; Chen, J.-W. Fatty-Acid-Binding Protein 4 as a Novel Contributor to Mononuclear Cell Activation and Endothelial Cell Dysfunction in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 9245. [Google Scholar] [CrossRef] [PubMed]
- Langlois, D.; Forcheron, F.; Li, J.-Y.; Del Carmine, P.; Neggazi, S.; Beylot, M. Increased Atherosclerosis in Mice Deficient in Perilipin1. Lipids Health Dis. 2011, 10, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debard, C.; Laville, M.; Berbe, V.; Loizon, E.; Guillet, C.; Morio-Liondore, B.; Boirie, Y.; Vidal, H. Expression of key genes of fatty acid oxidation, including adiponectin receptors, in skeletal muscle of Type 2 diabetic patients. Diabetologia 2004, 47, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, C.; Yang, H.; Liu, S.; Lu, Y.; Fu, P.; Liu, J. Metabolomics reveal mitochondrial and fatty acid metabolism disorders that contribute to the development of DKD in T2DM patients. Mol. BioSyst. 2017, 13, 2392–2400. [Google Scholar] [CrossRef]
- Ljubkovic, M.; Gressette, M.; Bulat, C.; Cavar, M.; Bakovic, D.; Fabijanic, D.; Grkovic, I.; Lemaire, C.; Marinovic, J. Disturbed Fatty Acid Oxidation, Endoplasmic Reticulum Stress, and Apoptosis in Left Ventricle of Patients With Type 2 Diabetes. Diabetes 2019, 68, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Mercier, C.; Rousseau, M.; Geraldes, P. Growth Factor Deregulation and Emerging Role of Phosphatases in Diabetic Peripheral Artery Disease. Front. Cardiovasc. Med. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Shintani, S.; Shimizu, Y.; Kondo, K.; Ishii, M.; Wu, H.; Murohara, T. Therapeutic angiogenesis by autologous adipose-derived regenerative cells: Comparison with bone marrow mononuclear cells. Am. J. Physiol. Circ. Physiol. 2014, 307, H869–H879. [Google Scholar] [CrossRef] [Green Version]
- Jude, E.B.; Oyibo, S.; Chalmers, N.; Boulton, A.J. Peripheral Arterial Disease in Diabetic and Nondiabetic Patients: A comparison of severity and outcome. Diabetes Care 2001, 24, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor α and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Hu, L.; Dai, S.-C.; Luan, X.; Chen, J.; Cannavicci, A. Dysfunction and Therapeutic Potential of Endothelial Progenitor Cells in Diabetes Mellitus. J. Clin. Med. Res. 2018, 10, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Yuan, R.; Geng, S.; Chen, K.; Diao, N.; Chu, H.W.; Li, L. Low-grade inflammatory polarization of monocytes impairs wound healing. J. Pathol. 2015, 238, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef] [Green Version]
- Prame, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-Z.; Lee, C.N.; Moreno-Luna, R.; Neumeyer, J.; Piekarski, B.; Zhou, P.; Moses, M.A.; Sachdev, M.; Pu, W.; Emani, S.; et al. Host non-inflammatory neutrophils mediate the engraftment of bioengineered vascular networks. Nat. Biomed. Eng. 2017, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Beyrau, M.; Bodkin, J.V.; Nourshargh, S. Neutrophil heterogeneity in health and disease: A revitalized avenue in inflammation and immunity. Open Biol. 2012, 2, 120134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.J.; O’Neill, C.L.; O’Doherty, T.M.; Knott, H.; Guduric-Fuchs, J.; Gardiner, T.A.; Stitt, A.W. Myeloid Angiogenic Cells Act as Alternative M2 Macrophages and Modulate Angiogenesis through Interleukin-8. Mol. Med. 2011, 17, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Jaguin, M.; Houlbert, N.; Fardel, O.; Lecureur, V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell. Immunol. 2013, 281, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, Y.; Yu, J.; Feng, L.; Hou, S.; Liu, Y.; Guo, M.; Xie, Y.; Meng, J.; Zhang, H.; et al. Targeting the Shift from M1 to M2 Macrophages in Experimental Autoimmune Encephalomyelitis Mice Treated with Fasudil. PLoS ONE 2013, 8, e54841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Nombela, F.; Vivancos, J.; Hamilton, J.A.; Corbí, Á.L.; Lizasoain, I.; Moro, M.A. N2 Neutrophils, Novel Players in Brain Inflammation After Stroke. Stroke 2013, 44, 3498–3508. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Culebras, A.; Durán-Laforet, V.; Peña-Martínez, C.; Moraga, A.; Ballesteros, I.; Cuartero, M.; De La Parra, J.; Palma-Tortosa, S.; Hidalgo, A.; Corbí, A.L.; et al. Role of TLR4 (Toll-Like Receptor 4) in N1/N2 Neutrophil Programming After Stroke. Stroke 2019, 50, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Kalka, C.; Masuda, H.; Takahashi, T.; Kalka-Moll, W.M.; Silver, M.; Kearney, M.; Li, T.; Isner, J.M.; Asahara, T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. USA 2000, 97, 3422–3427. [Google Scholar] [CrossRef]
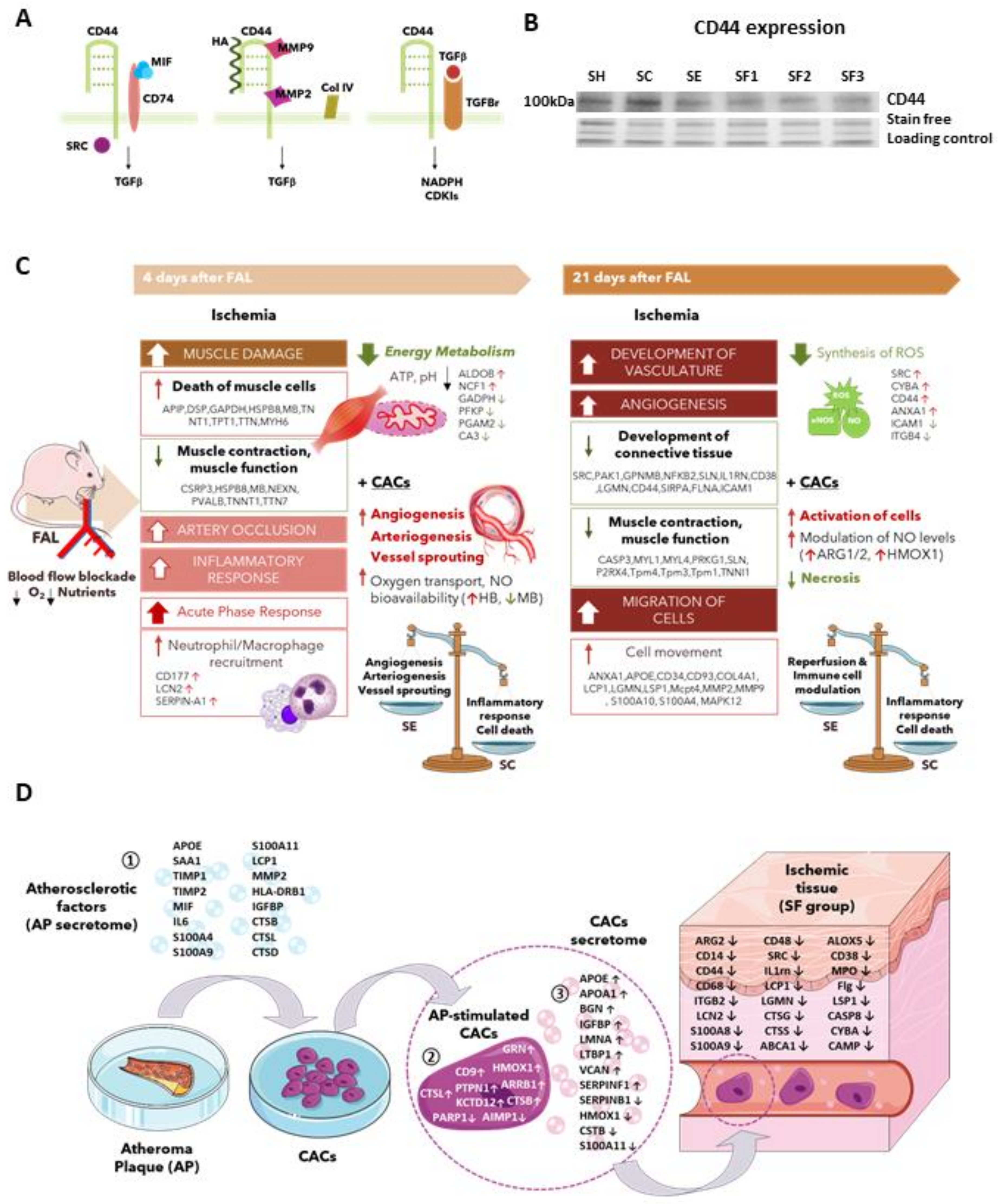

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Functions (p-Value) | Activation z-Score | Molecules | Proteins |
|---|---|---|---|---|
| Cardiovascular System Development and Function | Vasculature development (1.29E-06) | 2.856 ▲ | ABI1↑, ADD1↑, ANXA1↑, APOE↑, CASP3↑, CD34↑, COL4A1↑, COMMD1↑, CYBA↑, CYBB↑, EMILIN1↑, F11R↑, FLNA↑, G6PD↑, GIT1↑, GLUL↑, HLA-DQB1↑, HSPB1↑, HSPB6↑, HSPB7↑, HSPG2↑, IGFBP7↑, LAMA4↑, LGALS3↑, LRP1↑, Mcpt4↑, MMP2↑, MMP9↑, NOS3↑, NPC2↑, P2RX4↑, PAK1↑, PLVAP↑, PLXNB2↑, POFUT1↑, PROCR↑, PTGES↑, S100A4↑, SERPINH1↑, SGPL1↑, STAT3↑, THRAP3↑, TKT↑, GTF2I↓, HRAS↓, ICAM1↓, SMOC2↓ | 47 |
| Angiogenesis (3.63E-06) | 2.855 ▲ | ABI1↑, ADD1↑, ANXA1↑, APOE↑, CASP3↑, CD34↑, COL4A1↑, COMMD1↑, CYBA↑, CYBB↑, EMILIN1↑, F11R↑, FLNA↑, G6PD↑, GLUL↑, HLA-DQB1↑, HSPB1↑, HSPB6↑, HSPG2↑, IGFBP7↑, LAMA4↑, LGALS3↑, LRP1↑, Mcpt4↑, MMP2↑, MMP9↑, NOS3↑, PAK1↑, PLVAP↑, POFUT1↑, PROCR↑, PTGES↑, S100A4↑, SERPINH1↑, SGPL1↑, STAT3↑, THRAP3↑, TKT↑, GTF2I↓, HRAS↓, ICAM1↓, SMOC2↓ | 42 | |
| Vasculogenesis (1.21E-05) | 2.575 ▲ | ABI1↑, ANXA1↑, APOE↑, CASP3↑, CD34↑, COL4A1↑, COMMD1↑, CYBA↑, CYBB↑, F11R↑, FLNA↑, G6PD↑, GLUL↑, HLA-DQB1↑, HSPG2↑, IGFBP7↑, LGALS3↑, LRP1↑, Mcpt4↑, MMP2↑, MMP9↑, NOS3↑, PAK1↑, PLVAP↑, POFUT1↑, PROCR↑, S100A4↑, SERPINH1↑, SGPL1↑, STAT3↑, THRAP3↑, TKT↑, HRAS↓, ICAM1↓, SMOC2↓ | 35 | |
| Cell Death and Survival | Apoptosis (1.56E-06) | −2.602 ▼ | ACACA↑, ACLY↑, ALDH3A1↑, ANXA1↑, APOE↑, ARF4↑, ARL6IP1↑, ASAH1↑, ASNS↑, BASP1↑, CASP3↑, CASP6↑, COL4A1↑, CTSZ↑, CYBA↑, CYBB↑, CYP2F1↑, DDX58↑, DPM3↑, DTYMK↑, ELMO1↑, EMILIN2↑, ERC1↑, Ewsr1↑, FAH↑, FASN↑, FLNA↑, G6PD↑, GMDS↑, HLA-DQB1↑, HRAS↓, HSPB1↑, HSPB6↑, HSPG2↑, IGFBP7↑, ILF3↑, IQGAP2↑, IRF2BP2↑, Irgm1↑, LAMA4↑, LGALS3↑, LGALS7/LGALS7B↑, LGMN↑, LIMS1↑, LRP1↑, LSP1↑, Mcpt4↑, MMP2↑, MMP9↑, Mt2↑, MTA2↑, NELFB↑, NFKB2↑, NOS3↑, P2RX4↑, PAK1↑, PEX11B↑, PPP1R9B↑, PRKAR2B↑, PRMT1↑, PROCR↑, PRTN3↑, PTGES↑, RAB32↑, S100A10↑, S100A4↑, SCD↑, SEL1L↑, SERPINB1↑, SERPINH1↑, SET↑, SGPL1↑, SIRPA↑, SLC25A10↑, SLC25A5↑, SNX1↑, SRI↑, SRSF2↑, STAT3↑, SWAP70↑, TARDBP↑, TRIM28↑, UBA2↑, WDR5↑, XRCC5↑, ICAM1↓, IFIT2↓, MAPK12↓, NQO1↓, PAFAH1B3↓, TIGAR↓ | 91 |
| Cell survival (7.27E-04) | 3.789 ▲ | ACACA↑, ACLY↑, APOE↑, ASAH1↑, ASNS↑, CASP3↑, CASP6↑, CYBA↑, CYBB↑, EMILIN2↑, FASN↑, FLNA↑, GALK2↑, GIT1↑, GPNMB↑, HLA-A↑, HNRNPU↑, HSPB1↑, HSPB6↑, IGFBP7↑, LGALS3↑, LGALS7/LGALS7B↑, LIMS1↑, LRP1↑, MCFD2↑, MGST1↑, MMP9↑, Mt2↑, NFKB2↑, NOS3↑, PAK1↑, POSTN↑, PRMT1↑, PRTN3↑, PSME3↑, S100A4↑, SCD↑, SEL1L↑, SET↑, SGPL1↑, SHMT2↑, STAT3↑, TARDBP↑, THRSP↑, TRIM28↑, XRCC5↑, HRAS↓, ICAM1↓, MAPK12↓, SMC1A↓ | 50 | |
| Cellular Movement | Cell movement (1.03E-09) | 4.746 ▲ | ABI1↑, ACACA↑, ADD1↑, ANXA1↑, APBB1IP↑, APOE↑, ARF4↑, ARFGAP3↑, ARPC1B↑, ASNS↑, BIN2↑, CAP1↑, CD34↑, CD93↑, CLIC1↑, COL4A1↑, COMMD5↑, CORO1C↑, CTSZ↑, CYBB↑, DDX58↑, ELMO1↑, EMILIN2↑, ERC1↑, EVL↑, F11R↑, FABP5↑, FASN↑, FHL3↑, FLNA↑, FLNC↑, FSCN1↑, G6PD↑, GIT1↑, GIT2↑, GLUL↑, GPNMB↑, HLA-A↑, HNRNPA2B1↑, HSPB1↑, HSPG2↑, IFITM3↑, IGFBP7↑, ILF3↑, Irgm1↑, LAMA4↑, LCP1↑, LGALS3↑, LGMN↑, LIMS1↑, LIPE↑, LRP1↑, LSP1↑, Mcpt4↑, MMP2↑, MMP9↑, MRC2↑, Mt2↑, NAAA↑, NDUFAF3↑, NFKB2↑, NOS3↑, P2RX4↑, PAK1↑, PEX11B↑, PLXNB2↑, POSTN↑, PPP1R9B↑, PRMT1↑, PROCR↑, PRTN3↑, PTGES↑, RGS10↑, S100A10↑, S100A4↑, SERPINB1↑, SERPINH1↑, SGPL1↑, SIRPA↑, STAT3↑, SWAP70↑, TARDBP↑, TKT↑, GTF2I↓, HRAS↓, ICAM1↓, IFIT2↓, MAPK12↓, NNMT↓, NQO1↓, SMOC2↓, Tpm1↓, TPM3↓ | 93 |
| Cell Migration (1.17E-07) | 4.562 ▲ | ACACA↑, ADD1↑, ANXA1↑, APBB1IP↑, APOE↑, ARF4↑, ARFGAP3↑, ASNS↑, CAP1↑, CD34↑, CD93↑, COL4A1↑, COMMD5↑, CORO1C↑, CTSZ↑, CYBB↑, DDX58↑, ELMO1↑, EMILIN2↑, ERC1↑, EVL↑, F11R↑, FABP5↑, FASN↑, FLNA↑, FLNC↑, FSCN1↑, G6PD↑, GIT1↑, GIT2↑, GLUL↑, GPNMB↑, HLA-A↑, HNRNPA2B1↑, HSPB1↑, IFITM3↑, IGFBP7↑, ILF3↑, Irgm1↑, LAMA4↑, LCP1↑, LGALS3↑, LGMN↑, LRP1↑, LSP1↑, Mcpt4↑, MMP2↑, MMP9↑, MRC2↑, NAAA↑, NFKB2↑, NOS3↑, P2RX4↑, PAK1↑, PEX11B↑, PLXNB2↑, POSTN↑, PPP1R9B↑, PRMT1↑, PROCR↑, PRTN3↑, PTGES↑, S100A10↑, S100A4↑, SERPINB1↑, SGPL1↑, SIRPA↑, STAT3↑, SWAP70↑, TARDBP↑, HRAS↓, ICAM1↓, IFIT2↓, MAPK12↓, NDUFAF3↓, NNMT↓, NQO1↓, SMOC2↓, Tpm1↓, TPM3↓ | 80 | |
| Chemotaxis (5.15E-07) | 2.773 ▲ | ANXA1↑, APOE↑, BIN2↑, CYBB↑, ELMO1↑, GIT1↑, GIT2↑, LCP1↑, LGALS3↑, LGMN↑, LRP1↑, LSP1↑, MMP2↑, MMP9↑, Mt2↑, NFKB2↑, NOS3↑, P2RX4↑, PAK1↑, PRTN3↑, PTGES↑, RGS10↑, S100A4↑, SERPINB1↑, SERPINH1↑, SIRPA↑, STAT3↑, SWAP70↑ HRAS↓, ICAM1↓ | 30 |
| SE/SC | SF/SC | Proteins | Functions |
|---|---|---|---|
| ↑ | ↑ | LSMEM1 | Necrosis ▲ Apoptosis ▲ Atherosclerosis ▬ Neovascularization ▬ Angiogenesis ▼ Vasculature Development ▼ Endothelial cell development ▲ Endothelial cell Proliferation▲ |
| ↓ | ↓ | AACS, ALDH3A1, ATP2B4, BPHL, BTF3L4, CASQ2, CAV1, CAV3, CD99, CDC5L, CILP, CKAP5, COX6A1, CREG1, FAU, FHL1, FLG, GAMT, GCDH, GM8210, GPC1, GPNMB, HDGFL2, HSPB6, HSPB7, IFI30, IRF2BPL, KLHL41, LAMP2, LGALS3, MAN2B2, MAP3K20, MCAM, MRPL12, MRPS5, MRPS27, MTCH1, MYL4, MYL6B, NES, PLOD2, PRKG1, RBBP6, RPLP1, RPL13, RPL17, RPL21, RPL35, RPL37A, RPS17, S100A11, SCD1, SPARC, SEC16A, SPRR1A, SLMAP, SERPINF1, SERPINB6A, SERF2, SNRPD2, STMN1, TGTP1, TNC, TMEM167, TPP1, XIRP1 | |
| ↑ | - | ABCA1, APAF1, APBB1IP, APOBR, ARG2, ARL6IP1, ASS1, BC017643, CAPZA1, CAR13, CASP8, CD14, CYBB, DCAKD, DDX39, DHFR, DR1, DTYMK, ELANE, ELMO1, EMB, EMILIN1, EMILIN2, GCN1L1, GFPT1, GIT2, H2AFY, HMOX1, HVM22, IFIH1, IFIT1, IFIT2, IGHV5-4, IGJ, IGKV12-44, IGKV12-47, IGKV14-111, IL1RN, IQGAP2, ITGB2, LGALS7, LGALS9, LOC433053, LSP1, MOGS, MPO, MSR1, NAPSA, NCF1, NCF2, NCKAP1L, PLBD1, PLIN2, PRPF40A, P2RX4, PRTN3, PSTPIP1, PTGES, PTPN1, RETNLG, ROD1, SAA4, SDF2L1, SEL1L, SKAP2, SNX18, SPCS3, SLC9A3R1, SRC, STFA1, STS, SUN2, SUPT16H, TAP1, TBL2, VAV1, WASF2 | Apoptosis ▲ Atherosclerosis ▲ Angiogenesis Vasculature Development ▬ Vasculogenesis ▬ Atherogenesis ▬ Activation of cells ▲ Cell movement ▲ Immune response of cells ▲ |
| ↓ | - | ABHD5, ACADS, ACAT2, ACLY, ALDH2, ALDH1A7, ALDH1L1, ALDH4A1, CAR5B, CES1D, CES1F, COMP, COX8B, 4931406C07RIK, CRABP1, CRYAB, CSAD, CSRP3, CYP2E1, DNAJA4, ECHDC1, EPHX2, FABP4, FAH, FASN, GADD45GIP1, G3BP2, GLUL, GNAI1, GSTA3, GSTZ1, HDHD3, KLHL40, KRT10, LIPE, ME1, MGLL, MRPL11, MRPL41, MRPL53, NRAP, PCBD1, PCX, PDLIM1, PTGES3L, PTMS, PLIN1, RBP7, SEPT4, SSC5D, SLC25A1, THRSP, THBS4, TST, YAP1 | |
| - | ↑ | None | Necrosis ▲ Apoptosis ▲ PAD ▬ Diabetes mellitus ▬ Cell Activation ▼ Cell Attachment ▼ Cell movement ▼ Cell Migration ▼ Cell Outgrowth ▼ Lipid Accumulation ▲ Quantity of IFN in blood ▲ |
| - | ↓ | ACOT9, ACOX1, ACP2, ACTB, ACTN1, ACTN2, ACTN4, ADIPOQ, ADPRHL2, AIF1, ALDH16A1, ANKFY1, ANO6, AMDHD2, AMY1, APCS, AP1M1, AP2M1, APOBEC2, APOD, APOE, ARHGDIB, ARPIN, ARPC5, ART3, BASP1, BGN, BIN2, B2M, CACNG1, CD44, CDH13, CFH, CKAP4, CLTC, CNN2, COA7, COL1A2, COL12A1, COL14A1, COLGALT1, COPA, COPB1, COPE, COPG1, COPG2, CORO1C, COTL1, CSK, CTSA, CTSC, CTSD, CTSH, CTSZ, DCLK1, DCUN1D2, DDOST, DDX5, DHX15, DNM3, DNPEP, DYNC1H1, EEF1AKMT1, EEF1B2, EFEMP1, EIF4A3, EIF3E, EIF3L, EFHD2, EHD2, EHD4, ERGIC3, ERO1LB, ERP29, ETF1, ETFDH, EVL, FKBP15, FN1, GC, GLA, GM5571, GMFB, GMPR2, GSTT1, GSTT3, GUSB, H2-AA, H2-AB1, H2AFV, HAL, H2-D1, H2-EB1, HEXB, HIST1H4A, HIST1H2BB, HNRNPC, HPRT, HRG, HSPB2, HYPK, IFI204, IFI211, IFI44L, IGHG, IGHG3, IGHV2-3, IIGP1B, IQGAP1, IRGM1, ISG15, ITGA6, ITGB4, JPT1, LAMTOR5, LCP1, LGALS3BP, LGMN, LIPA, LMNA, LRRC59, LXN, MAN2A1, MAN2B1, MARCKSL1, MBL1, ME2, MFF, MMP9, MPZ, MYH7, MYL6, MYO1C, NAA15, NAA20, NAP1L1, NCAM1, NDUFB6, NGLY1, NGP, ORM2, OST4, PACSIN3, PAPSS2, PCNA, PDXK, PEA15A, PIGS, PMP2, POSTN, PPIC, PP1R14B, PPP1R14C, PRCP, PRDX4, PRX, PSMB10, PSMC1, PTRH2, PYCARD, RAB12, RACK1, RCC2, RCN3, RENBP, RNF213, RPA3, RPL3, RPL4, RPL5, RPL6, RPL7, RPL8, RPL9, RPL10A, RPL11, RPL12, RPL13A, RPL14, RPL15, RPL7A, RPL18, RPL22, RPL23, RPL23A, RPL24, RPL26, RPL27, RPL27A, RPL28, RPL36, RPLP0, RPLP2, RPL32P, RPL10-PS1, RPL19-PS11, RPL34-PS1, RPL36A-PS3, RPS2, RPS3, RPS5, RPS7, RPS8, RPS9, RPS10, RPS11, RPS14, RPS18, RPS19, RPS20, RPS21, RPS25, RPS28, RPS29, RPS15A, RPSA, RPS3A1, RPS2-PS13, RPS13-PS1, RPS26-PS1, RPS6-PS4, RPS16-PS2, RPS4X, RTCB, S100A4, SCAMP1, SCAMP3, SEPT5, SF3B5, SF3B6, SFN, SH3BGRL3, SIRPA, SLN, SNRPF, SH3BGRL, SNRPD3, SPATA7, SRSF2, SRSF1, SRSF5, SSR4, STARD7, STEAP3, STOM, SUB1, SULT1A1, SYNC, SYNGR2, TANGO2, TAPT1, THY1, TNNC1, TNNT3, TMED9, TM9SF3, TPD52, TRIM72, TUBA1A, TUBB2A, TUBB5, TWF2, UCHL1, UFM1, UFSP2, UPP1, VAT1, VPS29, WDR82, ZBP1 | |
| ↑ | ↓ | ADGRE1, ALDH1B1, ALOX5AP, AOAH, AP3D1, ARG1, CAD, C5AR1, CD48, CD68, CD180, CEP170, CPT1A, C1QTNF3, C1S1, CTSG, CTSS, CYBA, DDX58, DNAJB11, DNASE2A, FYB, GALK2, GSDMD, H13, HEXA, HK3, IFI44, ITGAM, KDELR2, LBR, MAGOHB, MFSD1, MPEG1, MYL12B, NCF4, OLFM4, PARVG, PHF11D, PLA2G15, PLD4, PLEK, PRPF8, PRRC2A, PTPN6, PTPRC, RAB32, S100A8, SEC11A, SEC61A1, SP100, SRGAP2, STAT1, STAT2, SYK, TAPBP, TAP2, TMED3, TOR3A, TOP2A, STFA3, UGT1A7C, U2SURP | Angiogenesis ▲ Diabetes mellitus ▬ Cell Activation ▲ Cells movement ▲ Immune response of cells ▲ Cell Degranulation ▲ |
| ↓ | ↑ | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltrán-Camacho, L.; Jiménez-Palomares, M.; Sanchez-Gomar, I.; Rosal-Vela, A.; Rojas-Torres, M.; Eslava-Alcon, S.; Alonso-Piñero, J.A.; González-Rovira, A.; Extremera-García, M.J.; Conejero, R.; et al. Long Term Response to Circulating Angiogenic Cells, Unstimulated or Atherosclerotic Pre-Conditioned, in Critical Limb Ischemic Mice. Biomedicines 2021, 9, 1147. https://doi.org/10.3390/biomedicines9091147
Beltrán-Camacho L, Jiménez-Palomares M, Sanchez-Gomar I, Rosal-Vela A, Rojas-Torres M, Eslava-Alcon S, Alonso-Piñero JA, González-Rovira A, Extremera-García MJ, Conejero R, et al. Long Term Response to Circulating Angiogenic Cells, Unstimulated or Atherosclerotic Pre-Conditioned, in Critical Limb Ischemic Mice. Biomedicines. 2021; 9(9):1147. https://doi.org/10.3390/biomedicines9091147
Chicago/Turabian StyleBeltrán-Camacho, Lucía, Margarita Jiménez-Palomares, Ismael Sanchez-Gomar, Antonio Rosal-Vela, Marta Rojas-Torres, Sara Eslava-Alcon, Jose Angel Alonso-Piñero, Almudena González-Rovira, Mª Jesús Extremera-García, Rosario Conejero, and et al. 2021. "Long Term Response to Circulating Angiogenic Cells, Unstimulated or Atherosclerotic Pre-Conditioned, in Critical Limb Ischemic Mice" Biomedicines 9, no. 9: 1147. https://doi.org/10.3390/biomedicines9091147