
Novel Coagulation Factor VIII Gene Therapy in a Mouse Model of Hemophilia A by Lipid-Coated Fe3O4 Nanoparticles

 ,
, 
 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Liver-Specific Promoter Construction
2.3. Dual Luciferase Assays
2.4. Gene Therapy Plasmid Construction and Production
2.5. Synthesis of DPPC-Fe3O4 Nanoparticles
2.6. Cell Culture and Gene Transfection
2.7. Physicochemical Analysis
2.8. Transmission Electron Microscopy (TEM)
2.9. Electrophoretic Gel Mobility Shift Assay (EMSA)
2.10. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
2.11. Gene Delivery via Tail Vein Injection
2.12. Flow Cytometric Analysis
2.13. Evaluation of Coagulation Phenotypic Restoration
2.14. Detection of Blood Biochemical Parameters
2.15. Histologic Section Analysis
2.16. Statistical Analysis
3. Results
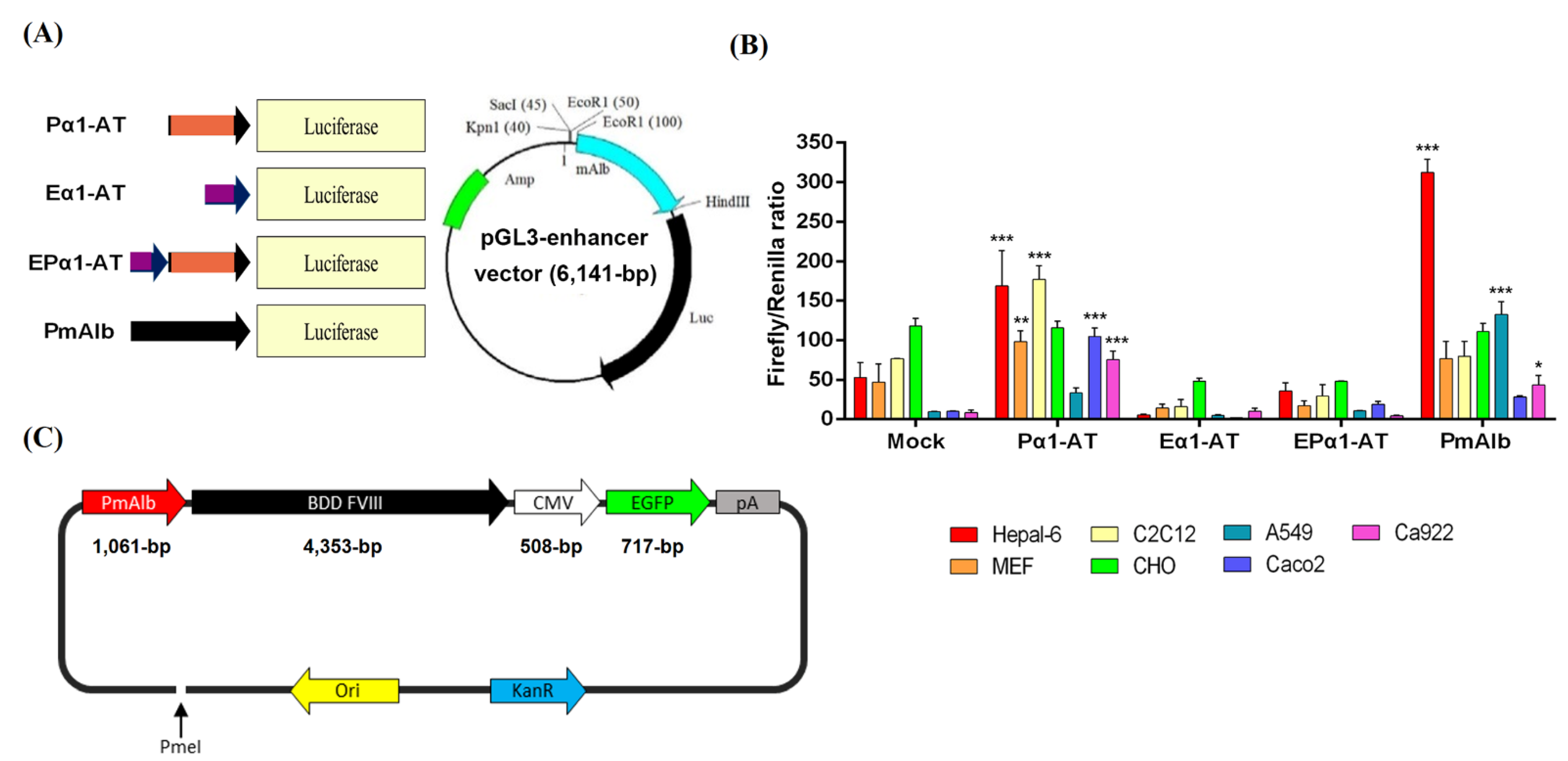
3.1. Liver-Specific Promoter Activity in Different Types of Cell Lines
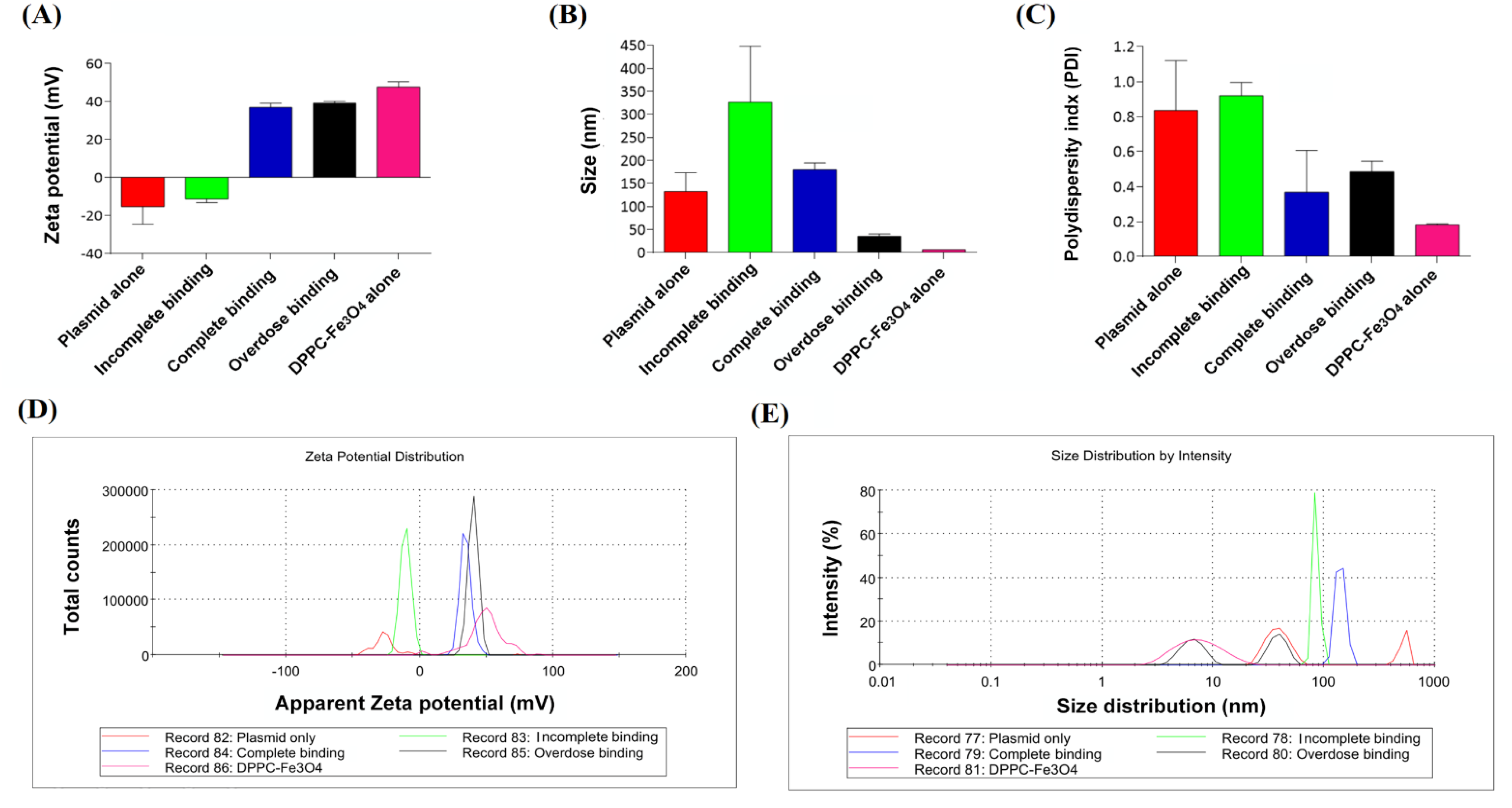
3.2. Characterization of DPPC-Fe3O4 Carrying Different Forms of Plasmid DNA
3.3. Characterization of Different Doses of DPPC-Fe3O4 with Circular Plasmid DNA
3.4. Evaluation of PmAlb-BDD-FVIII Circular Plasmid Expression in Mouse Hepatocytes
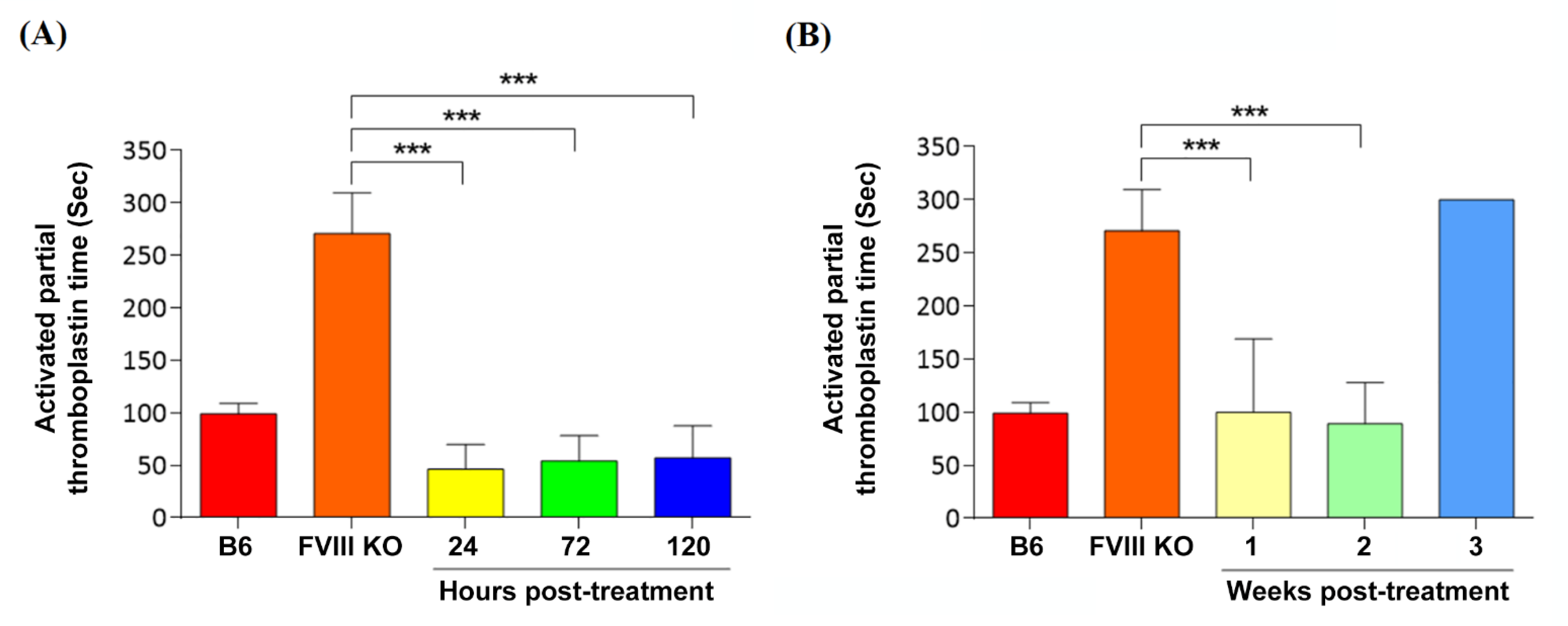
3.5. Coagulation Phenotypic Correction by Nanoparticle Gene Delivery
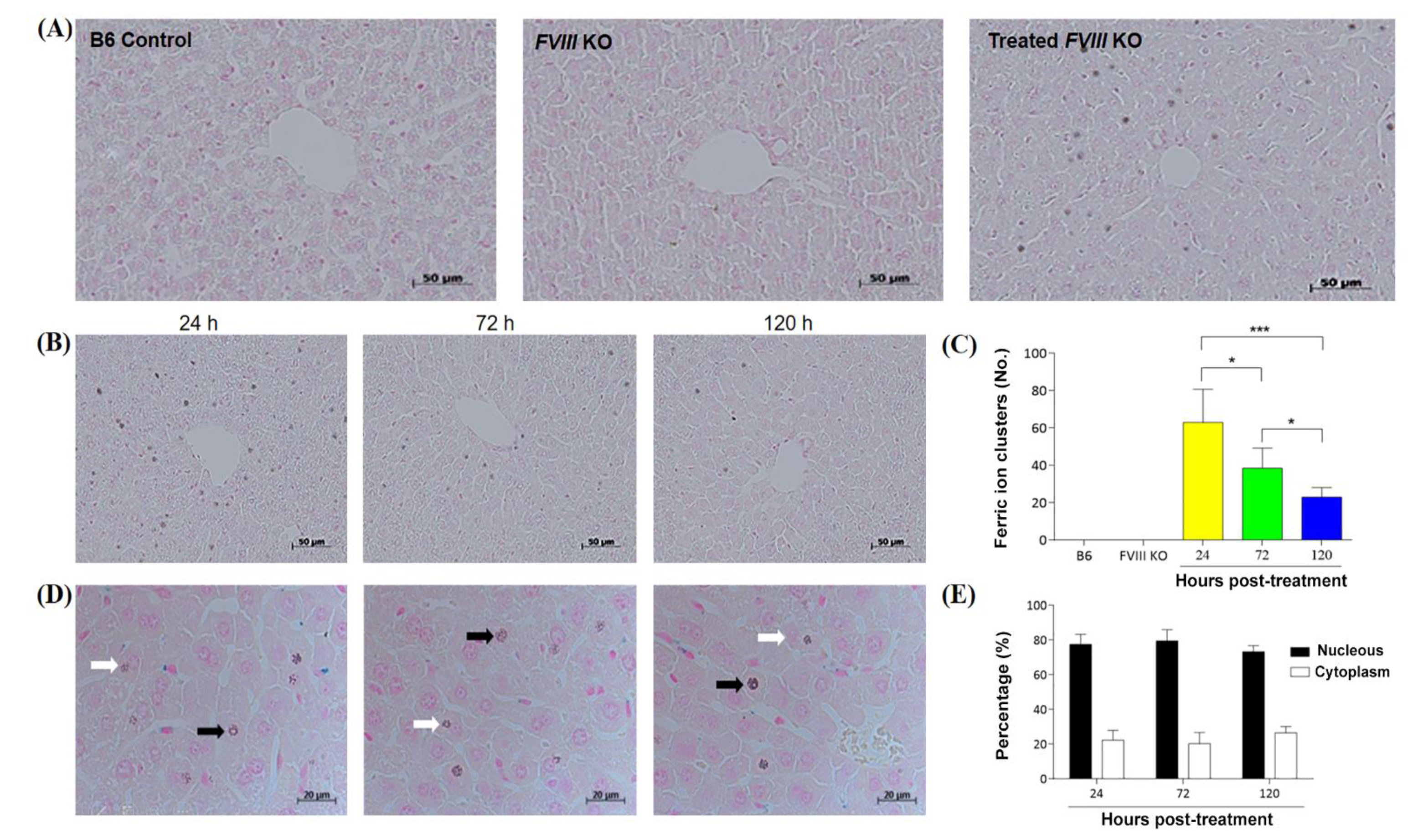
3.6. Distribution of DPPC-Fe3O4–Plasmid Nanoparticles in the Liver Tissue of Hemophilic Mice
3.7. Safety Validation after Delivery of the DPPC-Fe3O4-Plasmid Complex
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oldenburg, J.; Ananyeva, N.M.; Saenko, E.L. Molecular basis of hemophilia A. Haemophilia 2004, 10, 133–139. [Google Scholar] [CrossRef]
- Goodeve, A.C.; Peake, I.R. The molecular basis of hemophilia A: Genotype-phenotype relationships and inhibitor development. Semin. Thromb. Hemost. 2003, 29, 23–30. [Google Scholar] [CrossRef]
- Ketterling, R.P.; Bottema, C.D.K.; Phillips, J.A.; Sommer, S.S. Evidence that descendants of three founders constitute about 25% of hemophilia B in the United States. Genomics 1991, 10, 1093–1096. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.; Pasi, K.J. Haemophilias A and B. Lancet 2003, 361, 1801–1809. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of hemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Tantawy, A.A.; Matter, R.M.; Hamed, A.A.; Shams El Din El Telbany, M.A. Platelet microparticles in immune thrombocytopenic purpura in pediatrics. Pediatr. Hematol. Oncol. 2010, 27, 283–296. [Google Scholar] [CrossRef]
- Gouw, S.C.; van der Bom, J.G.; Ljung, R.; Escuriola, C.; Cid, A.R.; Claeyssens-Donadel, S.; van Geet, C.; Kenet, G.; Mäkipernaa, A.; Molinari, A.C.; et al. Factor VIII products and inhibitor development in severe hemophilia A. N. Engl. J. Med. 2013, 368, 231–239. [Google Scholar] [CrossRef]
- Preissner, K.T. Physiology of blood coagulation and fibrinolysis. Hamostaseologie 2008, 28, 259–271. [Google Scholar] [CrossRef]
- Gringeri, A.; Mantovani, L.G.; Scalone, L.; Mannucci, P.M.; COCIS Study Group. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: The COCIS Study Group. Blood 2003, 102, 2358–2363. [Google Scholar] [CrossRef] [Green Version]
- Mannucci, P.M. Hemophilia: Treatment options in the twenty-first century. J. Thromb. Haemost. 2003, 1, 1349–1355. [Google Scholar] [CrossRef]
- Mahlangu, J.; Oldenburg, J.; Paz-Priel, I.; Negrier, C.; Niggli, M.; Mancuso, M.E.; Schmitt, C.; Jiménez-Yuste, V.; Kempton, C.; Dhalluin, C.; et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N. Engl. J. Med. 2018, 379, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Donfield, S.M.; DiMichele, D.M.; Gringeri, A.; Gilbert, S.A.; Waters, J.; Berntorp, E.; FENOC Study Group. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: The FEIBA NovoSeven Comparative (FENOC) Study. Blood 2007, 109, 546–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Weyand, A.C.; Shavit, J.A. Novel treatments for hemophilia through rebalancing of the coagulation cascade. Pediatric Blood Cancer 2021, 68, e28934. [Google Scholar] [CrossRef] [PubMed]
- Mikaelsson, M.; Oswaldsson, U.; Jankowski, M.A. Measurement of factor VIII activity of B-domain deleted recombinant factor VIII. Semin. Hematol. 2001, 38, 13–23. [Google Scholar] [CrossRef]
- Chen, C.M.; Wang, C.H.; Wu, S.C.; Lin, C.C.; Lin, S.H.; Cheng, W.T.K. Temporal and spatial expression of biologically active human factor VIII in the milk of transgenic mice driven by mammary-specific bovine α-lactalbumin regulation sequences. Transgenic Res. 2002, 11, 257–268. [Google Scholar] [CrossRef]
- Lusher, J.M.; Lee, C.A.; Kessler, C.M.; Bedrosian, C.L. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe hemophilia A. Haemophilia 2003, 9, 38–49. [Google Scholar] [CrossRef]
- Ren, X.; Gong, X.; Cai, Q.; Guo, X.; Xu, M.; Ren, Z.; Zeng, Y. Efficient stabilization of recombinant human coagulation factor VIII in the milk of transgenic mice using hFVIII and vWF co-expression vector transduction. Biotechnol. Lett. 2015, 37, 1187–1194. [Google Scholar] [CrossRef]
- Meeks, S.L.; Josephson, C.D. Should hemophilia treaters switch to albumin-free recombinant factor VIII concentrates. Curr. Opin. Hematol. 2006, 13, 457–461. [Google Scholar] [CrossRef]
- Parti, R.; Schoppmann, A.; Lee, H.; Yang, L. Stability of lyophilized and reconstituted plasma/albumin-free recombinant human factor VIII (ADVATE RAHF-PFM). Haemophilia 2005, 11, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Bardi, E.; Astermark, J. Genetic risk factors for inhibitors in hemophilia A. Eur. J. Haematol. 2015, 94, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Astermark, J. FVIII inhibitors: Pathogenesis and avoidance. Blood 2015, 125, 2045–2051. [Google Scholar] [CrossRef] [Green Version]
- VandenDriessche, T.; Thorrez, L.; Naldini, L.; Follenzi, A.; Moons, L.; Berneman, Z.; Collen, D.; Chuah, M.K.L. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood 2002, 100, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Kattenhorn, L.M.; Tipper, C.H.; Stoica, L.; Geraghty, D.S.; Wright, T.L.; Clark, K.R.; Wadsworth, S.C. Adeno-associated virus gene therapy for liver disease. Hum. Gene Ther. 2016, 27, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.; Handyside, B.; Sihn, C.R.; Liu, S.; Zhang, L.; Xie, L.; Murphy, R.; Galicia, N.; Yates, B.; Minto, W.C.; et al. Induction of ER stress response by an AAV5 BDD FVIII construct is dependent on the strength of the hepatic-specific promoter. Mol. Ther. Methods Clin. Dev. 2020, 18, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Cristofolini, L.; Berzina, T.; Erokhina, S.; Konovalov, O.; Erokhin, V. Structural study of the DNA dipalmitoylphosphatidyl-choline complex at the air−water interface. Biomacromolecules 2007, 8, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta 1997, 1329, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Jenkins, G.J.S.; Asadi, R.; Doak, S.H. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010, 1, 5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, D.; Hyeon, T. Chemical design of biocompatible iron oxide nanoparticles for medical applications. Small 2013, 9, 1450–1466. [Google Scholar] [CrossRef] [PubMed]
- Stefaniu, C.; Brezesinski, G.; Möhwald, H. Polymer-capped magnetite nanoparticles change the 2D structure of DPPC model membranes. Soft Matter 2012, 8, 7952–7959. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Chen, B.; Ding, J.; Xia, G.; Gao, C.; Cheng, J.; Jin, N.; Zhou, Y.; Li, X.; et al. Pharmacokinetic parameters and tissue distribution of magnetic Fe3O4 nanoparticles in mice. Int. J. Nanomed. 2010, 5, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Ou-Yang, H.; Wu, S.C.; Sung, L.Y.; Yang, S.H.; Yang, S.H.; Chong, K.Y.; Chen, C.M. STAT3 is an upstream regulator of granzyme G in the maternal-to-zygotic transition of mouse embryos. Int. J. Mol. Sci. 2021, 22, 460. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.C.; Chang, W.H.; Tung, M.C.; Chen, H.L.; Liu, H.C.; Liao, C.H.; Lan, Y.W.; Chong, K.Y.; Yang, S.H.; Chen, C.M. Lactoferrin protects hyperoxia-induced lung and kidney systemic inflammation in an in vivo imaging model of NF-κB/luciferase transgenic mice. Mol. Imaging Biol. 2020, 22, 526–538. [Google Scholar] [CrossRef]
- Tsai, S.W.; Wu, H.S.; Chen, I.A.; Chen, H.L.; Chang, G.R.; Fan, H.C.; Chen, C.M. Recombinant porcine myostatin propeptide generated by the Pichia pastoris elevates myoblast growth and ameliorates high-fat diet-induced glucose intolerance. Res. Vet. Sci. 2019, 124, 200–211. [Google Scholar] [CrossRef]
- Lehnera, R.; Wanga, X.; Hunziker, P. Plasmid linearization changes shape and efficiency of transfection complexes. Eur. J. Nanomed. 2013, 5, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.Y.M.; Uludağ, H. Effects of size and topology of DNA molecules on intracellular delivery with non-viral gene carriers. BMC Biotechnol. 2008, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.W.; Yang, J.C.; Yen, C.C.; Huang, T.T.; Chen, Y.C.; Chen, H.L.; Chong, K.Y.; Chen, C.M. Predifferentiated amniotic fluid mesenchymal stem cells enhance lung alveolar epithelium regeneration and reverse elastase-induced pulmonary emphysema. Stem Cell Res. Ther. 2019, 10, 163. [Google Scholar] [CrossRef] [Green Version]
- Tsou, Y.A.; Chang, W.C.; Lin, C.D.; Chang, R.L.; Tsai, M.H.; Shih, L.C.; Staniczek, T.; Wu, T.F.; Hsu, H.Y.; Chang, W.D.; et al. Metformin increases survival in hypopharyngeal cancer patients with diabetes mellitus: Retrospective cohort study and cell-based analysis. Pharmaceuticals 2021, 14, 191. [Google Scholar] [CrossRef]
- Torres-Torronteras, J.; Gomez, A.; Eixarch, H.; Palenzuela, L.; Pizzorno, G.; Hirano, M.; Andreu, A.L.; Barquinero, J.; Martí, R. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 2011, 18, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Pérez, R.; Vila-Julià, F.; Hirano, M.; Mingozzi, F.; Torres-Torronteras, J.; Martí, R. Alpha-1-antitrypsin promoter improves the efficacy of an adeno-associated virus vector for the treatment of mitochondrial neurogastrointestinal encephalomyopathy. Hum. Gene Ther. 2019, 30, 985–998. [Google Scholar] [CrossRef]
- Xu, Z.; Hao, C.; Xie, B.; Sun, R. Effect of Fe3O4 nanoparticles on mixed POPC/DPPC monolayers at air-water interface. Scanning 2019, 2019, 5712937. [Google Scholar] [CrossRef]
- Chen, C.Y.; Tran, D.M.; Cavedon, A.; Cai, X.; Rajendran, R.; Lyle, M.J.; Martini, P.G.V.; Miao, C.H. Treatment of hemophilia A using Factor VIII messenger RNA lipid nanoparticles. Mol. Ther. Nucleic Acids 2020, 20, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Loring, H.S.; ElMallah, M.K.; Flotte, T.R. Development of RAAV2-CFTR: History of the first rAAV vector product to be used in humans. Hum. Gene Ther. Methods 2016, 27, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, B.; Cornell Sookdeo, C.; Berthelette, P.; Jackson, R.; Piraino, S.; Burnham, B.; Nass, S.; Souza, D.; O’Riordan, C.R.; Vincent, K.A.; et al. Characteristics of minimally oversized adeno-associated virus vectors encoding human factor VIII generated using producer cell lines and triple transfection. Hum. Gene Ther. Methods 2017, 28, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, L.L.; Galstyan, A.; Holler, E.; Ljubimova, J.Y. Magnetic iron oxide nanoparticles for imaging, Targeting and treatment of primary and metastatic tumors of the brain. J. Control. Release 2020, 320, 45–62. [Google Scholar] [CrossRef]
- Cao, W.; Dong, B.; Horling, F.; Firrman, J.A.; Lengler, J.; Klugmann, M.; de la Rosa, M.; Wu, W.; Wang, Q.; Wei, H. Minimal essential human factor VIII alterations enhance secretion and gene therapy efficiency. Mol. Ther. Methods Clin. Dev. 2020, 19, 486–495. [Google Scholar] [CrossRef]
- Wuerth, M.E.; Cragerud, R.K.; Spiegel, P.C. Structure of the human factor VIII C2 domain in complex with the 3E6 inhibitory antibody. Sci. Rep. 2015, 5, 17216. [Google Scholar] [CrossRef] [Green Version]
- Markovitz, R.C.; Healey, J.F.; Parker, E.T.; Meeks, S.L.; Lollar, P. The diversity of the immune response to the A2 domain of human factor VIII. Blood 2013, 121, 2785–2795. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Chen, S.; Noguchi, H.; Matsumoto, S.; Grayburn, P.A. In vivo non-viral gene delivery of human vascular endothelial growth factor improves revascularisation and restoration of euglycaemia after human islet transplantation into mouse liver. Diabetologia 2010, 53, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boletta, A.; Benigni, A.; Lutz, J.; Remuzzi, G.; Soria, M.R.; Monaco, L. Nonviral gene delivery to the rat kidney with polyethylenimine. Hum. Gene Ther. 1997, 8, 1243–1251. [Google Scholar] [CrossRef]
- Liu, F.; Huang, L. Noninvasive gene delivery to the liver by mechanical massage. Hepatology 2002, 35, 1314–1319. [Google Scholar] [CrossRef]
- Inoh, Y.; Nagai, M.; Matsushita, K.; Nakanishi, M.; Furuno, T. Gene transfection efficiency into dendritic cells is influenced by the size of cationic liposomes/DNA complexes. Eur. J. Pharm. Sci. 2017, 102, 230–236. [Google Scholar] [CrossRef]
- Betzer, O.; Shilo, M.; Opochinsky, R.; Barnoy, E.; Motiei, M.; Okun, E.; Yadid, G.; Popovtzer, R. The effect of nanoparticle size on the ability to cross the blood–brain barrier: An in vivo study. Nanomedicine 2017, 12, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Ren, Y.; Vandermark, J.; Archang, M.M.; Williford, J.-M.; Liu, H.-W.; Lee, J.; Wang, T.-H.; Mao, H.-Q. Continuous production of discrete plasmid DNA-polycation nanoparticles using flash nanocomplexation. Small 2016, 12, 6214–6222. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xiang, P.; Li, Q. Investigations of the effect of DNA size in transient transfection assay using dual luciferase system. Anal. Biochem. 2005, 346, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Troyanovsky, B.; Bitko, V.; Pastukh, V.; Fouty, B.; Solodushko, V. The functionality of minimal piggyBac transposons in mammalian cells. Mol. Ther. Nucleic Acids 2016, 5, e369. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′ to 3′) | Tm (°C) | Amplicon Size (bp) |
|---|---|---|---|
| Pα1-AT | F: 5′-ACCGAGGCACAGAGAGGTT-3′ R: 5′-GCCATTCACAAGGATACTGT-3′ | 67 | 581 |
| Eα1-AT | F: 5′-CTTCAGCATCAGGCATTTTGG-3′ R: 5′-TCTCCAGAACCTCTCGCAGT-3′ | 63 | 473 |
| PmAlb | F: 5′-TCACTCAAAAGAGTCCTGAA-3′ R: 5′-AGAAAGACTCGCTCTAATATAC-3′ | 57 | 1060 |
| EGFP | F: 5′-GACTTCTTCAAGTCCGCCATGC-3′ R: 5′-CTCCAGCAGGACCATGTGAT-3′ | 55 | 432 |
| BDD-hFVIII | F: 5′-CAGACTTTCGGAACAGAGGCA-3′ R: 5′-ATCTTTTTCCAGGTCAACATCA-3′ | 55 | 752 |
| β-actin | F: 5′-CCGTCTTCCCCTCCATCGTGGG-3′ R: 5′-AGATCATTGTAGAAGGTGTGG-3′ | 68 | 199 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, Y.-T.; Chen, Y.-T.; Fan, H.-C.; Tsai, T.-C.; Cheng, S.-N.; Lai, P.-S.; Chen, J.-K.; Chen, C.-M. Novel Coagulation Factor VIII Gene Therapy in a Mouse Model of Hemophilia A by Lipid-Coated Fe3O4 Nanoparticles. Biomedicines 2021, 9, 1116. https://doi.org/10.3390/biomedicines9091116
Kao Y-T, Chen Y-T, Fan H-C, Tsai T-C, Cheng S-N, Lai P-S, Chen J-K, Chen C-M. Novel Coagulation Factor VIII Gene Therapy in a Mouse Model of Hemophilia A by Lipid-Coated Fe3O4 Nanoparticles. Biomedicines. 2021; 9(9):1116. https://doi.org/10.3390/biomedicines9091116
Chicago/Turabian StyleKao, Yung-Tsung, Yen-Ting Chen, Hueng-Chuen Fan, Tung-Chou Tsai, Shin-Nan Cheng, Ping-Shan Lai, Jen-Kun Chen, and Chuan-Mu Chen. 2021. "Novel Coagulation Factor VIII Gene Therapy in a Mouse Model of Hemophilia A by Lipid-Coated Fe3O4 Nanoparticles" Biomedicines 9, no. 9: 1116. https://doi.org/10.3390/biomedicines9091116
APA StyleKao, Y.-T., Chen, Y.-T., Fan, H.-C., Tsai, T.-C., Cheng, S.-N., Lai, P.-S., Chen, J.-K., & Chen, C.-M. (2021). Novel Coagulation Factor VIII Gene Therapy in a Mouse Model of Hemophilia A by Lipid-Coated Fe3O4 Nanoparticles. Biomedicines, 9(9), 1116. https://doi.org/10.3390/biomedicines9091116