
Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis

 ,
,
Abstract
:1. Introduction
2. Developmental Aspects of Brain Metabolism and Effects of ELS
3. Neurogenesis and Cerebral Angiogenesis: Search for a Common Regulator of Metabolic Reprogramming
4. Conclusions and Further Prospects: Strategies Aimed to Restore Metabolic Plasticity of Brain Cells for Efficient Neurogenesis and Angiogenesis
Author Contributions
Funding
Conflicts of Interest
References
- Fogelman, N.; Canli, T. Early life stress, physiology, and genetics: A review. Front. Psychol. 2019, 10, 1668. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Panina, Y.A.; Malinovskaya, N.A.; Salmina, A.B. Early life stress and brain plasticity: From molecular alterations to aberrant memory and behavior. Rev. Neurosci. 2021, 32, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Malinovskaya, N.A.; Morgun, A.; Lopatina, O.L.; Panina, Y.; Volkova, V.V.; Gasymly, E.L.; Taranushenko, T.E.; Salmina, A.B. Early life stress: Consequences for the development of the brain. Neurosci. Behav. Physiol. 2018, 48, 233–250. [Google Scholar] [CrossRef]
- Ehuang, L.-T. Early-life stress impacts the developing hippocampus and primes seizure occurrence: Cellular, molecular, and epigenetic mechanisms. Front. Mol. Neurosci. 2014, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, H.; Merisaari, H.; Karlsson, L.; Scheinin, N.M.; Parkkola, R.; Saunavaara, J.; Lähdesmäki, T.; Lehtola, S.J.; Keskinen, M.; Pelto, J.; et al. Association of cumulative paternal early life stress with white matter maturation in newborns. JAMA Netw. Open 2020, 3, e2024832. [Google Scholar] [CrossRef]
- Grundwald, N.J.; Brunton, P.J. Prenatal stress programs neuroendocrine stress responses and affective behaviors in second generation rats in a sex-dependent manner. Psychoneuroendocrinology 2015, 62, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Spyrka, J.; Gugula, A.; Rak, A.; Tylko, G.; Hess, G.; Blasiak, A. Early life stress-induced alterations in the activity and morphology of ventral tegmental area neurons in female rats. Neurobiol. Stress 2020, 13, 100250. [Google Scholar] [CrossRef]
- Kronman, H.; Torres-Berrío, A.; Sidoli, S.; Issler, O.; Godino, A.; Ramakrishnan, A.; Mews, P.; Lardner, C.K.; Parise, E.M.; Walker, D.M.; et al. Long-term behavioral and cell-type-specific molecular effects of early life stress are mediated by H3K79me2 dynamics in medium spiny neurons. Nat. Neurosci. 2021, 24, 667–676. [Google Scholar] [CrossRef]
- Kaufman, D.; Banerji, M.A.; Shorman, I.; Smith, E.L.; Coplan, J.D.; Rosenblum, L.A.; Kral, J.G. Early-life stress and the development of obesity and insulin resistance in juvenile bonnet macaques. Diabetes 2007, 56, 1382–1386. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.; Junco, M.; Gomez, C.; Lajud, N. Early life stress increases metabolic risk, HPA axis reactivity, and depressive-like behavior when combined with postweaning social isolation in rats. PLoS ONE 2016, 11, e0162665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, Y.-M.; Tsai, T.-C.; Lin, Y.-T.; Chen, C.-C.; Huang, C.-C.; Hsu, K.-S. Early life stress dampens stress responsiveness in adolescence: Evaluation of neuroendocrine reactivity and coping behavior. Psychoneuroendocrinology 2016, 67, 86–99. [Google Scholar] [CrossRef]
- Savignac, H.M.; Dinan, T.G.; Cryan, J.F. Resistance to early-life stress in mice: Effects of genetic background and stress duration. Front. Behav. Neurosci. 2011, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E. Neurogenesis and brain aging. Rev. Neurosci. 2019, 30, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, T.J.; Gould, E. Stress, stress hormones, and adult neurogenesis. Exp. Neurol. 2012, 233, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Lucassen, P.J.; Oomen, C.A.; Naninck, E.F.; Fitzsimons, C.P.; Van Dam, A.-M.; Czeh, B.; Korosi, A. Regulation of adult neurogenesis and plasticity by (early) stress, glucocorticoids, and inflammation. Cold Spring Harb. Perspect. Biol. 2015, 7, a021303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbink, M.R.; Naninck, E.F.G.; Lucassen, P.J.; Korosi, A. Early-life stress diminishes the increase in neurogenesis after exercise in adult female mice. Hippocampus 2017, 27, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Naninck, E.; Hoeijmakers, L.; Kakava-Georgiadou, N.; Meesters, A.; Lazic, S.E.; Lucassen, P.J.; Korosi, A. Chronic early life stress alters developmental and adult neurogenesis and impairs cognitive function in mice. Hippocampus 2014, 25, 309–328. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, P.; Abbink, M.R.; Davidson, A.R.; Reijner, N.; Abiega, O.; Lucassen, P.J.; Korosi, A.; Fitzsimons, C.P. Early life stress decreases cell proliferation and the number of putative adult neural stem cells in the adult hypothalamus. Stress 2021, 24, 189–195. [Google Scholar] [CrossRef]
- Daun, K.A.; Fuchigami, T.; Koyama, N.; Maruta, N.; Ikenaka, K.; Hitoshi, S. Early Maternal and social deprivation expands neural stem cell population size and reduces hippocampus/amygdala-dependent fear memory. Front. Neurosci. 2020, 14, 22. [Google Scholar] [CrossRef]
- Haukvik, U.K.; McNeil, T.; Lange, E.H.; Melle, I.; Dale, A.M.; Andreassen, O.A.; Agartz, I. Pre- and perinatal hypoxia associated with hippocampus/amygdala volume in bipolar disorder. Psychol. Med. 2013, 44, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, E.L.; Kühn, F.; Schmitt, A.; Gattaz, W.F.; Gruber, O.; Schneider-Axmann, T.; Falkai, P.; Schmitt, A. Increased cell proliferation in the rat anterior cingulate cortex following neonatal hypoxia: Relevance to schizophrenia. J. Neural Transm. 2013, 120, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paparelli, A.; Iwata, K.; Wakuda, T.; Iyegbe, C.; Murray, R.; Takei, N. Perinatal asphyxia in rat alters expression of novel schizophrenia risk genes. Front. Mol. Neurosci. 2017, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Howell, K.; Pillai, A. Effects of prenatal hypoxia on schizophrenia-related phenotypes in heterozygous reeler mice: A gene × environment interaction study. Eur. Neuropsychopharmacol. 2014, 24, 1324–1336. [Google Scholar] [CrossRef] [Green Version]
- Katsel, P.; Roussos, P.; Pletnikov, M.; Haroutunian, V. Microvascular anomaly conditions in psychiatric disease. Schizophrenia—angiogenesis connection. Neurosci. Biobehav. Rev. 2017, 77, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.; Soares, R.; Coelho, R.; Figueiredo-Braga, M. Angiogenesis in the pathophysiology of schizophrenia—A comprehensive review and a conceptual hypothesis. Life Sci. 2015, 128, 79–93. [Google Scholar] [CrossRef]
- Wilson, R.S.; Arnold, S.E.; Schneider, J.A.; Kelly, J.F.; Tang, Y.; Bennett, D.A. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology 2006, 27, 143–153. [Google Scholar] [CrossRef]
- Hoeijmakers, L.; Amelianchik, A.; Verhaag, F.; Kotah, J.; Lucassen, P.J.; Korosi, A. Early-life stress does not aggravate spatial memory or the process of hippocampal neurogenesis in adult and middle-aged APP/PS1 mice. Front. Aging Neurosci. 2018, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Hirai, S.; Hosokawa, M.; Saito, T.; Sakuma, H.; Saido, T.; Hasegawa, M.; Okado, H. Early-life stress induces the development of Alzheimer’s disease pathology via angiopathy. Exp. Neurol. 2021, 337, 113552. [Google Scholar] [CrossRef]
- Theodoridou, D.; Christodoulides, P.; Zakopoulou, V.; Syrrou, M. Developmental dyslexia: Environment matters. Brain Sci. 2021, 11, 782. [Google Scholar] [CrossRef]
- Guidi, L.G.; Velayos-Baeza, A.; Martinez-Garay, I.; Monaco, A.P.; Paracchini, S.; Bishop, D.V.M.; Molnár, Z. The neuronal migration hypothesis of dyslexia: A critical evaluation 30 years on. Eur. J. Neurosci. 2018, 48, 3212–3233. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Haigh, K.; Haigh, J.J.; Vasudevan, A. Endothelial VEGF sculpts cortical cytoarchitecture. J. Neurosci. 2013, 33, 14809–14815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oomen, C.A.; Girardi, C.; Cahyadi, R.; Verbeek, E.C.; Krugers, H.; Joels, M.; Lucassen, P.J. Opposite effects of early maternal deprivation on neurogenesis in male versus female rats. PLoS ONE 2009, 4, e3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, M.; Ruhrberg, C. Cross-talk between blood vessels and neural progenitors in the developing brain. Neuronal Signal. 2018, 2, NS20180139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozhilenkova, E.; Lopatina, O.L.; Komleva, Y.K.; Salmin, V.V.; Salmina, A.B. Blood-brain barrier-supported neurogenesis in healthy and diseased brain. Rev. Neurosci. 2017, 28, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, L.; Palazzo, O.; La Rosa, C.; Piumatti, M. Do large brains of long-living mammals prefer non-newly generated, immature neurons? Neural Regen. Res. 2018, 13, 633–634. [Google Scholar] [CrossRef]
- Subburaju, S.; Kaye, S.; Choi, Y.K.; Baruah, J.; Datta, D.; Ren, J.; Kumar, A.S.; Szabo, G.; Fukumura, D.; Jain, R.K.; et al. NAD+-mediated rescue of prenatal forebrain angiogenesis restores postnatal behavior. Sci. Adv. 2020, 6, eabb9766. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.C.; Yamaguchi, N.; Nakata, M.; Eogawa, S. Modification of female and male social behaviors in estrogen receptor beta knockout mice by neonatal maternal separation. Front. Neurosci. 2014, 8, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunton, P.J. Programming the brain and behaviour by early-life stress: A focus on neuroactive steroids. J. Neuroendocrinol. 2015, 27, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Malinovskaya, N.A.; Komleva, Y.K.; Salmin, V.V.; Morgun, A.; Shuvaev, A.N.; Panina, Y.; Boitsova, E.B.; Salmina, A.B. Endothelial progenitor cells physiology and metabolic plasticity in brain angiogenesis and blood-brain barrier modeling. Front. Physiol. 2016, 7, 599. [Google Scholar] [CrossRef] [Green Version]
- Salmina, A.B.; Morgun, A.; Kuvacheva, N.V.; Lopatina, O.L.; Komleva, Y.K.; Malinovskaya, N.A.; Pozhilenkova, E. Establishment of neurogenic microenvironment in the neurovascular unit: The connexin 43 story. Rev. Neurosci. 2014, 25, 97–111. [Google Scholar] [CrossRef]
- Gapp, K.; Corcoba, A.; Van Steenwyk, G.; Mansuy, I.M.; Duarte, J.M. Brain metabolic alterations in mice subjected to postnatal traumatic stress and in their offspring. Br. J. Pharmacol. 2016, 37, 2423–2432. [Google Scholar] [CrossRef]
- Hoeijmakers, L.; Lesuis, S.L.; Krugers, H.; Lucassen, P.J.; Korosi, A. A preclinical perspective on the enhanced vulnerability to Alzheimer’s disease after early-life stress. Neurobiol. Stress 2018, 8, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Blazey, T.; Snyder, A.Z.; Goyal, M.S.; Vlassenko, A.G.; Raichle, M.E. A systematic meta-analysis of oxygen-to-glucose and oxygen-to-carbohydrate ratios in the resting human brain. PLoS ONE 2018, 13, e0204242. [Google Scholar] [CrossRef]
- Magistretti, P.J. Imaging brain aerobic glycolysis as a marker of synaptic plasticity. Proc. Natl. Acad. Sci. USA 2016, 113, 7015–7016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018, 217, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Dienel, G.; Jacob, Z.; Lee, H.; Makaryus, R.; Gjedde, A.; Hyder, F.; Rothman, D.L. Trajectories of brain lactate and Re-visited oxygen-glucose index calculations do not support elevated non-oxidative metabolism of glucose across childhood. Front. Neurosci. 2018, 12, 631. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.; Morris, J.C.; Raichle, M.E. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef]
- Takahashi, S. Lactate and ketone bodies act as energy substrates as well as signal molecules in the brain. In Psychology and Patho-Physiological Outcomes of Eating [Working Title]; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Riske, L.; Thomas, R.K.; Baker, G.B.; Dursun, S.M. Lactate in the brain: An update on its relevance to brain energy, neurons, glia and panic disorder. Ther. Adv. Psychopharmacol. 2017, 7, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Lust, W.D.; Pundik, S.; Zechel, J.; Zhou, Y.; Buczek, M.; Selman, W.R. Changing metabolic and energy profiles in fetal, neonatal, and adult rat brain. Metab. Brain Dis. 2003, 18, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; D’Angelo, M. Neuronal cells rearrangement during aging and neurodegenerative disease: Metabolism, oxidative stress and organelles dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.; Blithikioti, C.; Ku, M.; et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.A.; Adamson, D.C. Neuronal-astrocyte metabolic interactions: Understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Omuro, H.; Liu, Y.-F.; Soya, M.; Shima, T.; McEwen, B.S.; Soya, H. Astrocytic glycogen-derived lactate fuels the brain during exhaustive exercise to maintain endurance capacity. Proc. Natl. Acad. Sci. USA 2017, 114, 6358–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Rinholm, J. Mitochondria in myelinating oligodendrocytes: Slow and out of breath? Metabolites 2021, 11, 359. [Google Scholar] [CrossRef]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte bioenergetics in health and disease. Neuroscientist 2018, 25, 334–343. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Childers, G.; Giridharan, S.; Hernandes, I.L. An association between mitochondria and microglia effector function: What do we think we know? Neuroimmunol. Neuroinflamm. 2020, 2020, 150–165. [Google Scholar] [CrossRef]
- Lauro, C.; Limatola, C. Metabolic Reprograming of microglia in the regulation of the innate inflammatory response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Caja, S.; Enríquez, J.A. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Busija, D.W.; Katakam, P.V. Mitochondrial mechanisms in cerebral vascular control: Shared signaling pathways with preconditioning. J. Vasc. Res. 2014, 51, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmina, A.B.; Kuvacheva, N.V.; Morgun, A.; Komleva, Y.K.; Pozhilenkova, E.A.; Lopatina, O.L.; Gorina, Y.V.; Taranushenko, T.E.; Petrova, L.L. Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. 2015, 64, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Moreira, P.I. Oxidative stress: A major player in cerebrovascular alterations associated to neurodegenerative events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef] [Green Version]
- Susan, W.S.L.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2021, 1–9. [Google Scholar] [CrossRef]
- Nwadozi, E.; Rudnicki, M.; Haas, T.L. Metabolic coordination of pericyte phenotypes: Therapeutic implications. Front. Cell Dev. Biol. 2020, 8, 77. [Google Scholar] [CrossRef]
- Folmes, C.; Dzeja, P.P.; Nelson, T.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Laaper, M. Regulation of neural stem cell fate decisions by mitochondrial dynamics. Neural Regen. Res. 2018, 13, 1548–1549. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Rivera, D.; Sarmiento-Soto, H.; López-Barneo, J.; Muñoz-Cabello, A.M. Mitochondrial complex i function is essential for neural stem/progenitor cells proliferation and differentiation. Front. Neurosci. 2019, 13, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, E.A.; Cheung, W.; Mikheev, A.M.; Sweet, I.R.; Bielas, J.H.; Zhang, J.; Rostomily, R.C.; Horner, P.J. Aging neural progenitor cells have decreased mitochondrial content and lower oxidative metabolism. J. Biol. Chem. 2011, 286, 38592–38601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; Bosch, A.V.D.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Salmina, A.B.; Yauzina, N.A.; Kuvacheva, N.V.; Petrova, M.M.; Taranushenko, T.Y.; Malinovskaya, N.A.; Lopatina, O.L.; Morgun, A.V.; Pozhilenkova, Y.A.; Okuneva, O.S.; et al. Insulin and insulin resistance: New molecule markers and target molecule for the diagnosis and therapy of diseases of the central nervous system. Bull. Sib. Med. 2013, 12, 104–118. [Google Scholar] [CrossRef]
- Gorina, Y.V.; Salmina, A.B.; Kuvacheva, N.V.; Komleva, Y.K.; Fedyukovich, L.V.; Uspenskaya, Y.A.; Morozova, G.A.; Demko, I.V.; Petrova, M.M. Neuroinflammation and insulin resistance in alzheimer’s disease. Sib. Med Rev. 2014, 11–19. [Google Scholar] [CrossRef]
- Komleva, Y.; Chernykh, A.; Lopatina, O.; Gorina, Y.; Lokteva, I.; Salmina, A.; Gollasch, M. Inflamm-aging and brain insulin resistance: New insights and role of life-style strategies on cognitive and social determinants in aging and neurodegeneration. Front. Neurosci. 2021, 14, 618395. [Google Scholar] [CrossRef] [PubMed]
- Gorina, Y.V.; Komleva, Y.K.; Lopatina, O.L.; Chernykh, A.I.; Salmina, A.B. The effect of insulin resistance on amygdale glucose metabolism alterations in experimental Alzheimer’s disease. Bull. Sib. Med. 2017, 16, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Diounou, H.; Olier, M.; Lencina, C.; Riba, A.; Barretto, S.; Nankap, M.; Sommer, C.; Guillou, H.; Ellero-Simatos, S.; Guzylack-Piriou, L.; et al. Early life stress induces type 2 diabetes-like features in ageing mice. Brain Behav. Immun. 2019, 80, 452–463. [Google Scholar] [CrossRef]
- Kasischke, K.A.; Vishwasrao, H.D.; Fisher, P.J.; Zipfel, W.R.; Webb, W.W. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 2004, 305, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P. Neuron-glia metabolic coupling: Role in plasticity and neuroprotection. J. Neurol. Sci. 2017, 381, 24. [Google Scholar] [CrossRef] [Green Version]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Descalzi, G. Cortical astrocyte-neuronal metabolic coupling emerges as a critical modulator of stress-induced hopelessness. Neurosci. Bull. 2021, 37, 132–134. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, Y.; Yu, Z.; Wang, W.; Cheng, X.; Ji, W.; Guo, S.; Zhou, Q.; Wu, N.; Chen, Y.; et al. Brain endothelial cells maintain lactate homeostasis and control adult hippocampal neurogenesis. Cell Stem Cell 2019, 25, 754–767.e9. [Google Scholar] [CrossRef] [PubMed]
- Khilazheva, E.D.; Kuvacheva, N.V.; Morgun, A.V.; Boitsova, E.B.; Malinovskaya, N.A.; Pozhilenkova, E.A.; Salmina, A.B. Modulation of lactate production, transport and reception by cells in the model of brain neurovasculr unit. Eksperimental’naia Klinicheskaia Farmakologiia 2016, 79, 7–12. [Google Scholar]
- Boitsova, E.B.; Morgun, A.V.; Osipova, E.D.; Pozhilenkova, E.A.; Martinova, G.P.; Frolova, O.V.; Olovannikova, R.Y.; Tohidpour, A.; Gorina, Y.V.; Panina, Y.; et al. The inhibitory effect of LPS on the expression of GPR81 lactate receptor in blood-brain barrier model in vitro. J. Neuroinflamm. 2018, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Khilazheva, E.D.; Pisareva, N.V.; Morgun, A.V.; Boitsova, E.B.; Taranushenko, T.E.; Frolova, O.V.; Salmina, A.B. Activation of GPR81 lactate receptors stimulates mitochondrial biogenesis in cerebral microvessel endothelial cells. Ann. Clin. Exp. Neurol. 2017, 11, 34–39. [Google Scholar] [CrossRef]
- Dumas, S.; García-Caballero, M.; Carmeliet, P. Metabolic signatures of distinct endothelial phenotypes. Trends Endocrinol. Metab. 2020, 31, 580–595. [Google Scholar] [CrossRef] [PubMed]
- Yetkin-Arik, B.; Vogels, I.M.C.; Neyazi, N.; Van Duinen, V.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells in vitro are less glycolytic and have a more flexible response to metabolic stress than non-tip cells. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ludikhuize, M.C.; Colman, M.J.R. Metabolic regulation of stem cells and differentiation: A forkhead box o transcription factor perspective. Antioxid. Redox Signal. 2020, 34, 1004–1024. [Google Scholar] [CrossRef]
- Knobloch, M.; Pilz, G.-A.; Ghesquière, B.; Kovacs, W.; Wegleiter, T.; Moore, D.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Jones, A.; Deeney, J.; Hur, S.K.; Bankaitis, V.A. Inborn errors of long-chain fatty acid β-oxidation link neural stem cell self-renewal to autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Malik, N.; Kim, Y.; He, Y.; Li, M.; Dubois, W.; Liu, H.; Peat, T.J.; Nguyen, J.T.; Tseng, Y.; et al. Fatty acid oxidation is required for embryonic stem cell survival during metabolic stress. EMBO Rep. 2021, 22, e52122. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Poplawski, J.; Radmilovic, A.; Montina, T.D.; Metz, G.A.S. Cardiorenal metabolic biomarkers link early life stress to risk of non-communicable diseases and adverse mental health outcomes. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Delpierre, C.; Fantin, R.; Barboza-Solis, C.; Lepage, B.; Darnaudéry, M.; Kelly-Irving, M. The early life nutritional environment and early life stress as potential pathways towards the metabolic syndrome in mid-life? A lifecourse analysis using the 1958 British Birth cohort. BMC Public Health 2016, 16, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detka, J.; Kurek, A.; Kucharczyk, M.; Głombik, K.; Basta-Kaim, A.; Kubera, M.; Lason, W.; Budziszewska, B. Brain glucose metabolism in an animal model of depression. Neuroscience 2015, 295, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Emmerzaal, T.L.; Preston, G.; Geenen, B.; Verweij, V.; Wiesmann, M.; Vasileiou, E.; Grüter, F.; De Groot, C.; Schoorl, J.; De Veer, R.; et al. Impaired mitochondrial complex I function as a candidate driver in the biological stress response and a concomitant stress-induced brain metabolic reprogramming in male mice. Transl. Psychiatry 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, L.A.; Schasfoort, E.M.; Steffens, A.B.; Korf, J. Effects of stress and exercise on rat hippocampus and striatum extracellular lactate. Am. J. Physiol. Integr. Comp. Physiol. 1990, 259, R773–R779. [Google Scholar] [CrossRef]
- Osborne, D.M.; Epearson-Leary, J.; McNay, E.C. The neuroenergetics of stress hormones in the hippocampus and implications for memory. Front. Neurosci. 2015, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, X.; Zhou, W.; Messina, J.L. Acute psychological stress results in the rapid development of insulin resistance. J. Endocrinol. 2013, 217, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Szyf, M. DNA methylation, behavior and early life adversity. J. Genet. Genom. 2013, 40, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.L.; Shutman, E.; Chinaka, C.; Deepika, K.; Pelaez, L.; Dabney, K.W. Aberrant epigenomic modulation of glucocorticoid receptor gene (nr3c1) in early life stress and major depressive disorder correlation: Systematic review and quantitative evidence synthesis. Int. J. Environ. Res. Public Health 2019, 16, 4280. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocr. 2018, 49, 72–85. [Google Scholar] [CrossRef]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath, K.; Nieves, G.M.; Goodwill, H. Early life stress accelerates behavioral and neural maturation of the hippocampus in male mice. Horm. Behav. 2016, 82, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Van der Kooij, M.; Grosse, J.; Zanoletti, O.; Papilloud, A.; Sandi, C. The effects of stress during early postnatal periods on behavior and hippocampal neuroplasticity markers in adult male mice. Neuroscience 2015, 311, 508–518. [Google Scholar] [CrossRef]
- Wei, L.; Hao, J.; Lacher, R.K.; Abbott, T.; Chung, L.; Colangelo, C.M.; Kaffman, A. Early-life stress perturbs key cellular programs in the developing mouse hippocampus. Dev. Neurosci. 2015, 37, 476–488. [Google Scholar] [CrossRef] [Green Version]
- Sousa, V.C.; Vital, J.; Costenla, A.R.; Batalha, V.; Sebastião, A.M.; Ribeiro, J.; Lopes, L.V. Maternal separation impairs long term-potentiation in CA1-CA3 synapses and hippocampal-dependent memory in old rats. Neurobiol. Aging 2014, 35, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Lemche, E. Early life stress and epigenetics in late-onset Alzheimer’s dementia: A systematic review. Curr. Genom. 2018, 19, 522–602. [Google Scholar] [CrossRef]
- Gorina, Y.V.; Komleva, Y.K.; Lopatina, O.L.; Volkova, V.V.; Gersog, G.E.; Popova, N.N.; Salmina, A.B. Features of molecule expression markers of insulin resistance in experimental Alzheimer’s disease. Probl. Endocrinol. 2015, 61, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Parr, L.A.; Boudreau, M.; Hecht, E.; Winslow, J.T.; Nemeroff, C.B.; Sánchez, M.M. Early life stress affects cerebral glucose metabolism in adult rhesus monkeys (Macaca mulatta). Dev. Cogn. Neurosci. 2012, 2, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Banqueri, M.; Gutiérrez-Menéndez, A.; Méndez, M.; Conejo, N.M.; Arias, J.L. Early life stress due to repeated maternal separation alters the working memory acquisition brain functional network. Stress 2021, 24, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Eagleson, K.L.; Villaneuva, M.; Southern, R.M.; Levitt, P. Proteomic and mitochondrial adaptations to early-life stress are distinct in juveniles and adults. Neurobiol. Stress 2020, 13, 100251. [Google Scholar] [CrossRef]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.; Sandi, C. Mitofusin-2 in the nucleus accumbens regulates anxiety and depression-like behaviors through mitochondrial and neuronal actions. Biol. Psychiatry 2021, 89, 1033–1044. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S. Psychological stress and mitochondria: A conceptual framework. Psychosom. Med. 2018, 80, 126–140. [Google Scholar] [CrossRef]
- Li, M.; Fu, X.; Xie, W.; Guo, W.; Li, B.; Cui, R.; Yang, W. Effect of early life stress on the epigenetic profiles in depression. Front. Cell Dev. Biol. 2020, 8, 867. [Google Scholar] [CrossRef]
- Wang, H.-T.; Huang, F.-L.; Hu, Z.-L.; Zhang, W.-J.; Qiao, X.-Q.; Huang, Y.-Q.; Dai, R.-P.; Li, F.; Li, C.-Q. Early-life social isolation-induced depressive-like behavior in rats results in microglial activation and neuronal histone methylation that are mitigated by minocycline. Neurotox. Res. 2017, 31, 505–520. [Google Scholar] [CrossRef]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Catale, C.; Bussone, S.; Iacono, L.L.; Viscomi, M.T.; Palacios, D.; Troisi, A.; Carola, V. Exposure to different early-life stress experiences results in differentially altered DNA methylation in the brain and immune system. Neurobiol. Stress 2020, 13, 100249. [Google Scholar] [CrossRef]
- Mueller, B.R.; Bale, T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.K.; Jang, M.-H.; Guo, J.U.; Kitabatake, Y.; Chang, M.-L.; Pow-Anpongkul, N.; Flavell, R.A.; Lu, B.; Ming, G.-L.; Song, H. Neuronal activity-induced gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 2009, 323, 1074–1077. [Google Scholar] [CrossRef] [Green Version]
- Tognini, P.; Napoli, D.; Pizzorusso, T. Dynamic DNA methylation in the brain: A new epigenetic mark for experience-dependent plasticity. Front. Cell. Neurosci. 2015, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lux, V. Epigenetic programming effects of early life stress: A dual-activation hypothesis. Curr. Genom. 2018, 19, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.A.; Ambrosini, S.; Lüscher, T.; Paneni, F.; Costantino, S. Epigenetic control of mitochondrial function in the vasculature. Front. Cardiovasc. Med. 2020, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Cuenin, C.; Chung, F.; Rodríguez-Aguilera, J.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A. Mitochondrial stability in diabetic retinopathy: Lessons learned from epigenetics. Diabetes 2019, 68, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, J.; Ryali, V.; Srivastava, K.; Bhat, P.S.; Shashikumar, R. Cognitive reserve: The warehouse within. Ind. Psychiatry J. 2011, 20, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Perosa, V.; Priester, A.; Ziegler, G.; Cardenas-Blanco, A.; Dobisch, L.; Spallazzi, M.; Assmann, A.; Maass, A.; Speck, O.; Oltmer, J.; et al. Hippocampal vascular reserve associated with cognitive performance and hippocampal volume. Brain 2020, 143, 622–634. [Google Scholar] [CrossRef]
- Kerr, A.; Steuer, E.; Pochtarev, V.; Swain, R. Angiogenesis but not neurogenesis is critical for normal learning and memory acquisition. Neuroscience 2010, 171, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Rypma, B.; Gratton, G.; Fabiani, M. Age-related changes in cerebrovascular health and their effects on neural function and cognition: A comprehensive review. Psychophysiology 2021, 58, e13796. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Ninomiya, I.; Kanazawa, M. Angiogenesis and neuronal remodeling after ischemic stroke. Neural Regen. Res. 2020, 15, 16–19. [Google Scholar] [CrossRef]
- Shaw, K.; Bell, L.; Boyd, K.; Grijseels, D.M.; Clarke, D.; Bonnar, O.; Crombag, H.S.; Hall, C.N. Neurovascular coupling and oxygenation are decreased in hippocampus compared to neocortex because of microvascular differences. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Wagenführ, L.; Meyer, A.K.; Marrone, L.; Storch, A. Oxygen tension within the neurogenic niche regulates dopaminergic neurogenesis in the developing midbrain. Stem Cells Dev. 2016, 25, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krabbe, C.; Bak, S.T.; Jensen, P.; von Linstow, C.; Serrano, A.M.; Hansen, C.; Meyer, M. Influence of oxygen tension on dopaminergic differentiation of human fetal stem cells of midbrain and forebrain origin. PLoS ONE 2014, 9, e96465. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Liang, X.; Chen, D.; Chen, Y.; Doycheva, D.; Tang, J.; Tang, J.; Zhang, J.H. Delayed hyperbaric oxygen therapy promotes neurogenesis through reactive oxygen species/hypoxia-inducible factor-1α/β-catenin pathway in middle cerebral artery occlusion rats. Stroke 2014, 45, 1807–1814. [Google Scholar] [CrossRef]
- Lange, C.; García, M.T.; Decimo, I.; Bifari, F.; Eelen, G.; Quaegebeur, A.; Boon, R.; Zhao, H.; Boeckx, B.; Chang, J.; et al. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J. 2016, 35, 924–941. [Google Scholar] [CrossRef]
- Shen, J.; Wang, D.; Wang, X.; Gupta, S.; Ayloo, B.; Wu, S.; Prasad, P.; Xiong, Q.; Xia, J.; Ge, S. Neurovascular coupling in the dentate gyrus regulates adult hippocampal neurogenesis. Neuron 2019, 103, 878–890.e3. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, J.; Chen, X.; Wan, J.; Wang, J.; Zhang, P.; Liu, Y. Hypoxia stimulates neural stem cell proliferation by increasing HIF-1α expression and activating Wnt/β-catenin signaling. Cell. Mol. Biol. 2017, 63, 12–19. [Google Scholar] [CrossRef]
- Pulga, A. Dynamics of the Cerebral Microvasculature during the Course of Memory Consolidation in the Rat: Physiological and Altered Conditions Induced by Hypertension and Hypergravity. Ph.D. Thesis, Neurons and Cognition Université de Bordeaux, Bordeaux, France, December 2016. [Google Scholar]
- Liu, X.; Kuzum, D. Hippocampal-Cortical memory trace transfer and reactivation through cell-specific stimulus and spontaneous background noise. Front. Comput. Neurosci. 2019, 13, 67. [Google Scholar] [CrossRef]
- Frankland, P.W.; Josselyn, S.A. Hippocampal neurogenesis and memory clearance. Neuropsychopharmacology 2015, 41, 382–383. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Büeler, H. Mitochondrial and autophagic regulation of adult neurogenesis in the healthy and diseased brain. Int. J. Mol. Sci. 2021, 22, 3342. [Google Scholar] [CrossRef] [PubMed]
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy metabolism regulates stem cell pluripotency. Front. Cell Dev. Biol. 2020, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario, K.; Shuttleworth, C.W.; Cunningham, L.A. Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J. Neurochem. 2013, 125, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Maegawa, H.; Fujita, T.; Takahara, N.; Egawa, K.; Kashiwagi, A.; Kikkawa, R. Insulin-induced c- jun N-terminal kinase activation is negatively regulated by protein kinase c? Endocrinology 2001, 142, 2669–2676. [Google Scholar] [CrossRef]
- Higashida, H.; Salmina, A.B.; Olovyannikova, R.Y.; Hashii, M.; Yokoyama, S.; Koizumi, K.; Jin, D.; Liu, H.-X.; Lopatina, O.; Amina, S.; et al. Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 2007, 51, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Salmina, A.; Olovyannikova, R.Y.; Noda, M.; Higashida, H. ADP-ribosyl cyclase as a therapeutic target for central nervous system diseases. Cent. Nerv. Syst. Agents Med. Chem. 2006, 6, 193–210. [Google Scholar] [CrossRef]
- Koch-Nolte, F.; Haag, F.; Guse, A.H.; Lund, F.; Ziegler, M. Emerging roles of NAD+ and its metabolites in cell signaling. Sci. Signal. 2009, 2, mr1. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Faini, A.C.; Morandi, F.; Bracci, C.; Lanza, F.; Giuliani, N.; Paulus, A.; Malavasi, F. The circular life of human CD38: From basic science to clinics and back. Molecules 2020, 25, 4844. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, L.R.; Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014, 33, 1321–1340. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.G.; Gardner, D.K.; Harvey, A.J. Nicotinamide adenine dinucleotide induces a bivalent metabolism and maintains pluripotency in human embryonic stem cells. Stem Cells 2020, 38, 624–638. [Google Scholar] [CrossRef]
- Yuan, X.; Liu, Y.; Bijonowski, B.M.; Tsai, A.-C.; Fu, Q.; Logan, T.M.; Ma, T.; Li, Y. NAD+/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Miura, M.; Williams, E.; Jaksch, F.; Kadowaki, T.; Yamauchi, T.; Guarente, L. NAD+ supplementation rejuvenates aged gut adult stem cells. Aging Cell 2019, 18, e12935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of ALS SOD1G93A mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Lautrup, S.H.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Kiss, T.; Balasubramanian, P.; Valcarcel-Ares, M.N.; Tarantini, S.; Yabluchanskiy, A.; Csipo, T.; Lipecz, A.; Reglodi, D.; Zhang, X.A.; Bari, F.; et al. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: A potential mechanism for the prevention of vascular cognitive impairment. Geroscience 2019, 41, 619–630. [Google Scholar] [CrossRef]
- Cuny, H.; Rapadas, M.; Gereis, J.; Martin, E.M.M.A.; Kirk, R.B.; Shi, H.; Dunwoodie, S.L. NAD deficiency due to environmental factors or gene–environment interactions causes congenital malformations and miscarriage in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 3738–3747. [Google Scholar] [CrossRef]
- Ear, P.H.; Chadda, A.; Gumusoglu, S.B.; Schmidt, M.; Vogeler, S.; Malicoat, J.; Kadel, J.; Moore, M.M.; Migaud, M.E.; Stevens, H.; et al. Maternal nicotinamide riboside enhances postpartum weight loss, juvenile offspring development, and neurogenesis of adult offspring. Cell Rep. 2019, 26, 969–983.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimenko, M.; Cherepanov, S.; Furuhara, K.; Lopatina, O.; Salmina, A.B.; Shabalova, A.A.; Tsuji, C.; Yokoyama, S.; Ishihara, K.; Brenner, C.; et al. Nicotinamide riboside supplementation corrects deficits in oxytocin, sociability and anxiety of CD157 mutants in a mouse model of autism spectrum disorder. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Iwata, R.; Vanderhaeghen, P. Mitochondria dynamics in postmitotic cells drives neurogenesis through Sirtuin-dependent chromatin remodeling. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial cell metabolism in health and disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Leaw, B.; Nair, S.; Lim, R.; Thornton, C.; Mallard, C.; Hagberg, H. Mitochondria, bioenergetics and excitotoxicity: New therapeutic targets in perinatal brain injury. Front. Cell. Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- De Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian rhythms in mitochondrial respiration. J. Mol. Endocrinol. 2018, 60, R115–R130. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.-Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, R.; Sridharan, P.; Nguyen, L.; Loren, A.; Williams, N.S.; Kettimuthu, K.P.; Cintrón-Pérez, C.J.; Vázquez-Rosa, E.; Pieper, A.A.; Stevens, H.E. Maternal P7C3-A20 treatment protects offspring from neuropsychiatric sequelae of prenatal stress. Antioxid. Redox Signal. 2021, 35, 511–530. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD + elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-A. Cooperative instruction of signaling and metabolic pathways on the epigenetic landscape. Mol. Cells 2018, 41, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Głombik, K.; Stachowicz, A.; Ślusarczyk, J.; Trojan, E.; Budziszewska, B.; Suski, M.; Kubera, M.; Lason, W.; Wędzony, K.; Olszanecki, R.; et al. Maternal stress predicts altered biogenesis and the profile of mitochondrial proteins in the frontal cortex and hippocampus of adult offspring rats. Psychoneuroendocrinology 2015, 60, 151–162. [Google Scholar] [CrossRef]
- Jia, L.; Wang, J.; Cao, H.; Zhang, X.; Rong, W.; Xu, Z. Activation of PGC-1α and mitochondrial biogenesis protects against prenatal hypoxic–ischemic brain injury. Neuroscience 2020, 432, 63–72. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. The mitochondrion as potential interface in early-life stress brain programming. Front. Behav. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Zitkovsky, E.K.; Daniels, T.E.; Tyrka, A.R. Mitochondria and early-life adversity. Mitochondrion 2021, 57, 213–221. [Google Scholar] [CrossRef]
- Leke, R.; Bak, L.K.; Iversen, P.; Sørensen, M.; Keiding, S.; Vilstrup, H.; Ott, P.; Portela, L.V.; Schousboe, A.; Waagepetersen, H.S. Synthesis of neurotransmitter GABA via the neuronal tricarboxylic acid cycle is elevated in rats with liver cirrhosis consistent with a high GABAergic tone in chronic hepatic encephalopathy. J. Neurochem. 2011, 117, 824–832. [Google Scholar] [CrossRef]
- Ben-Ari, Y. GABA excites and sculpts immature neurons well before delivery: Modulation by GABA of the development of ventricular progenitor cells. Epilepsy Curr. 2007, 7, 167–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, H.; Asrican, B.; Li, W.; Gu, B.; Wen, Z.; Lim, S.-A.; Haniff, I.; Ramakrishnan, C.; Deisseroth, K.; Philpot, B.; et al. Long-range GABAergic inputs regulate neural stem cell quiescence and control adult hippocampal neurogenesis. Cell Stem Cell 2017, 21, 604–617.e5. [Google Scholar] [CrossRef]
- Giachino, C.; Barz, M.; Tchorz, J.S.; Tomé, M.; Gassmann, M.; Bischofberger, J.; Bettler, B.; Taylor, V. GABA suppresses neurogenesis in the adult hippocampus through GABAB receptors. Development 2014, 141, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.; Sailor, K.A.; Ming, G.-L.; Song, H. Synaptic integration and plasticity of new neurons in the adult hippocampus. J. Physiol. 2008, 586, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Pallotto, M.; Deprez, F. Regulation of adult neurogenesis by GABAergic transmission: Signaling beyond GABAA-receptors. Front. Cell. Neurosci. 2014, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Kumar, P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabó, G.; et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2017, 28, 221–248. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Malinovskaya, N.A.; Komleva, Y.K.; Gorina, Y.V.; Shuvaev, A.N.; Olovyannikova, R.Y.; Belozor, O.; Belova, O.A.; Higashida, H.; Salmina, A.B. Excitation/inhibition imbalance and impaired neurogenesis in neurodevelopmental and neurodegenerative disorders. Rev. Neurosci. 2019, 30, 807–820. [Google Scholar] [CrossRef]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hübner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisseroth, K.; Singla, S.; Toda, H.; Monje, M.; Palmer, T.; Malenka, R.C. Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 2004, 42, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Derks, N.A.V.; Krugers, H.J.; Hoogenraad, C.; Joels, M.; Sarabdjitsingh, R.A. Effects of early life stress on synaptic plasticity in the developing hippocampus of male and female rats. PLoS ONE 2016, 11, e0164551. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; Sarabdjitsingh, R.A.; Van Der Weerd, N.; Feenstra, E.; Damsteegt, R.; Joëls, M. Age-dependent shift in spontaneous excitation-inhibition balance of infralimbic prefrontal layer II/III neurons is accelerated by early life stress, independent of forebrain mineralocorticoid receptor expression. Neuropharmacology 2020, 180, 108294. [Google Scholar] [CrossRef]
- Ohta, K.-I.; Suzuki, S.; Warita, K.; Sumitani, K.; Tenkumo, C.; Ozawa, T.; Ujihara, H.; Kusaka, T.; Miki, T. The effects of early life stress on the excitatory/inhibitory balance of the medial prefrontal cortex. Behav. Brain Res. 2020, 379, 112306. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Morstein, J.; Novak, A.J.E.; Trauner, D.; Xiong, Q.; Yu, Y.; Ge, S. Metabolic tuning of inhibition regulates hippocampal neurogenesis in the adult brain. Proc. Natl. Acad. Sci. USA 2020, 117, 25818–25829. [Google Scholar] [CrossRef] [PubMed]
- Yatomi, Y.; Ozaki, Y.; Ohmori, T.; Igarashi, Y. Sphingosine 1-phosphate: Synthesis and release. Prostaglandins Other Lipid Mediat. 2001, 64, 107–122. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood–brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.-P.; Yamashita, T.; Proia, R. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.-X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.-S.; Lee, M.-J.; et al. Edg-1, the G protein–coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Xiang, H.; Chen, R.; Yang, J.; Yang, X.; Zhou, J.; Liu, H.; Zhao, S.; Xiao, J.; Chen, P.; et al. S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 2019, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fugio, L.B.; Coeli-Lacchini, F.; Leopoldino, A.M. Sphingolipids and mitochondrial dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moruno-Manchon, J.F.; Uzor, N.-E.; Dabaghian, Y.; Furr-Stimming, E.E.; Finkbeiner, S.; Tsvetkov, A.S. Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci. Rep. 2015, 5, 15213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 2011, 31, 6850–6857. [Google Scholar] [CrossRef] [Green Version]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Harada, J.; Foley, M.; Moskowitz, M.A.; Waeber, C. Sphingosine-1-phosphate induces proliferation and morphological changes of neural progenitor cells. J. Neurochem. 2004, 88, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Callihan, P.; Alqinyah, M.; Hooks, S.B. Sphingosine-1-phosphate (s1p) signaling in neural progenitors. Adv. Struct. Saf. Stud. 2017, 1697, 141–151. [Google Scholar] [CrossRef]
- Alfonso, J.; Penkert, H.; Duman, C.; Zuccotti, A.; Monyer, H. Downregulation of sphingosine 1-phosphate receptor 1 promotes the switch from tangential to radial migration in the ob. J. Neurosci. 2015, 35, 13659–13672. [Google Scholar] [CrossRef] [Green Version]
- Blanc, C.A.; Grist, J.J.; Rosen, H.; Sears-Kraxberger, I.; Steward, O.; Lane, T.E. Sphingosine-1-phosphate receptor antagonism enhances proliferation and migration of engrafted neural progenitor cells in a model of viral-induced demyelination. Am. J. Pathol. 2015, 185, 2819–2832. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Watanabe, T.; Sato, K.; Kon, J.; Tomura, H.; Tamama, K.; Kuwabara, A.; Kanda, T.; Kobayashi, I.; Ohta, H.; et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem. J. 2000, 348, 71–76. [Google Scholar] [CrossRef]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Laviña, B.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nyström, S.; Rymo, S.; Chen, L.L.; et al. The sphingosine-1-phosphate receptor s1pr1 restricts sprouting angiogenesis by regulating the interplay between VE-Cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Zhang, Y.; D’Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat. Commun. 2016, 7, 12086. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, C.; Liu, P.; Zhao, J.; Xu, W. Sphingosine 1-phosphate (S1P) promotes mitochondrial biogenesis in Hep G2 cells by activating Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Cell Stress Chaperon. 2013, 19, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-survival lipid sphingosine-1-phosphate metabolically programs t cells to limit anti-tumor activity. Cell Rep. 2019, 28, 1879–1893.e7. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [Green Version]
- Kalkhoran, S.B.; Hernandez-Resendiz, S.; Crespo-Avilan, G.; Ramachandra, C.; Ong, S.-G.; Hausenloy, D. Pharmacological modulators of mitochondrial dynamics as novel therapeutics for cardiovascular and neurological diseases. Cond. Med. 2020, 3, 144–159. [Google Scholar]
- Georgakopoulos, N.D.; Wells, G.; Campanella, N.D.G.M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Burmistrova, O.; Olias-Arjona, A.; Lapresa, R.; Jimenez-Blasco, D.; Eremeeva, T.; Shishov, D.; Romanov, S.; Zakurdaeva, K.; Almeida, A.; Fedichev, P.O.; et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Sessions, E.H.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD+ with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, E.L.; Baune, B.T. Modulation of early stress-induced neurobiological changes: A review of behavioural and pharmacological interventions in animal models. Transl. Psychiatry 2014, 4, e390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.; Worrell, T.R.; Zimnisky, R.; Schmauss, C. Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiol. Dis. 2012, 45, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Fournier, N.M.; Duman, R.S. Role of vascular endothelial growth factor in adult hippocampal neurogenesis: Implications for the pathophysiology and treatment of depression. Behav. Brain Res. 2012, 227, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashiri, H.; Houwing, D.J.; Homberg, J.R.; Salari, A.-A. The combination of fluoxetine and environmental enrichment reduces postpartum stress-related behaviors through the oxytocinergic system and HPA axis in mice. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 2018, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Peyton, L.; Oliveros, A.; Choi, D.-S.; Jang, M.-H. Hippocampal regenerative medicine: Neurogenic implications for addiction and mental disorders. Exp. Mol. Med. 2021, 53, 358–368. [Google Scholar] [CrossRef]
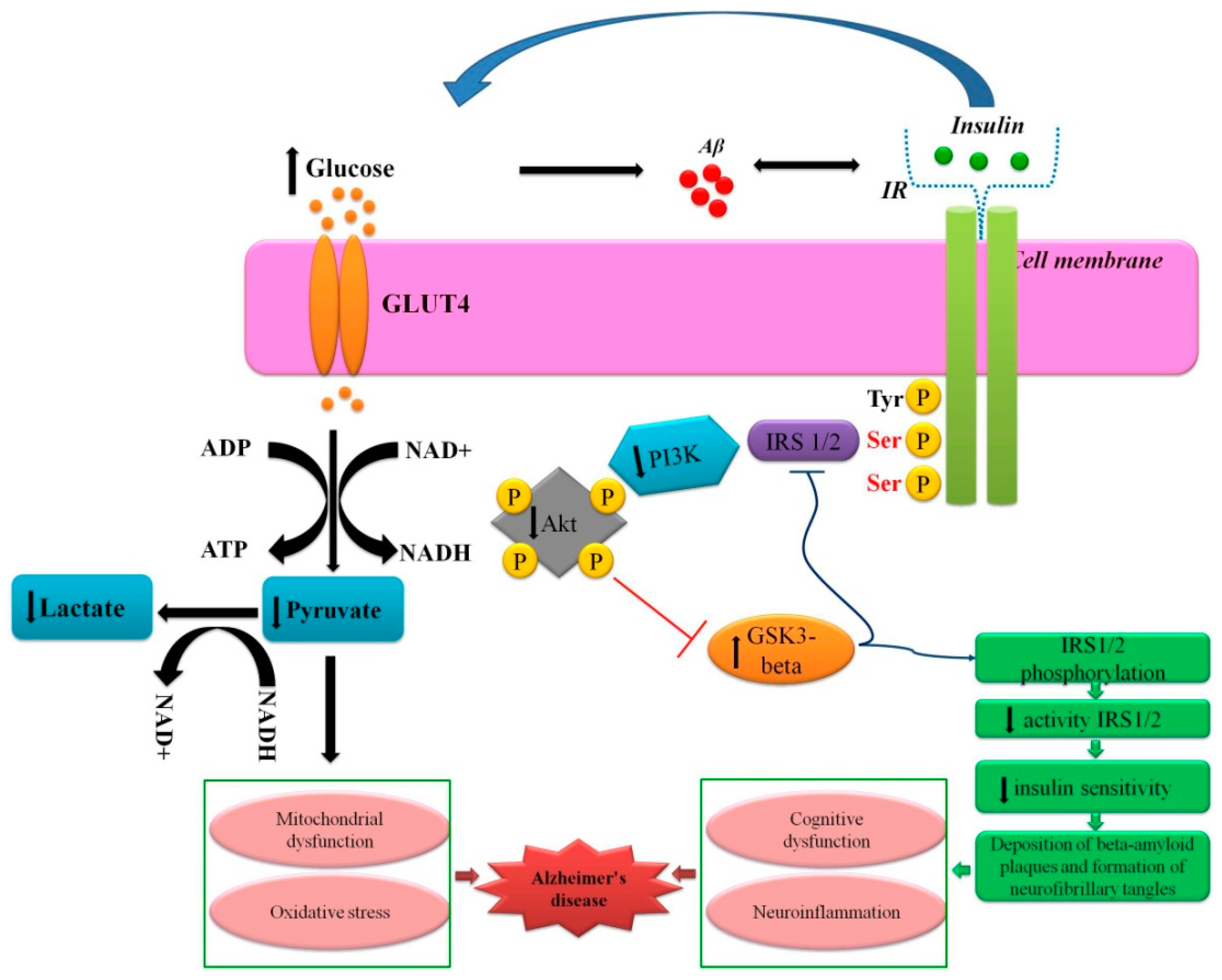



{kind=link}
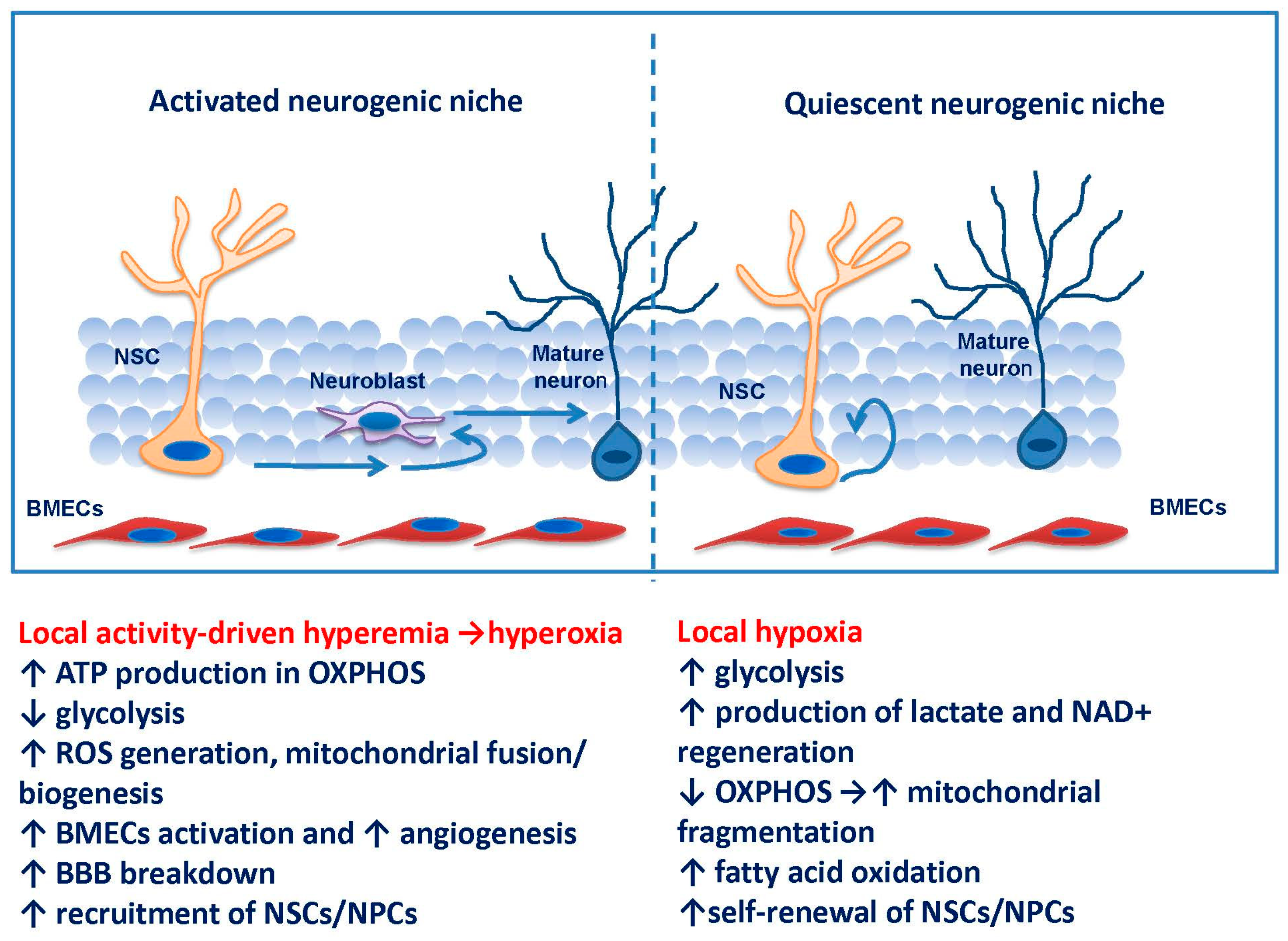{kind=link}
{kind=link}
| Type of Cells | Metabolic Characteristics | References |
|---|---|---|
| Neurons | Oxidative phosphorylation (OXPHOS) dominates. Perisynaptic localization of mitochondria. Under stress and aging, they redistribute mitochondria in the cell and demonstrate impairment in glycose utilization and decline in mitochondrial function. Poorly utilize fatty acids. | [53,54,55,56] |
| Astrocytes | Glycolysis dominates. They support the metabolism of neurons with lactate. Capable of depositing the glycogen. Mitochondrial activity determines the efficiency of glutamate uptake. Serve as donors of mitochondria when neurons are damaged. | [57,58,59,60] |
| Oligodendrocytes | Myelination is under the control of OXPHOS (to a greater extent) and glycolysis, at the end of the myelination program, glycolysis dominates, and lactate maintains axonal vitality. | [61,62,63] |
| Microglia | Resting microglia uses OXPHOS, activated microglia uses glycolysis. Microglia activation is accompanied by mitochondrial fragmentation. | [64,65,66] |
| BMECs | Have a higher content of mitochondria than endothelial cells in other tissues. Angiogenesis is accompanied by increased glycolysis and OXPHOS, fatty acid oxidation. Disturbances in mitochondrial dynamics are characteristic of damage to the BBB. Various reactive oxygen species (ROS)-generating enzymes are expressed. | [67,68,69,70,71] |
| Pericytes | Glycolysis dominates; however, these cells might donate mitochondria for damaged perivascular astroglia. | [72] |
| NSCs/NPCs | Self-maintenance and proliferation of NSCs/NPCs require glycolysis, oxidation of fatty acids. Differentiation is accompanied by the prevalence of OXPHOS, and significant changes in the shape of mitochondria from fragmented to elongated. Able to deliver functional mitochondria to target cells. Aging NSCs/NPCs demonstrate lower oxidative metabolism. | [73,74,75,76,77,78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmina, A.B.; Gorina, Y.V.; Komleva, Y.K.; Panina, Y.A.; Malinovskaya, N.A.; Lopatina, O.L. Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis. Biomedicines 2021, 9, 1092. https://doi.org/10.3390/biomedicines9091092
Salmina AB, Gorina YV, Komleva YK, Panina YA, Malinovskaya NA, Lopatina OL. Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis. Biomedicines. 2021; 9(9):1092. https://doi.org/10.3390/biomedicines9091092
Chicago/Turabian StyleSalmina, Alla B., Yana V. Gorina, Yulia K. Komleva, Yulia A. Panina, Natalia A. Malinovskaya, and Olga L. Lopatina. 2021. "Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis" Biomedicines 9, no. 9: 1092. https://doi.org/10.3390/biomedicines9091092
APA StyleSalmina, A. B., Gorina, Y. V., Komleva, Y. K., Panina, Y. A., Malinovskaya, N. A., & Lopatina, O. L. (2021). Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis. Biomedicines, 9(9), 1092. https://doi.org/10.3390/biomedicines9091092