
Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model

 , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Isolation of Mitochondrial Fraction
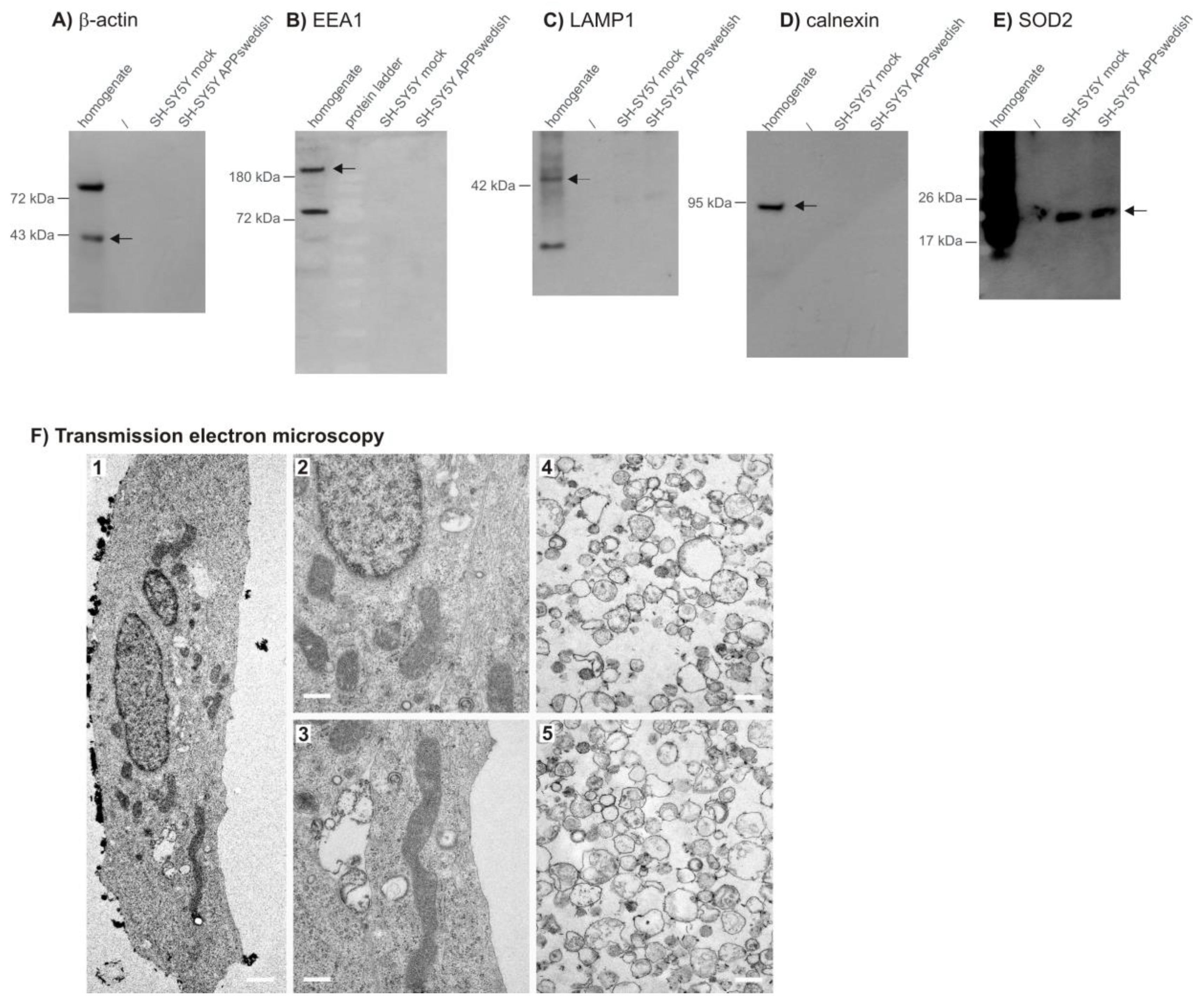
2.4. Western Blot Analysis
2.5. Verification of the Mitochondrial Sample by Transmission Electron Microscopy
2.6. Lipid Extraction and Mass Spectrometry
2.7. Detection of Cardiolipin
2.8. Statistical Analysis
3. Results
3.1. Lipid Analysis in Isolated Mitochondria Fraction and Total Cell Homogenate of APPswedish Transfected Human Neuroblastoma Cells
3.2. Mass Spectrometry Analysis of Phosphatidylcholine (PCaa) in Mitochondria and Total Cell Homogenate of APPswedish Transfected SH-SY5Y Cells
3.3. Alterations in Phosphatidylcholine Plasmalogen Species (PCae) in Mitochondria and Total Cell Homogenate of SH-SY5Y Cells Stably Expressing APPswedish
3.4. Lyso-Phosphatidylcholine (Lyso-PC) Species
3.5. Alterations in Phosphatidylethanolamine Species (PEaa) in Mitochondria and Total Cell Homogenate of APPswedish Transfected Cells
3.6. Analysis of Phosphatidylethanolamine Plasmalogens (PEae) in Mitochondria and Total Cell Homogenates of SH-SY5Y APPswedish Cells
3.7. Alterations in Lyso-Phosphatidylethanolmine (lyso-PE) Species in Mitochondria and Total Cell Homogenate of APPswedish Transfected SH-SY5Y Cells
3.8. Analysis of the Carnitine Carrier System in Total Cell Homogenate
3.9. Analysis of Cardiolipin and Phosphatidyl-Glycerol in Total Cell Homogenate
3.10. Alterations in Mitochondrial Lipid Composition of the Cellular AD Model Compared to Neuroblastoma Cells Expressing Wildtype APP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 2012, 425, 534–539. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C. Take five-bace and the gamma-secretase quartet conduct alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Grimm, M.O.; Tomic, I.; Hartmann, T. Potential external source of a beta in biological samples. Nat. Cell Biol. 2002, 4, E164–E165. [Google Scholar] [CrossRef]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium signaling and amyloid toxicity in alzheimer disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Fedrizzi, L.; Carafoli, E. Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. Biofactors 2011, 37, 189–196. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Gotz, J. Mitochondrial dysfunction—the beginning of the end in alzheimer’s disease? Separate and synergistic modes of tau and amyloid-beta toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes alzheimer’s pathology in female mouse model of alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction, and alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S487–S498. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in alzheimer’s and related diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Small, D.H.; Gasperini, R.; Vincent, A.J.; Hung, A.C.; Foa, L. The role of abeta-induced calcium dysregulation in the pathogenesis of alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 225–233. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. High mitochondrial calcium levels precede neuronal death in vivo in alzheimer’s disease. Cell Stress 2020, 4, 187–190. [Google Scholar] [CrossRef]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of atp alternations in an alzheimer’s disease transgenic mouse model. J. Alzheimer’s Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.Y.; Cho, H.J.; Kim, C.; Jung, Y.O.; Kang, M.J.; Murray, M.E.; Hong, H.S.; Choi, Y.J.; Choi, H.; Kim, D.K.; et al. Mitochondrial atp synthase activity is impaired by suppressed o-glcnacylation in alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 6492–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Anandatheerthavarada, H.K. Mitochondrial trafficking of app and alpha synuclein: Relevance to mitochondrial dysfunction in alzheimer’s and parkinson’s diseases. Biochim. Biophys. Acta 2010, 1802, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Pagani, L.; Eckert, A. Amyloid-beta interaction with mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Canevari, L.; Clark, J.B.; Bates, T.E. Beta-amyloid fragment 25–35 selectively decreases complex iv activity in isolated mitochondria. FEBS Lett. 1999, 457, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic abeta peptides increase oxidative stress in vivo through nmda-receptor and nitric-oxide-synthase mechanisms, and inhibit complex iv activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef]
- Robinson, A.; Grosgen, S.; Mett, J.; Zimmer, V.C.; Haupenthal, V.J.; Hundsdorfer, B.; Stahlmann, C.P.; Slobodskoy, Y.; Muller, U.C.; Hartmann, T.; et al. Upregulation of pgc-1alpha expression by alzheimer’s disease-associated pathway: Presenilin 1/amyloid precursor protein (app)/intracellular domain of app. Aging Cell 2014, 13, 263–272. [Google Scholar] [CrossRef]
- Slomnicki, L.P.; Lesniak, W. A putative role of the amyloid precursor protein intracellular domain (aicd) in transcription. Acta Neurobiol. Exp. 2008, 68, 219–228. [Google Scholar]
- Grimm, M.O.; Hundsdorfer, B.; Grosgen, S.; Mett, J.; Zimmer, V.C.; Stahlmann, C.P.; Haupenthal, V.J.; Rothhaar, T.L.; Lehmann, J.; Patzold, A.; et al. Ps dependent app cleavage regulates glucosylceramide synthase and is affected in alzheimer’s disease. Cell Physiol. Biochem. 2014, 34, 92–110. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Grosgen, S.; Haupenthal, V.J.; Blumel, T.; Hundsdorfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. App intracellular domain derived from amyloidogenic beta- and gamma-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The app intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Zinser, E.G.; Grosgen, S.; Hundsdorfer, B.; Rothhaar, T.L.; Burg, V.K.; Kaestner, L.; Bayer, T.A.; Lipp, P.; Muller, U.; et al. Amyloid precursor protein (app) mediated regulation of ganglioside homeostasis linking alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE 2012, 7, e34095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.; Grosgen, S.; Rothhaar, T.L.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Muller, U.; Fassbender, K.; Riemenschneider, M.; et al. Intracellular app domain regulates serine-palmitoyl-coa transferase expression and is affected in alzheimer’s disease. Int. J. Alzheimer’s Dis. 2011, 2011, 695413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.; Kuchenbecker, J.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Burg, V.K.; Friess, P.; Muller, U.; Grimm, H.S.; Riemenschneider, M.; et al. Plasmalogen synthesis is regulated via alkyl-dihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in alzheimer’s disease. J. Neurochem. 2011, 116, 916–925. [Google Scholar] [CrossRef]
- Cai, X.D.; Golde, T.E.; Younkin, S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 1993, 259, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, N.D.; Kellett, K.A.; Beckett, C.; Makova, N.Z.; Revett, T.J.; Nalivaeva, N.N.; Hooper, N.M.; Turner, A.J. The transcriptionally active amyloid precursor protein (app) intracellular domain is preferentially produced from the 695 isoform of app in a {beta}-secretase-dependent pathway. J. Biol. Chem. 2010, 285, 41443–41454. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid. Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in alzheimer’s disease brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grosgen, S.; Riemenschneider, M.; Tanila, H.; Grimm, H.S.; Hartmann, T. From brain to food: Analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in alzheimer’s disease human post mortem brains and mice model via mass spectrometry. J. Chromatogr. A 2011, 1218, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Mankidy, R.; Ritchie, S.; Heath, D.; Wood, J.A.; Flax, J.; Goodenowe, D.B. Circulating plasmalogen levels and alzheimer disease assessment scale-cognitive scores in alzheimer patients. J. Psychiatry Neurosci. 2010, 35, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Su, X.Q.; Wang, J.; Sinclair, A.J. Plasmalogens and alzheimer’s disease: A review. Lipids Health Dis. 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhaar, T.L.; Grosgen, S.; Haupenthal, V.J.; Burg, V.K.; Hundsdorfer, B.; Mett, J.; Riemenschneider, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O. Plasmalogens inhibit app processing by directly affecting gamma-secretase activity in alzheimer’s disease. Sci. World J. 2012, 2012, 141240. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Liu, X.; Wang, M.; Liu, L.; Sun, X.; Ma, L.; Xie, W.; Wang, C.; Tang, S.; Wang, D.; et al. Lysophosphatidylcholine and amide as metabolites for detecting alzheimer disease using ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry-based metabonomics. J. Neuropathol Exp. Neurol 2014, 73, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in alzheimer’s disease. J. Neural. Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Li, N.J.; Liu, W.T.; Li, W.; Li, S.Q.; Chen, X.H.; Bi, K.S.; He, P. Plasma metabolic profiling of alzheimer’s disease by liquid chromatography/mass spectrometry. Clin. Biochem. 2010, 43, 992–997. [Google Scholar] [CrossRef]
- Sanchez-Mejia, R.O.; Mucke, L. Phospholipase a2 and arachidonic acid in alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyrd-Hansen, M.; Darding, M.; Miasari, M.; Santoro, M.M.; Zender, L.; Xue, W.; Tenev, T.; da Fonseca, P.C.; Zvelebil, M.; Bujnicki, J.M.; et al. Iaps contain an evolutionarily conserved ubiquitin-binding domain that regulates nf-kappab as well as cell survival and oncogenesis. Nat. Cell Biol. 2008, 10, 1309–1317. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, D.T.; Lemere, C.A.; Selkoe, D.J.; Clemens, J.A. Cytosolic phospholipase a2 (cpla2) immunoreactivity is elevated in alzheimer’s disease brain. Neurobiol. Dis. 1996, 3, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Gentile, M.T.; Reccia, M.G.; Sorrentino, P.P.; Vitale, E.; Sorrentino, G.; Puca, A.A.; Colucci-D’Amato, L. Role of cytosolic calcium-dependent phospholipase a2 in alzheimer’s disease pathogenesis. Mol. Neurobiol. 2012, 45, 596–604. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Blumel, T.; Stotzel, H.; Grimm, H.S.; Hartmann, T. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem. Cell Biol. 2016, 94, 534–542. [Google Scholar] [CrossRef]
- Lauer, A.A.; Janitschke, D.; Dos Santos Guilherme, M.; Nguyen, V.T.T.; Bachmann, C.M.; Qiao, S.; Schrul, B.; Boehm, U.; Grimm, H.S.; Hartmann, T.; et al. Shotgun lipidomics of liver and brain tissue of alzheimer’s disease model mice treated with acitretin. Sci. Rep. 2021, 11, 15301. [Google Scholar] [CrossRef]
- Kim, H.W.; Rapoport, S.I.; Rao, J.S. Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Mol. Psychiatry 2011, 16, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.H.; Pelleieux, S.; Vitale, N.; Olivier, J.L. Dietary arachidonic acid as a risk factor for age-associated neurodegenerative diseases: Potential mechanisms. Biochimie 2016, 130, 168–177. [Google Scholar] [CrossRef]
- Fukuyama, H.; Ogawa, M.; Yamauchi, H.; Yamaguchi, S.; Kimura, J.; Yonekura, Y.; Konishi, J. Altered cerebral energy metabolism in alzheimer’s disease: A pet study. J. Nucl. Med. 1994, 35, 1–6. [Google Scholar]
- Hunt, A.; Schonknecht, P.; Henze, M.; Seidl, U.; Haberkorn, U.; Schroder, J. Reduced cerebral glucose metabolism in patients at risk for alzheimer’s disease. Psychiatry Res. 2007, 155, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sasaki, M.; Kitagaki, H.; Yamaji, S.; Sakamoto, S.; Matsuda, K.; Mori, E. Reduction of cerebellar glucose metabolism in advanced alzheimer’s disease. J. Nucl. Med. 1997, 38, 925–928. [Google Scholar] [PubMed]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, mitochondria, and neurological disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 1375–1392. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.Z.; Soderberg, M.; Sindelar, P.; Edlund, C. Content and fatty acid composition of cardiolipin in the brain of patients with alzheimer’s disease. Neurochem. Int. 1994, 25, 295–300. [Google Scholar] [CrossRef]
- Scherer, M.; Schmitz, G.; Liebisch, G. Simultaneous quantification of cardiolipin, bis(monoacylglycero)phosphate and their precursors by hydrophilic interaction lc-ms/ms including correction of isotopic overlap. Anal. Chem. 2010, 82, 8794–8799. [Google Scholar] [CrossRef]
- Bird, S.S.; Marur, V.R.; Sniatynski, M.J.; Greenberg, H.K.; Kristal, B.S. Lipidomics profiling by high-resolution lc-ms and high-energy collisional dissociation fragmentation: Focus on characterization of mitochondrial cardiolipins and monolysocardiolipins. Anal. Chem. 2011, 83, 940–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. App processing in alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Buttner, S. Mitochondrial lipids in neurodegeneration. Cell Tissue Res. 2017, 367, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [Green Version]
- Burg, V.K.; Grimm, H.S.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Haupenthal, V.J.; Zimmer, V.C.; Mett, J.; Weingartner, O.; Laufs, U.; et al. Plant sterols the better cholesterol in alzheimer’s disease? A mechanistical study. J. Neurosci. 2013, 33, 16072–16087. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the alzheimer amyloid precursor protein (app). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Pennington, E.R.; Funai, K.; Brown, D.A.; Shaikh, S.R. The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1039–1052. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in alzheimer’s disease prefrontal cortex. J. Alzheimer’s Dis. 2011, 24, 507–517. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Chiang, J.; Cipolla, M.; Hale, J.; Diallo, F.; Chirino, A.; Arakaki, X.; Harrington, M.G. Alterations in cerebrospinal fluid glycerophospholipids and phospholipase a2 activity in alzheimer’s disease. J. Lipid Res. 2013, 54, 2884–2897. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of alzheimer’s disease, parkinson’s disease, and huntington’s disease: A mini review. Oxid. Med. Cell Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Lin, C.Y.; Li, J.S.; Lou, Y.I. Wheat germ agglutinin-conjugated liposomes incorporated with cardiolipin to improve neuronal survival in alzheimer’s disease treatment. Int. J. Nanomed. 2017, 12, 1757–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez-Gutierrez, L.; Re, F.; Bereczki, E.; Ioja, E.; Gregori, M.; Andersen, A.J.; Anton, M.; Moghimi, S.M.; Pei, J.J.; Masserini, M.; et al. Repeated intraperitoneal injections of liposomes containing phosphatidic acid and cardiolipin reduce amyloid-beta levels in app/ps1 transgenic mice. Nanomedicine 2015, 11, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Soderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in alzheimer’s disease. Lipids 1991, 26, 421–425. [Google Scholar] [CrossRef]
- Tully, A.M.; Roche, H.M.; Doyle, R.; Fallon, C.; Bruce, I.; Lawlor, B.; Coakley, D.; Gibney, M.J. Low serum cholesteryl ester-docosahexaenoic acid levels in alzheimer’s disease: A case-control study. Br. J. Nutr. 2003, 89, 483–489. [Google Scholar] [CrossRef]
- Pan, Y.; Khalil, H.; Nicolazzo, J.A. The impact of docosahexaenoic acid on alzheimer’s disease: Is there a role of the blood-brain barrier? Curr. Clin. Pharmacol. 2015, 10, 222–241. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.Q.; Zhou, Y.; Halabisky, B.; Cisse, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase a2 reduction ameliorates cognitive deficits in a mouse model of alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Chalbot, S.; Zetterberg, H.; Blennow, K.; Fladby, T.; Grundke-Iqbal, I.; Iqbal, K. Cerebrospinal fluid secretory ca2+-dependent phospholipase a2 activity is increased in alzheimer disease. Clin. Chem. 2009, 55, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, J.Y.; Holopainen, J.M.; Kinnunen, P.K. Activation of phospholipase a2 by amyloid beta-peptides in vitro. Biochemistry 1996, 35, 9407–9414. [Google Scholar] [CrossRef]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and nmda induce ros from nadph oxidase and aa release from cytosolic phospholipase a2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef]
- Korenberg, J.R.; Pulst, S.M.; Neve, R.L.; West, R. The alzheimer amyloid precursor protein maps to human chromosome 21 bands q21.105-q21.05. Genomics 1989, 5, 124–127. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Epitope | Dilution | 2nd Antibody |
|---|---|---|---|
| A5441 (Merck, Darmstadt, Germany) | β-actin | 1:5000 | anti-mouse IgG HRP (P0260; Dako, Glostrup, Denmark) |
| ab2900 (Abcam, Berlin, Germany) | EEA1 | 1:2000 | anti-rabbit IgG HRP (W4011; Promega, Walldorf, Germany) |
| ab25630 (Abcam, Berlin, Germany) | Lamp-1 | 1:2000 | anti-mouse IgG HRP (P0260; Dako, Glostrup, Denmark) |
| C4721 (Merck, Darmstadt, Germany) | calnexin | 1:2000 | anti-rabbit IgG HRP (W4011; Promega, Walldorf, Germany) |
| 24127 (proteintech, Manchester, United Kingdom) | SOD2 | 1:5000 | anti-rabbit IgG HRP (W4011; Promega, Walldorf, Germany) |
| W02 | APP/Aβ | 5 µg/mL | anti-mouse IgG HRP (P0260; Dako, Glostrup, Denmark) |
| Name | Vendor |
|---|---|
| lyso-phosphatidylcholine (19:0 Lyso-PC) | Avanti Polar Lipids (855776P) |
| phosphatidylcholine (06:0 PC (DHPC)) | Avanti Polar Lipids (850305P) |
| phosphatidylcholine plasmalogens (12:0 Diether PC) | Avanti Polar Lipids (999994P) |
| phosphatidylethanolamine (08:0 PE) | Avanti Polar Lipids (850699C) |
| phosphatidylglycerol (17:0–14:1 PG) | Avanti Polar Lipids (LM1204) |
| octanoyl-L-carnitine d3 | Supelco Analytical (53230) |
| palmitoyl-L-carnitine d3 | Supelco Analytical (55107) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurokin, I.; Lauer, A.A.; Janitschke, D.; Winkler, J.; Theiss, E.L.; Griebsch, L.V.; Pilz, S.M.; Matschke, V.; van der Laan, M.; Grimm, H.S.; et al. Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model. Biomedicines 2021, 9, 1062. https://doi.org/10.3390/biomedicines9081062
Kurokin I, Lauer AA, Janitschke D, Winkler J, Theiss EL, Griebsch LV, Pilz SM, Matschke V, van der Laan M, Grimm HS, et al. Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model. Biomedicines. 2021; 9(8):1062. https://doi.org/10.3390/biomedicines9081062
Chicago/Turabian StyleKurokin, Irina, Anna Andrea Lauer, Daniel Janitschke, Jakob Winkler, Elena Leoni Theiss, Lea Victoria Griebsch, Sabrina Melanie Pilz, Veronika Matschke, Martin van der Laan, Heike Sabine Grimm, and et al. 2021. "Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model" Biomedicines 9, no. 8: 1062. https://doi.org/10.3390/biomedicines9081062
APA StyleKurokin, I., Lauer, A. A., Janitschke, D., Winkler, J., Theiss, E. L., Griebsch, L. V., Pilz, S. M., Matschke, V., van der Laan, M., Grimm, H. S., Hartmann, T., & Grimm, M. O. W. (2021). Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model. Biomedicines, 9(8), 1062. https://doi.org/10.3390/biomedicines9081062