
Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Synthetic Peptides
2.2. HTRA2 Protease Activity Assay
2.3. Retina Extraction and Homogenization
2.4. Co-Immunoprecipitation (Co-IP) of HTRA2 Protein Interaction Partners
2.5. 1D SDS-PAGE and In-Gel Trypsin Digestion
2.6. LC-MS/MS Analysis
2.7. Protein Identification and Quantification
2.8. Data Analysis and Statistics
2.9. Pathway and Enrichment Analysis
2.10. Targeted MS Analysis
3. Results
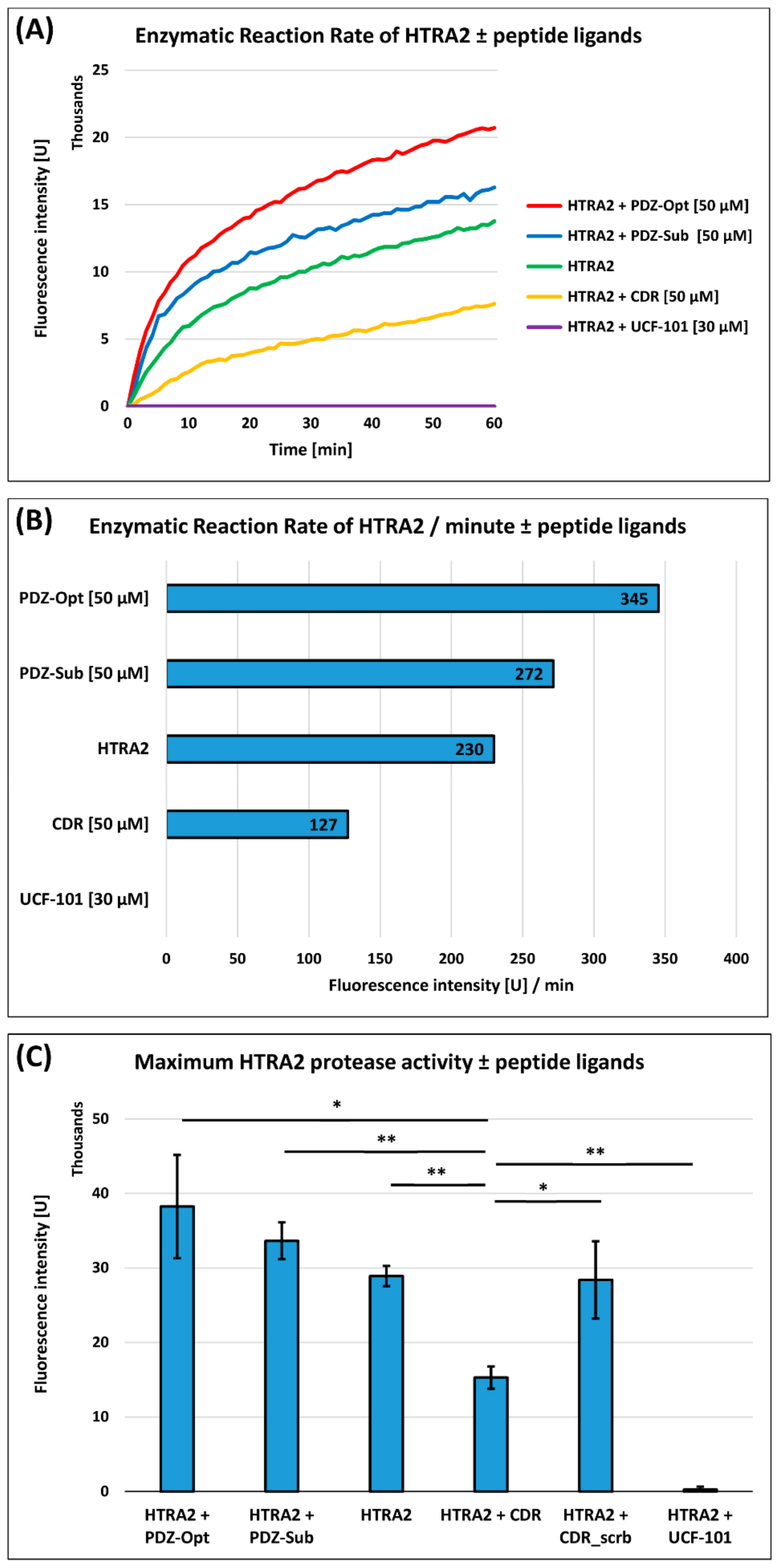
3.1. HTRA2 Protease Activity Assay
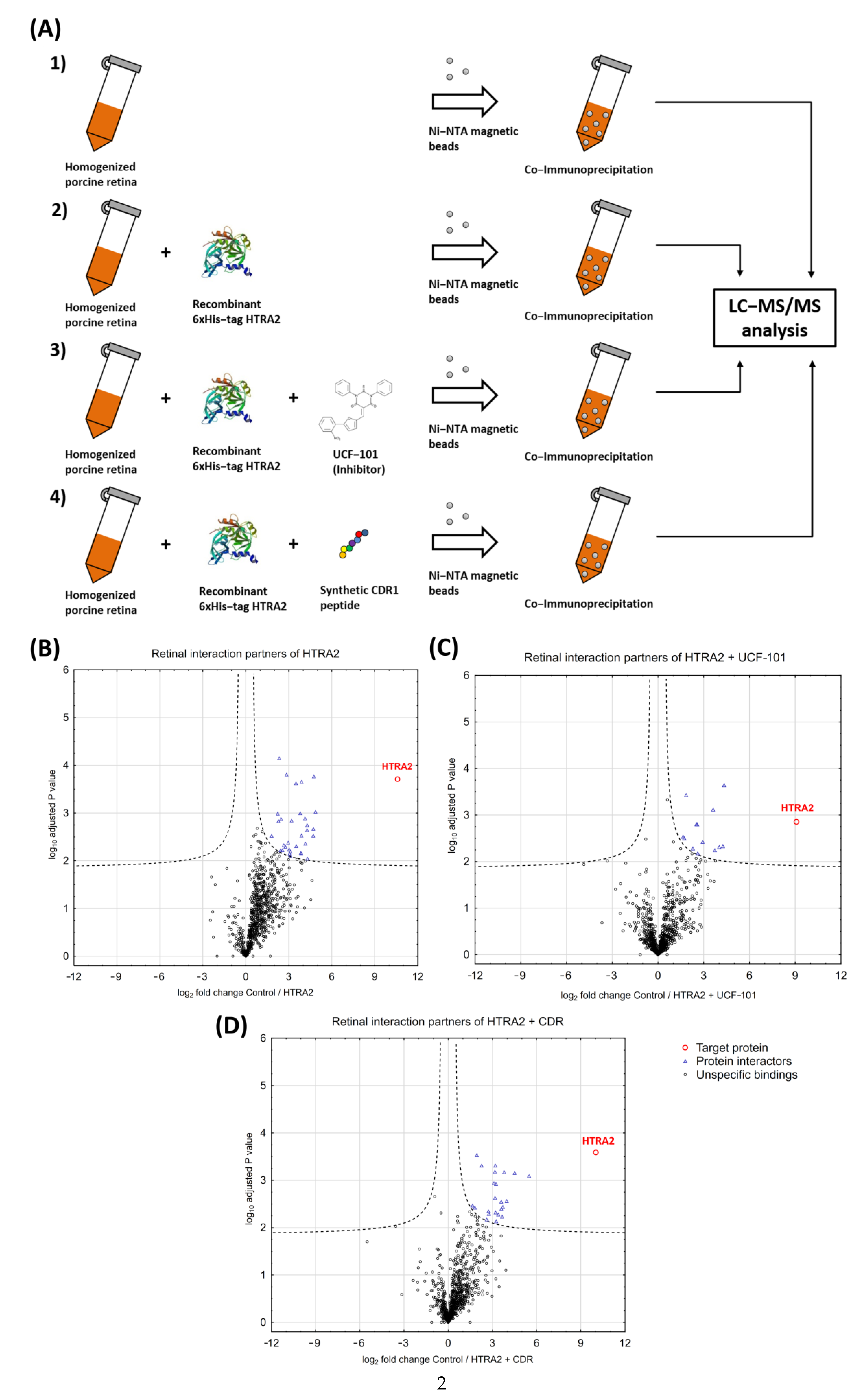
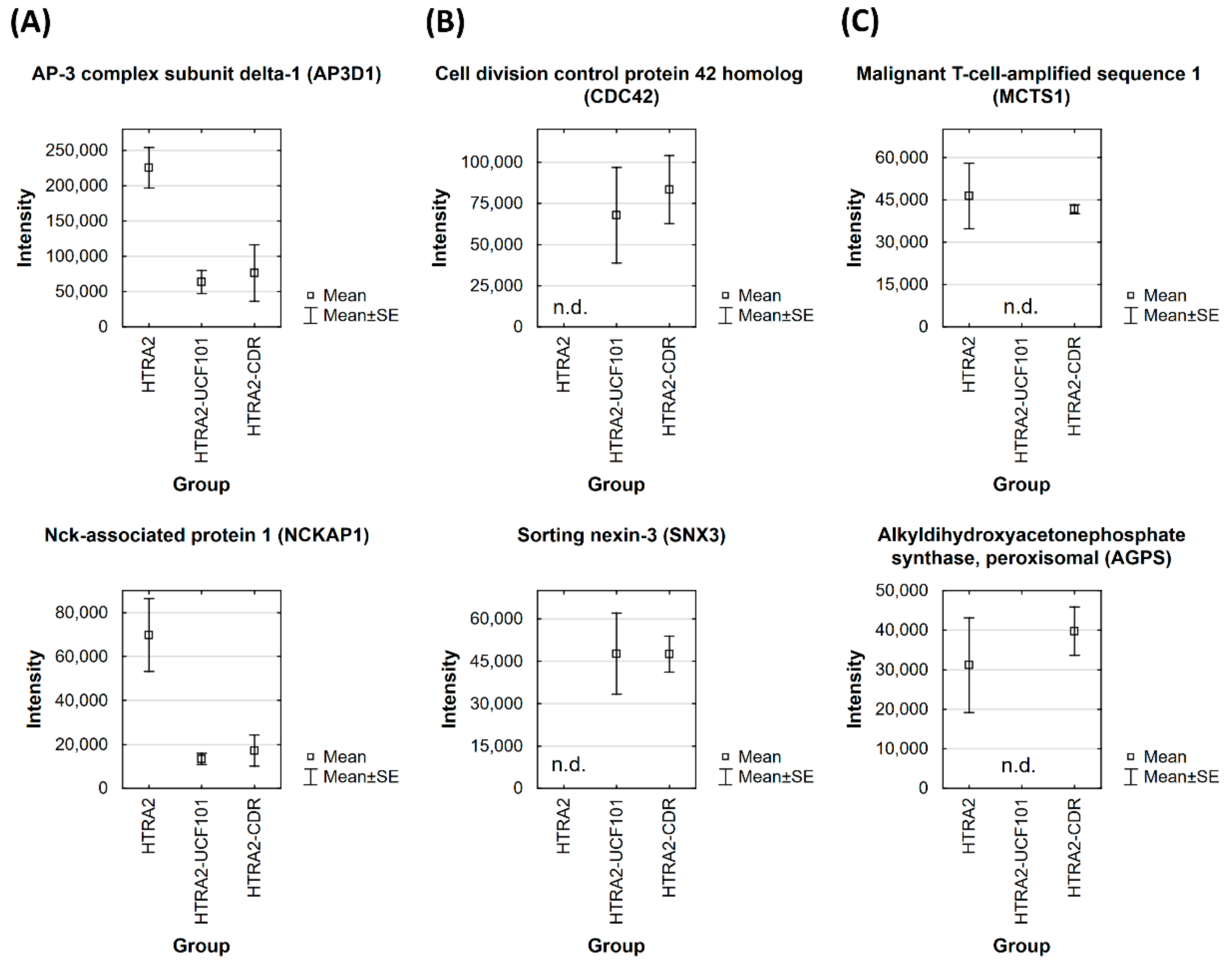
3.2. Identification of HTRA2-Specific Protein Interaction Partners in Retinal Tissues
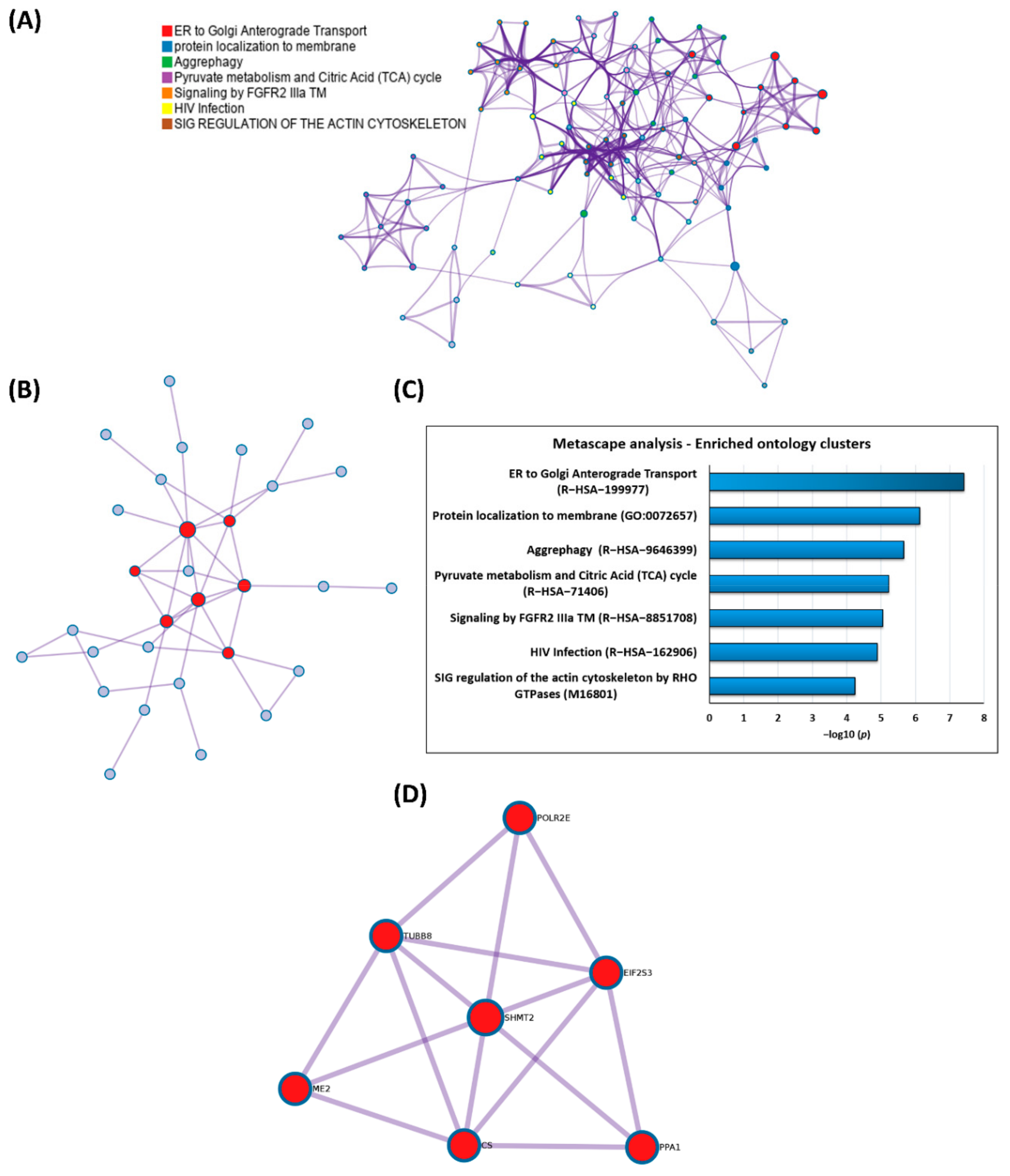
3.3. GO Enrichment and Pathway Analysis of the HTRA2-Specific Retinal Interactome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins, L.M.; Turk, B.E.; Cowling, V.; Borg, A.; Jarrell, E.T.; Cantley, L.C.; Downward, J. Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J. Biol. Chem. 2003, 278, 49417–49427. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Appleton, B.A.; Wu, P.; Wiesmann, C.; Sidhu, S.S. Structural and functional analysis of the ligand specificity of the HtrA2/Omi PDZ domain. Protein Sci. 2007, 16, 1738–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeleń, F.; Oleksy, A.; Smietana, K.; Otlewski, J. PDZ domains—Common players in the cell signaling. Acta Biochim. Pol. 2003, 50, 985–1017. [Google Scholar] [CrossRef] [Green Version]
- Faccio, L.; Fusco, C.; Chen, A.; Martinotti, S.; Bonventre, J.V.; Zervos, A.S. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J. Biol. Chem. 2000, 275, 2581–2588. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Alnemri, E.S. A novel role for the mitochondrial HTRA2/OMI protease in aging. Autophagy 2013, 9, 420–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, Y.-M.; Choi, J.-Y.; Park, H.-J.; Kim, K.-J.; Ahn, S.-G.; Seong, G.-H.; Kim, I.-K.; Kang, S.; Rhim, H. Autocatalytic processing of HtrA2/Omi is essential for induction of caspase-dependent cell death through antagonizing XIAP. J. Biol. Chem. 2004, 279, 37588–37596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Cilenti, L.; Lee, Y.; Hess, S.; Srinivasula, S.; Park, K.M.; Junqueira, D.; Davis, H.; Bonventre, J.V.; Alnemri, E.S.; Zervos, A.S. Characterization of a Novel and Specific Inhibitor for the Pro-apoptotic Protease Omi/HtrA2. J. Biol. Chem. 2003, 278, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.M.; Datta, P.; Srinivasula, S.M.; Ji, W.; Gupta, S.; Zhang, Z.; Davies, E.; Hajnoczky, G.; Saunders, T.L.; van Keuren, M.L.; et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003, 425, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Hu, Q.; Wang, H.; Man, N.; Ren, H.; Wen, L.; Nukina, N.; Fei, E.; Wang, G. Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ. 2010, 17, 1773–1784. [Google Scholar] [CrossRef]
- Xu, J.; Jiao, K.; Liu, X.; Sun, Q.; Wang, K.; Xu, H.; Zhang, S.; Wu, Y.; Wu, L.; Liu, D.; et al. Omi/HtrA2 Participates in Age-Related Autophagic Deficiency in Rat Liver. Aging Dis. 2018, 9, 1031. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, M.; Behbahani, H.; Gellhaar, S.; Forsell, C.; Belin, A.C.; Anvret, A.; Zettergren, A.; Nissbrandt, H.; Lind, C.; Sydow, O.; et al. Altered enzymatic activity and allele frequency of OMI/HTRA2 in Alzheimer’s disease. FASEB J. 2011, 25, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Darreh-Shori, T.; Rezaeianyazdi, S.; Lana, E.; Mitra, S.; Gellerbring, A.; Karami, A.; Bogdanovic, N.; Lithner, C.U.; Winblad, B.; Behbahani, H. Increased Active OMI/HTRA2 Serine Protease Displays a Positive Correlation with Cholinergic Alterations in the Alzheimer’s Disease Brain. Mol. Neurobiol. 2019, 56, 4601–4619. [Google Scholar] [CrossRef] [Green Version]
- Schmelter, C.; Fomo, K.N.; Perumal, N.; Manicam, C.; Bell, K.; Pfeiffer, N.; Grus, F.H. Synthetic Polyclonal-Derived CDR Peptides as an Innovative Strategy in Glaucoma Therapy. J. Clin. Med. 2019, 8, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelter, C.; Perumal, N.; Funke, S.; Bell, K.; Pfeiffer, N.; Grus, F.H. Peptides of the variable IgG domain as potential biomarker candidates in primary open-angle glaucoma (POAG). Hum. Mol. Genet. 2017, 26, 4451–4464. [Google Scholar] [CrossRef]
- Bell, K.; Wilding, C.; Funke, S.; Perumal, N.; Beck, S.; Wolters, D.; Holz-Muller, J.; Pfeiffer, N.; Grus, F.H. Neuroprotective effects of antibodies on retinal ganglion cells in an adolescent retina organ culture. J. Neurochem. 2016, 139, 256–269. [Google Scholar] [CrossRef]
- Perumal, N.; Strassburger, L.; Schmelter, C.; Gericke, A.; Pfeiffer, N.; Grus, F.H.; Manicam, C. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. J. Vis. Exp. 2019, 2, e59140. [Google Scholar] [CrossRef] [Green Version]
- Schmelter, C.; Funke, S.; Treml, J.; Beschnitt, A.; Perumal, N.; Manicam, C.; Pfeiffer, N.; Grus, F.H.; Grus, F. Comparison of Two Solid-Phase Extraction (SPE) Methods for the Identification and Quantification of Porcine Retinal Protein Markers by LC-MS/MS. Int. J. Mol. Sci. 2018, 19, 3847. [Google Scholar] [CrossRef] [Green Version]
- Perumal, N.; Funke, S.; Pfeiffer, N.; Grus, F.H. Characterization of lacrimal proline-rich protein 4 (PRR4) in human tear proteome. Proteomics 2014, 14, 1698–1709. [Google Scholar] [CrossRef]
- Perumal, N.; Straßburger, L.; Herzog, D.P.; Müller, M.B.; Pfeiffer, N.; Grus, F.H.; Manicam, C. Bioenergetic shift and actin cytoskeleton remodelling as acute vascular adaptive mechanisms to angiotensin II in murine retina and ophthalmic artery. Redox Biol. 2020, 34, 101597. [Google Scholar] [CrossRef] [PubMed]
- Perumal, N.; Funke, S.; Wolters, D.; Pfeiffer, N.; Grus, F.H. Characterization of human reflex tear proteome reveals high expression of lacrimal proline-rich protein 4 (PRR4). Proteomics 2015, 15, 3370–3381. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, E.; Danielsen, M.; Larsen, K.; Bendixen, C. Advances in porcine genomics and proteomics—A toolbox for developing the pig as a model organism for molecular biomedical research. Brief. Funct. Genom. 2010, 9, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Guard, S.E.; Ebmeier, C.C.; Old, W.M. Label-Free Immunoprecipitation Mass Spectrometry Workflow for Large-scale Nuclear Interactome Profiling. J. Vis. Exp. 2019, 153, e60432. [Google Scholar] [CrossRef]
- Bach, M.; Lehmann, A.; Brunnert, D.; Vanselow, J.T.; Hartung, A.; Bargou, R.C.; Holzgrabe, U.; Schlosser, A.; Chatterjee, M. Ugi Reaction-Derived alpha-Acyl Aminocarboxamides Bind to Phosphatidylinositol 3-Kinase-Related Kinases, Inhibit HSF1-Dependent Heat Shock Response, and Induce Apoptosis in Multiple Myeloma Cells. J. Med. Chem. 2017, 60, 4147–4160. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Keshishian, H.; Chang, B.; Addona, T.A.; Gillette, M.A.; Carr, S.A. Accurate inclusion mass screening: A bridge from unbiased discovery to targeted assay development for biomarker verification. Mol. Cell. Proteom. 2008, 7, 1952–1962. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A Serine Protease, HtrA2, Is Released from the Mitochondria and Interacts with XIAP, Inducing Cell Death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- Su, X.J.; Huang, L.; Qu, Y.; Mu, D. Progress in research on the role of Omi/HtrA2 in neurological diseases. Rev. Neurosci. 2019, 30, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.; Siegelin, M.D.; Dehghani, F.; Cilenti, L.; Zervos, A.S.; Rami, A. The serine protease Omi/HtrA2 is involved in XIAP cleavage and in neuronal cell death following focal cerebral ischemia/reperfusion. Neurochem. Int. 2007, 50, 172–180. [Google Scholar] [CrossRef]
- Bejugam, P.R.; Kuppili, R.R.; Singh, N.; Gadewal, N.; Chaganti, L.K.; Sastry, G.M.; Bose, K. Allosteric regulation of serine protease HtrA2 through novel non-canonical substrate binding pocket. PLoS ONE 2013, 8, e55416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuiyan, M.S.; Fukunaga, K. Inhibition of HtrA2/Omi ameliorates heart dysfunction following ischemia/reperfusion injury in rat heart in vivo. Eur. J. Pharmacol. 2007, 557, 168–177. [Google Scholar] [CrossRef]
- Zhang, C.; He, A.; Liu, S.; He, Q.; Luo, Y.; He, Z.; Chen, Y.; Tao, A.; Yan, J. Inhibition of HtrA2 alleviated dextran sulfate sodium (DSS)-induced colitis by preventing necroptosis of intestinal epithelial cells. Cell Death Dis. 2019, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Klupsch, K.; Downward, J. The protease inhibitor Ucf-101 induces cellular responses independently of its known target, HtrA2/Omi. Cell Death Differ. 2006, 13, 2157–2159. [Google Scholar] [CrossRef] [PubMed]
- Zurawa-Janicka, D.; Skorko-Glonek, J.; Lipinska, B. HtrA proteins as targets in therapy of cancer and other diseases. Expert Opin. Ther. Targets 2010, 14, 665–679. [Google Scholar] [CrossRef]
- Vande Walle, L.; van Damme, P.; Lamkanfi, M.; Saelens, X.; Vandekerckhove, J.; Gevaert, K.; Vandenabeele, P. Proteome-wide Identification of HtrA2/Omi Substrates. J. Proteome Res. 2007, 6, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.A.; Oorschot, V.; Hesser, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.; Seong, E.; Burmeister, M.; Faundez, V. Neuronal and non-neuronal functions of the AP-3 sorting machinery. J. Cell Sci. 2007, 120, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, A.J.; Yacoubian, T.A.; Slone, S.R.; Caldwell, K.A.; Caldwell, G.A. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci. 2012, 32, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- Griffin, E.F.; Yan, X.; Caldwell, K.A.; Caldwell, G.A. Distinct functional roles of Vps41-mediated neuroprotection in Alzheimer’s and Parkinson’s disease models of neurodegeneration. Hum. Mol. Genet. 2018, 27, 4176–4193. [Google Scholar] [CrossRef]
- Ammann, S.; Schulz, A.; Krägeloh-Mann, I.; Dieckmann, N.M.G.; Niethammer, K.; Fuchs, S.; Eckl, K.M.; Plank, R.; Werner, R.; Altmüller, J. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood 2016, 127, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baguma-Nibasheka, M.; Kablar, B. Altered retinal cell differentiation in the AP-3 delta mutant (Mocha) mouse. Int. J. Dev. Neurosci. 2009, 27, 701–708. [Google Scholar] [CrossRef]
- Yamamoto, A.; Behl, C. Human Nck-associated protein1 and its binding protein affect the metabolism of beta-amyloid precursor protein with Swedish mutation. Neurosci. Lett. 2001, 316, 50–54. [Google Scholar] [CrossRef]
- Yamamoto, A.; Suzuki, T.; Sakaki, Y. Isolation of hNap1BP which interacts with human Nap1 (NCKAP1) whose expression is down-regulated in Alzheimer’s disease. Gene 2001, 271, 159–169. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Liao, H.; Chen, H.; Deng, S.; Jia, Y.; Deng, C.; Lin, J.; Ge, J.; Zhuo, Y. Elevated intraocular pressure induces amyloid-β deposition and tauopathy in the lateral geniculate nucleus in a monkey model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5434–5443. [Google Scholar] [CrossRef] [Green Version]
- Noh, M.-Y.; Kwon, M.-S.; Oh, K.-W.; Nahm, M.; Park, J.; Kim, Y.-E.; Ki, C.-S.; Jin, H.K.; Bae, J.; Kim, S.H. Defective phagocytic function of induced microglia-like cells is correlated with rapid progression of sporadic ALS. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Higham, J.P.; Malik, B.R.; Buhl, E.; Dawson, J.M.; Ogier, A.S.; Lunnon, K.; Hodge, J.J.L. Alzheimer’s disease associated genes ankyrin and tau cause shortened lifespan and memory loss in Drosophila. Front. Cell. Neurosci. 2019, 13, 260. [Google Scholar] [CrossRef]
- Wang, H.Y.; Bakshi, K.; Frankfurt, M.; Stucky, A.; Goberdhan, M.; Shah, S.M.; Burns, L.H. Reducing amyloid-related Alzheimer’s disease pathogenesis by a small molecule targeting filamin A. J. Neurosci. 2012, 32, 9773–9784. [Google Scholar] [CrossRef] [Green Version]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Des, G.P.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S Proteasomes in Mouse Brain Neurons Causes Neurodegeneration and Lewy-Like Inclusions Resembling Human Pale Bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Graham, K.; Foote, M.; Liang, F.; Rizkallah, R.; Hurt, M.; Wang, Y.; Wu, Y.; Zhou, Y. 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 2013, 126, 4173–4186. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, C.; Zhang, H.; Wu, R. Autophagy in glaucoma: Crosstalk with apoptosis and its implications. Brain Res. Bull. 2015, 117, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adornetto, A.; Parisi, V.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G.; Tonin, P.; Russo, R. The Role of Autophagy in Glaucomatous Optic Neuropathy. Front. Cell Dev. Biol. 2020, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [Green Version]
- Govek, E.-E.; Wu, Z.; Acehan, D.; Molina, H.; Rivera, K.; Zhu, X.; Fang, Y.; Tessier-Lavigne, M.; Hatten, M.E. Cdc42 Regulates Neuronal Polarity during Cerebellar Axon Formation and Glial-Guided Migration. iScience 2018, 1, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Wirth, A.; Ponimaskin, E. Cdc42: An important regulator of neuronal morphology. Int. J. Biochem. Cell Biol. 2012, 44, 447–451. [Google Scholar] [CrossRef]
- Heynen, S.R.; Meneau, I.; Caprara, C.; Samardzija, M.; Imsand, C.; Levine, E.M.; Grimm, C. CDC42 is required for tissue lamination and cell survival in the mouse retina. PLoS ONE 2013, 8, e53806. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Baek, J.I.; Zuo, X.; Kim, S.H.; Dunaief, J.L.; Lipschutz, J.H. Cdc42 and sec10 Are Required for Normal Retinal Development in Zebrafish. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3361–3370. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Wu, K.; Lin, X.; Liu, Q.; Ye, Y.; Yu, M. Dexamethasone increases Cdc42 expression in human TM-1 cells. Curr. Eye Res. 2015, 40, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Shei, W.; Chan, A.S.; Chua, B.-T.; Goh, S.-R.; Chong, Y.-F.; Hilmy, M.H.; Nongpiur, M.E.; Baskaran, M.; Khor, C.-C.; et al. Primary angle closure glaucoma (PACG) susceptibility gene PLEKHA7 encodes a novel Rac1/Cdc42 GAP that modulates cell migration and blood-aqueous barrier function. Hum. Mol. Genet. 2017, 26, 4011–4027. [Google Scholar] [CrossRef]
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular matrix in the trabecular meshwork: Intraocular pressure regulation and dysregulation in glaucoma. Exp. Eye Res. 2015, 133, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Tanna, A.P.; Johnson, M. Rho Kinase Inhibitors as a Novel Treatment for Glaucoma and Ocular Hypertension. Ophthalmology 2018, 125, 1741–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrino, E.; Supuran, C.T. Rho-kinase inhibitors in the management of glaucoma. Expert Opin. Ther. Pat. 2019, 29, 817–827. [Google Scholar] [CrossRef]
- Lucas, M.; Gershlick, D.C.; Vidaurrazaga, A.; Rojas, A.L.; Bonifacino, J.S.; Hierro, A. Structural mechanism for cargo recognition by the retromer complex. Cell 2016, 167, 1623–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, N.; Bessa, C.; Rodrigues, A.J.; Marques, P.; Chan, F.-Y.; Carvalho, A.X.d.; Correia-Neves, M.; Sousa, N. Sorting nexin 3 mutation impairs development and neuronal function in Caenorhabditis elegans. Cell. Mol. Life Sci. 2018, 75, 2027–2044. [Google Scholar] [CrossRef]
- Xu, S.; Nigam, S.M.; Brodin, L. Overexpression of SNX3 decreases amyloid-β peptide production by reducing internalization of amyloid precursor protein. Neurodegener. Dis. 2018, 18, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K. Serine catabolism by SHMT2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Chen, G.; Aras, S.; Wang, J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci. Alliance 2018, 1, e201800036. [Google Scholar] [CrossRef]
- Ning, S.; Ma, S.; Saleh, A.Q.; Guo, L.; Zhao, Z.; Chen, Y. SHMT2 overexpression predicts poor prognosis in intrahepatic cholangiocarcinoma. Gastroenterol. Res. Pract. 2018, 2018, 4369253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Krieger, J.R.; Taylor, P.; Bagshaw, R.; Kang, J.; Jeedigunta, S.; Wybenga-Groot, L.E.; Zhang, W.; Badr, H.; Mirhadi, S. Cancer proteome and metabolite changes linked to SHMT2. PLoS ONE 2020, 15, e0237981. [Google Scholar] [CrossRef] [PubMed]
- Escande-Beillard, N.; Loh, A.; Saleem, S.N.; Kanata, K.; Hashimoto, Y.; Altunoglu, U.; Metoska, A.; Grandjean, J.; Ng, F.M.; Pomp, O.; et al. Loss of PYCR2 Causes Neurodegeneration by Increasing Cerebral Glycine Levels via SHMT2. Neuron 2020, 107, 82–94. [Google Scholar] [CrossRef] [PubMed]
- García-Cazorla, À.; Verdura, E.; Juliá-Palacios, N.; Anderson, E.N.; Goicoechea, L.; Planas-Serra, L.; Tsogtbaatar, E.; Dsouza, N.R.; Schlüter, A.; Urreizti, R. Impairment of the mitochondrial one-carbon metabolism enzyme SHMT2 causes a novel brain and heart developmental syndrome. Acta Neuropathol. 2020, 140, 971–975. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Burchell, V.S.; Holmström, K.M.; Yao, Z.; Deas, E.; Cain, K.; Fedele, V.; Moisoi, N.; Campanella, M.; Miguel Martins, L.; et al. HtrA2 deficiency causes mitochondrial uncoupling through the F1F0-ATP synthase and consequent ATP depletion. Cell Death Dis. 2012, 3, e335. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Major Protein ID | Protein Name | Gene Name | Score | Peptides | Unique Peptides | Sequence Coverage [%] | Mol. Weight [kDa] | HTRA2 | HTRA2 + UCF-101 | HTRA2 + CDR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Q01484 | Ankyrin-2 | ANK2 | 48.2 | 14 | 14 | 4.7 | 433.7 | √ | - | - |
| 2 | O95782 | AP-2 complex subunit alpha-1 | AP2A1 | 18.1 | 8 | 4 | 8.9 | 107.5 | √ | - | - |
| 3 | O14617 | AP-3 complex subunit delta-1 | AP3D1 | 47.0 | 12 | 12 | 13.9 | 130.2 | √ | - | - |
| 4 | P00889 | Citrate synthase, mitochondrial | CS | 6.4 | 3 | 3 | 7.8 | 51.6 | √ | - | - |
| 5 | Q96EY1 | DnaJ homolog subfamily A member 3, mitochondrial | DNAJA3 | 22.6 | 3 | 3 | 6 | 52.5 | √ | - | - |
| 6 | Q8TEA8 | D-aminoacyl-tRNA deacylase 1 | DTD1 | 4.6 | 1 | 1 | 7.2 | 23.4 | √ | - | - |
| 7 | P41091 | Eukaryotic translation initiation factor 2 subunit 3 | EIF2S3 | 6.4 | 4 | 4 | 13.3 | 51.1 | √ | - | - |
| 8 | Q9NZY2 | Putative uncharacterized protein FAM30A | FAM30A | 3.6 | 1 | 1 | 10.4 | 14.6 | √ | - | - |
| 9 | P21333 | Filamin-A | FLNA | 10.8 | 3 | 3 | 1.7 | 280.7 | √ | - | - |
| 10 | P36915 | Guanine nucleotide-binding protein-like 1 | GNL1 | 25.8 | 6 | 6 | 11.2 | 68.7 | √ | - | - |
| 11 | Q16775 | Hydroxyacylglutathione hydrolase, mitochondrial | HAGH | 3.6 | 2 | 2 | 6.5 | 33.8 | √ | - | - |
| 12 | Q9Y4L1 | Hypoxia up-regulated protein 1 | HYOU1 | 24.9 | 6 | 6 | 7.9 | 111.3 | √ | - | - |
| 13 | P23368 | NAD-dependent malic enzyme, mitochondrial | ME2 | 7.2 | 3 | 3 | 6.5 | 65.4 | √ | - | - |
| 14 | Q9Y2A7 | Nck-associated protein 1 | NCKAP1 | 33.0 | 13 | 13 | 12.7 | 128.8 | √ | - | - |
| 15 | Q6VY07 | Phosphofurin acidic cluster sorting protein 1 | PACS1 | 8.3 | 2 | 2 | 2.5 | 104.9 | √ | - | - |
| 16 | Q01082 | Spectrin beta chain, non-erythrocytic 1 | SPTBN1 | 38.1 | 9 | 9 | 5 | 274.6 | √ | - | - |
| 17 | Q6S8J3 | POTE ankyrin domain family member E | POTEE | 3.6 | 9 | 1 | 10.4 | 121.4 | √ | - | - |
| 18 | P62191 | 26S proteasome regulatory subunit 4 | PSMC1 | 3.0 | 1 | 1 | 2.7 | 49.2 | √ | n.d. | - |
| 19 | P46405 | 40S ribosomal protein S12 | RPS12 | 7.1 | 5 | 5 | 49.2 | 14.5 | √ | n.d. | - |
| 20 | P55735 | Protein SEC13 homolog | SEC13 | 4.5 | 2 | 2 | 8.1 | 35.5 | √ | n.d. | n.d. |
| 21 | P11493 | Serine/threonine-protein phosphatase 2A catalytic subunit beta isoform (Fragment) | PPP2CB | 3.8 | 1 | 1 | 3.8 | 33.6 | √ | n.d. | n.d. |
| 22 | Q92777 | Synapsin-2 | SYN2 | 9.3 | 6 | 4 | 12 | 63.0 | √ | n.d. | n.d. |
| 23 | P34897 | Serine hydroxymethyltransferase, mitochondrial | SHMT2 | 3.6 | 2 | 2 | 4.4 | 56.0 | √ | - | n.d. |
| 24 | P13984 | General transcription factor IIF subunit 2 | GTF2F2 | 9.7 | 4 | 4 | 20.1 | 28.4 | - | √ | - |
| 25 | Q95250 | Membrane-associated progesterone receptor component 1 | PGRMC1 | 10.8 | 2 | 2 | 17 | 21.6 | - | √ | - |
| 26 | Q15181 | Inorganic pyrophosphatase | PPA1 | 47.5 | 5 | 5 | 21.1 | 32.7 | - | √ | - |
| 27 | O14579 | Coatomer subunit epsilon | COPE | 3.5 | 1 | 1 | 5.5 | 34.5 | n.d. | √ | - |
| 28 | Q99747 | Gamma-soluble NSF attachment protein | NAPG | 22.6 | 5 | 5 | 18.9 | 34.7 | - | - | √ |
| 29 | O43660 | Pleiotropic regulator 1 | PLRG1 | 3.9 | 4 | 4 | 11.1 | 57.2 | - | - | √ |
| 30 | P19388 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | POLR2E | 21.5 | 2 | 2 | 12.4 | 24.6 | - | - | √ |
| 31 | Q8N474 | Secreted frizzled-related protein 1 | SFRP1 | 10.2 | 3 | 3 | 14 | 35.4 | - | - | √ |
| 32 | Q9BQE3 | Tubulin alpha-1C chain | TUBA1C | 2.6 | 22 | 1 | 64.6 | 49.9 | - | - | √ |
| 33 | Q5T653 | 39S ribosomal protein L2, mitochondrial | MRPL2 | 4.5 | 1 | 1 | 8.2 | 33.3 | n.d. | - | √ |
| 34 | Q2M3V2 | Ankyrin repeat domain-containing protein SOWAHA | SOWAHA | 2.5 | 1 | 1 | 1.5 | 57.4 | n.d. | - | √ |
| 35 | Q3ZCM7 | Tubulin beta-8 chain | TUBB8 | 14.5 | 11 | 2 | 28.2 | 49.8 | n.d. | - | √ |
| 36 | O43464 | Serine protease HTRA2, mitochondrial | HTRA2 | 323.3 | 18 | 18 | 52.8 | 48.8 | √ | √ | √ |
| 37 | Q15185 | Prostaglandin E synthase 3 | PTGES3 | 4.3 | 2 | 2 | 15.6 | 18.7 | √ | √ | √ |
| 38 | Q13404 | Ubiquitin-conjugating enzyme E2 variant 1 | UBE2V1 | 3.0 | 2 | 1 | 12.9 | 16.5 | √ | √ | √ |
| 39 | P09038 | Fibroblast growth factor 2 | FGF2 | 5.4 | 3 | 3 | 9.7 | 30.8 | - | √ | √ |
| 40 | P19130 | Ferritin heavy chain | FTH1 | 10.6 | 3 | 2 | 21.5 | 21.0 | - | √ | √ |
| 41 | Q6QA76 | PDZ domain-containing protein 11 | PDZD11 | 11.6 | 3 | 3 | 35.7 | 16.2 | - | √ | √ |
| 42 | Q007T2 | Cell division control protein 42 homolog | CDC42 | 19.3 | 4 | 4 | 22 | 21.3 | n.d. | √ | √ |
| 43 | O60493 | Sorting nexin-3 | SNX3 | 5.0 | 3 | 2 | 18.5 | 18.8 | n.d. | √ | √ |
| 44 | Q9Y5L4 | Mitochondrial import inner membrane translocase subunit Tim13 | TIMM13 | 7.1 | 1 | 1 | 14.7 | 10.5 | n.d. | √ | √ |
| 45 | P61088 | Ubiquitin-conjugating enzyme E2 N | UBE2N | 3.3 | 2 | 2 | 16.4 | 17.1 | - | √ | √ |
| 46 | Q13825 | Methylglutaconyl-CoA hydratase, mitochondrial | AUH | 8.2 | 5 | 5 | 19.2 | 35.6 | √ | - | √ |
| 47 | Q29290 | Cystatin-B | CSTB | 2.5 | 1 | 1 | 12.2 | 11.1 | √ | - | √ |
| 48 | P31150 | Rab GDP dissociation inhibitor alpha | GDI1 | 4.7 | 3 | 2 | 11.4 | 50.6 | √ | - | √ |
| 49 | Q04760 | Lactoylglutathione lyase | GLO1 | 3.3 | 2 | 2 | 12.5 | 20.8 | √ | - | √ |
| 50 | O00116 | Alkyldihydroxyacetonephosphate synthase, peroxisomal | AGPS | 10.3 | 1 | 1 | 2.6 | 72.9 | √ | n.d. | √ |
| 51 | Q9ULC4 | Malignant T-cell-amplified sequence 1 | MCTS1 | 4.5 | 1 | 1 | 10.5 | 20.6 | √ | n.d. | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmelter, C.; Fomo, K.N.; Perumal, N.; Pfeiffer, N.; Grus, F.H. Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues. Biomedicines 2021, 9, 1013. https://doi.org/10.3390/biomedicines9081013
Schmelter C, Fomo KN, Perumal N, Pfeiffer N, Grus FH. Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues. Biomedicines. 2021; 9(8):1013. https://doi.org/10.3390/biomedicines9081013
Chicago/Turabian StyleSchmelter, Carsten, Kristian Nzogang Fomo, Natarajan Perumal, Norbert Pfeiffer, and Franz H. Grus. 2021. "Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues" Biomedicines 9, no. 8: 1013. https://doi.org/10.3390/biomedicines9081013
APA StyleSchmelter, C., Fomo, K. N., Perumal, N., Pfeiffer, N., & Grus, F. H. (2021). Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues. Biomedicines, 9(8), 1013. https://doi.org/10.3390/biomedicines9081013