
IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications?


Abstract
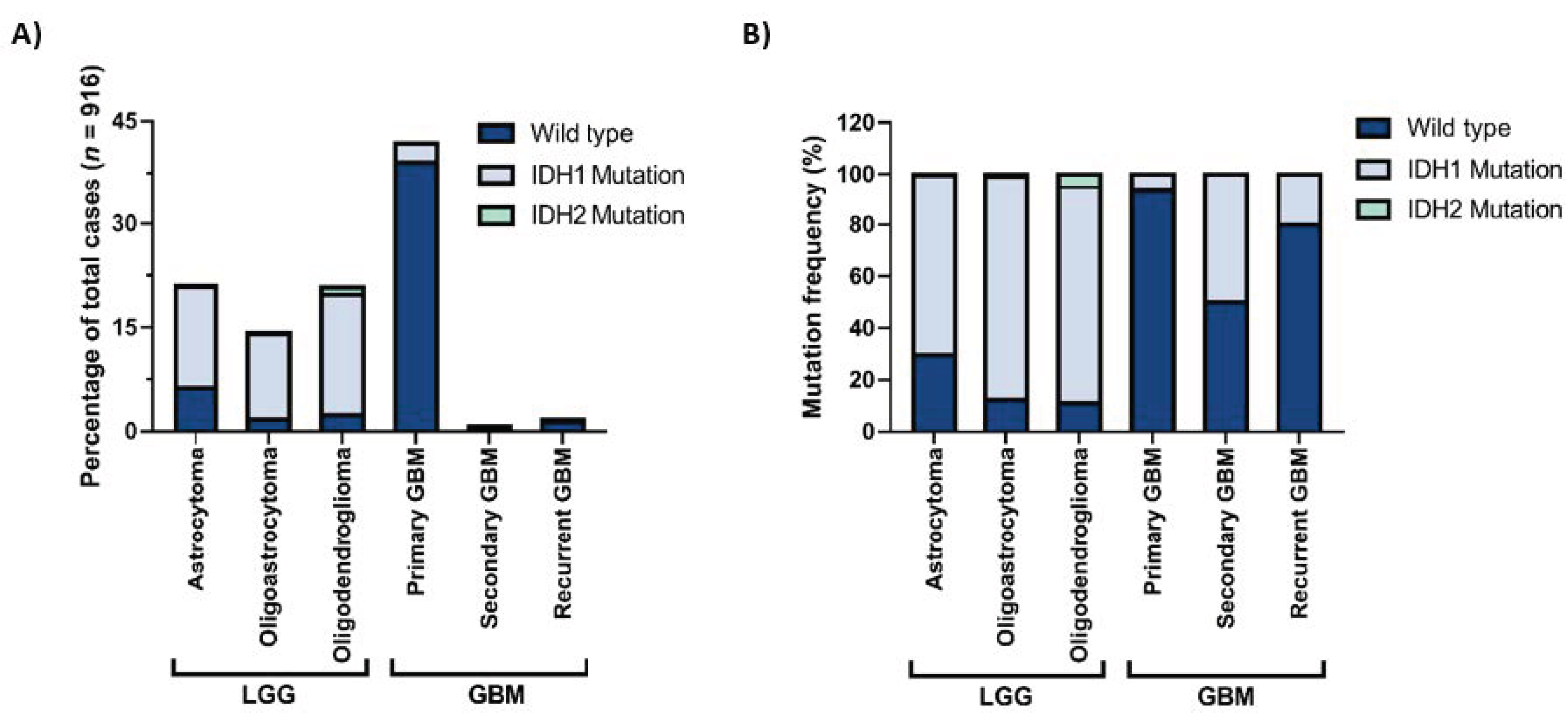
1. Introduction
2. Potential Therapeutic Approaches
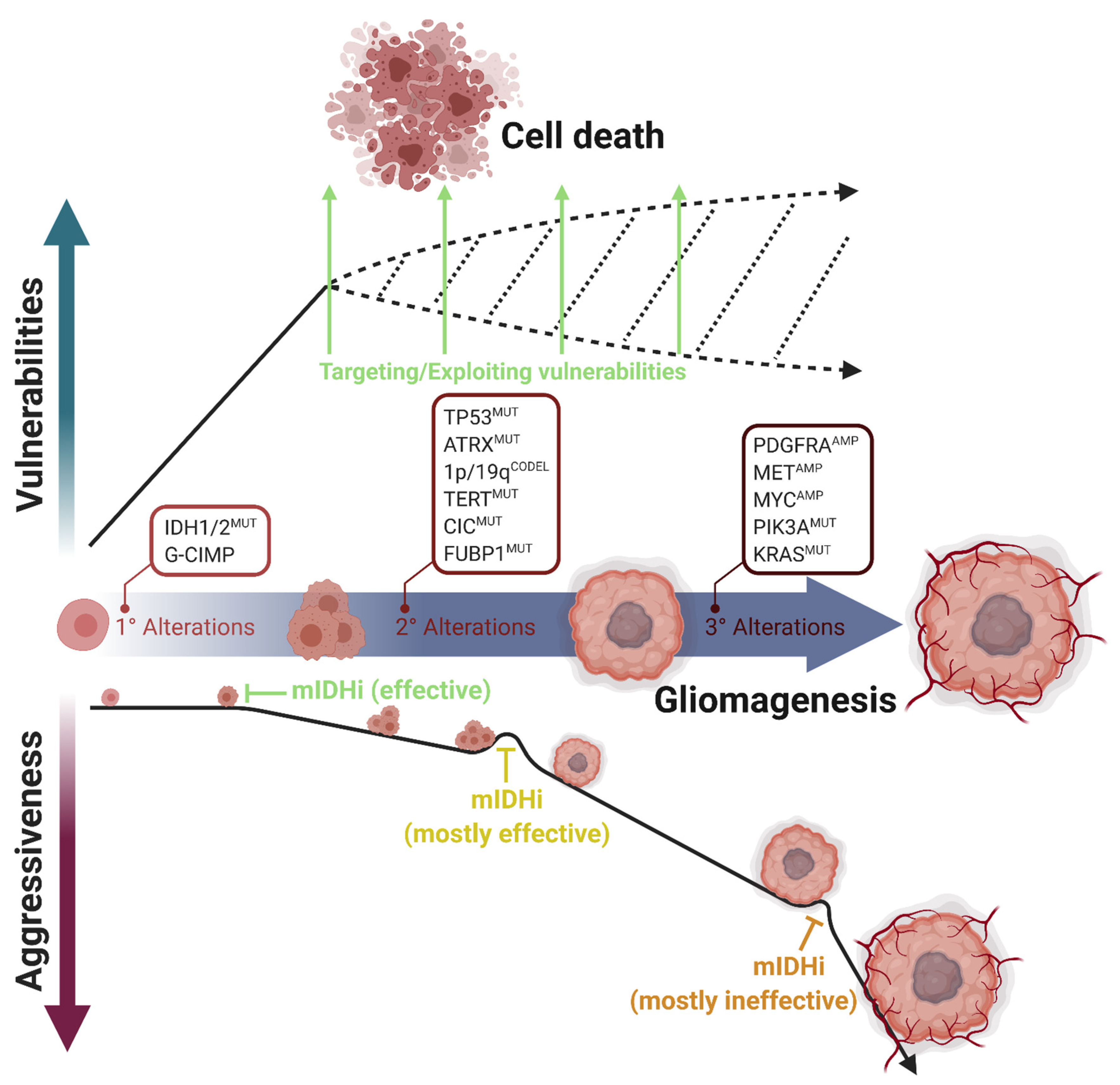
2.1. Mutant IDH Inhibitors
2.2. Targeting Metabolic Deficiencies
2.2.1. Lipid Metabolism
2.2.2. Amino Acid Metabolism (Glutamate/Glutamine/Glucose)
2.2.3. NAD+ Metabolism
2.2.4. Mitochondrial Metabolism and Oxidative Stress
2.2.5. Mammalian Target of Rapamycin (mTOR) Signaling
2.2.6. ER Stress
2.2.7. Hypoxia
2.3. Targeting the DNA Damage Pathway
2.3.1. Conventional Therapies (Temozolomide and Irradiation)
2.3.2. PARP-mediated DNA Repair
2.4. Immunotherapy
2.4.1. Peptide Vaccines
2.4.2. Immune Checkpoint Inhibitors
2.4.3. CAR T Cell Therapy
2.5. Epigenetic Approaches
2.5.1. DNA Demethylation
2.5.2. BET Inhibitors
2.5.3. Combination Treatments
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro-Oncology 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Hurley, J.H.; Dean, A.M.; Koshland, D.E.; Stroud, R.M. Catalytic Mechanism of NADP+-Dependent Isocitrate Dehydrogenase: Implications from the Structures of Magnesium-Isocitrate and NADP + Complexes. Biochemistry 1991, 30, 8671–8678. [Google Scholar] [CrossRef]
- Lee, S.M.; Koh, H.J.; Park, D.C.; Song, B.J.; Huh, T.L.; Park, J.W. Cytosolic NADP+-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Ramachandran, N.; Colman, R.F. Chemical characterization of distinct subunits of pig heart DPN-specific isocitrate dehy-drogenase. J. Biol. Chem. 1980, 255, 8859–8864. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.L.; Zervos, P.R.; Plaut, G.W.E. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and sub-strate concentrations approximating conditions in mitochondria. Metabolism 1986, 35, 661–667. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef]
- Ye, D.; Xiong, Y.; Guan, K.-L. The mechanisms of IDH mutations in tumorigenesis. Cell Res. 2012, 22, 1102–1104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Noorani, I. Genetically engineered mouse models of gliomas: Technological developments for translational discoveries. Cancers 2019, 11, 1335. [Google Scholar] [CrossRef]
- Philip, B.; Yu, D.X.; Silvis, M.R.; Shin, C.H.; Robinson, J.P.; Robinson, G.L.; Welker, A.E.; Angel, S.N.; Tripp, S.R.; Sonnen, J.A.; et al. Mutant IDH1 Promotes Glioma Formation In Vivo. Cell Rep. 2018, 23, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 Contribute to Human Oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Butterfield, Y.S.; Morozova, O.; Chittaranjan, S.; Blough, M.D.; An, J.; Birol, I.; Chesnelong, C.; Chiu, R.; Chuah, E.; et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J. Pathol. 2012, 226, 7–16. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Metellus, P.; Coulibaly, B.; Colin, C.; de Paula, A.M.; Vasiljevic, A.; Taieb, D.; Barlier, A.; Boisselier, B.; Mokhtari, K.; Wang, X.W.; et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010, 120, 719–729. [Google Scholar] [CrossRef]
- Noorani, I.; de la Rosa, J.; Choi, Y.H.; Strong, A.; Ponstingl, H.; Vijayabaskar, M.S.; Lee, J.; Lee, E.; Richard-Londt, A.; Frie-drich, M.; et al. PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas. Genome Biol. 2020, 21, 181. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of sec-ondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komi-sopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Okoye-Okafor, U.C.; Bartholdy, B.; Cartier, J.; Gao, E.N.; Pietrak, B.; Rendina, A.R.; Rominger, C.; Quinn, C.; Smallwood, A.; Wiggall, K.J.; et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.R.; Engstrom, A.; Zoeller, E.L.; Liu, X.; Shanks, J.R.; Zhang, X.; Johns, M.A.; Vertino, P.M.; Fu, H.; Cheng, X. Char-acterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J. Biol. Chem. 2016, 291, 2631–2646. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-Oncology 2014, 16, 686–695. [Google Scholar] [CrossRef]
- Kayabolen, A.; Sahin, G.N.; Seker-Polat, F.; Cingoz, A.; Isik, B.; Acar, S.; Wakimoto, H.; Cahill, D.P.; Solaroglu, I.; Cribbs, A.; et al. Combined inhibition of KDM6A/B and HDACs exacerbates integrated stress response and mediates therapeutic effects in IDH1-mutant glioma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kopinja, J.; Sevilla, R.S.; Levitan, D.; Dai, D.; Vanko, A.; Spooner, E.; Ware, C.; Forget, R.; Hu, K.; Kral, A.; et al. A Brain Pen-etrant Mutant IDH1 Inhibitor Provides in Vivo Survival Benefit. Sci. Rep. 2017, 7, 13853. [Google Scholar] [CrossRef] [PubMed]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.-C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A.; Turcan, S.; et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decita-bine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Luo, l.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.-L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pol-lyea, D.A.; et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.-C.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; de la Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in Isocitrate Dehydrogenase 1-Mutated Advanced Glioma. J. Clin. Oncol. 2020, 38, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Wakabayashi, T.; Miyakita, Y.; Narita, Y.; Mineharu, Y.; Arakawa, Y.; Yamasaki, F.; Sugiyama, K.; Hata, N.; Muragaki, Y.; et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. J. Clin. Oncol. 2019, 37, 2004. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schimmer, A.D.; Yee, K.W.L.; Hochhaus, A.; Kraemer, A.; Carvajal, R.D.; Janku, F.; Bedard, P.; Carpio, C.; Wick, A.; et al. A Phase I Study of IDH305 in Patients with Advanced Malignancies Including Relapsed/Refractory AML and MDS That Harbor IDH1R132 Mutations. Blood 2016, 128, 1073. [Google Scholar] [CrossRef]
- de la Fuente, M.I.; Colman, H.; Rosenthal, M.; van Tine, B.A.; Levaci, D.; Walbert, T.; Gan, H.K.; Vieito, M.; Milhem, M.M.; Lipford, K.; et al. A phase Ib/II study of olutasidenib in patients with relapsed/refractory IDH1 mutant gliomas: Safety and efficacy as single agent and in combination with azacitidine. J. Clin. Oncol. 2020, 38, 2505. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Erridge, S.; Vogelbaum, M.A.; Nowak, A.K.; Sanson, M.; Brandes, A.A.; Wick, W.; Clement, P.M.; Baurain, J.-F.; Mason, W.P.; et al. Second interim and first molecular analysis of the EORTC randomized phase III intergroup CAT-NON trial on concurrent and adjuvant temozolomide in anaplastic glioma without 1p/19q codeletion. J. Clin. Oncol. 2019, 37, 2000. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zecchini, V.; Frezza, C. Metabolic synthetic lethality in cancer therapy. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 723–731. [Google Scholar] [CrossRef]
- Jo, S.H.; Son, M.K.; Koh, H.J.; Lee, S.M.; Song, I.H.; Kim, Y.O.; Lee, Y.S.; Jeong, K.S.; Kim, W.B.; Park, J.W.; et al. Control of Mitochondrial Redox Balance and Cellular Defense against Oxidative Damage by Mitochondrial NADP+-dependent Isocitrate Dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.W.; Park, D.C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.L.; Metallo, C.M. Onco-genic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibro-sarcoma Sells. Cell Rep. 2018, 25, 1018–1026.e4. [Google Scholar] [CrossRef] [PubMed]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 Mutations Induce Organelle Defects Via Dysregulated Phospholipids. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dowdy, T.; Zhang, L.; Celiku, O.; Movva, S.; Lita, A.; Ruiz-Rodado, V.; Gilbert, M.R.; Larion, M. Sphingolipid pathway as a source of vulnerability in IDH1mut glioma. Cancers 2020, 12, 2910. [Google Scholar] [CrossRef]
- Newsholme, P.; Procopio, J.; Lima, M.M.R.; Pithon-Curi, T.C.; Curi, R. Glutamine and glutamate?their central role in cell metabolism and function. Cell Biochem. Funct. 2003, 21, 1–9. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Salamanca-Cardona, L.; Shah, H.; Poot, A.J.; Correa, F.M.; di Gialleonardo, V.; Lui, H.; Miloushev, V.Z.; Granlund, K.L.; Tee, S.S.; Cross, J.R.; et al. In Vivo Imaging of Glutamine Metabolism to the Oncometabolite 2-Hydroxyglutarate in IDH1/2 Mu-tant Tumors. Cell Metab. 2017, 26, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rab-inowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzi-vassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1 R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef]
- Burki, F.; Kaessmann, H. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet. 2004, 36, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Shashidharan, P.; Plaitakis, A. The discovery of human of GLUD2 glutamate dehydrogenase and its implications for cell function in health and disease. Neurochem. Res. 2014, 39, 460–470. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.A.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1 -Mutant Cancers and Poten-tiates NAD + Depletion–Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.J.; Fink, A.; Banagis, J.A.; Nagashima, H.; Subramanian, M.; Lee, C.K.; Melamed, L.; Tummala, S.S.; Tateishi, K.; Wakimoto, H.; et al. Sirtuin activation targets IDH-mutant tumors. Neuro-Oncology 2020, 23, 53–62. [Google Scholar] [CrossRef]
- Nagashima, H.; Lee, C.K.; Tateishi, K.; Higuchi, F.; Subramanian, M.; Rafferty, S.; Melamed, L.; Miller, J.J.; Wakimoto, H.; Cahill, D.P. Poly(ADP-ribose) Glycohydrolase Inhibition Sequesters NAD+ to Potentiate the Metabolic Lethality of Alkyl-ating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov. 2020, 10, 1672–1689. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.-T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef]
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-hydroxyglutarate inhibits ATP synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Zhang, Y.; Shu, C.; Tsujiuchi, T.; Banu, M.A.; Garcia, F.; Roth, K.A.; Bruce, J.N.; et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat. Commun. 2017, 8, 1067. [Google Scholar] [CrossRef]
- Carbonneau, M.; Gagne, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Batsios, G.; Viswanath, P.; Subramani, E.; Najac, C.; Gillespie, A.M.; Santos, R.D.; Molloy, A.R.; Pieper, R.O.; Ronen, S.M. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci. Rep. 2019, 9, 10521. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable Signaling Pathway Mutations Are Associated with Malignant Phenotype in IDH-Mutant Gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef]
- Fan, Q.-W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 ki-nase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Puli, S.; Jain, A.; Lai, J.C.K.; Bhushan, A. Effect of combination treatment of rapamycin and isoflavones on mtor pathway in human glioblastoma (U87) cells. Neurochem. Res. 2010, 35, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; Mccracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Ong, W.Q.; Luchman, H.A.; Cairncross, J.G.; Huang, B.; Pieper, R.O.; Phil-lips, J.J.; Ronen, S.M. 2-Hydroxyglutarate-Mediated Autophagy of the Endoplasmic Reticulum Leads To an Unusual Down-regulation of Phospholipid Biosynthesis in Mutant IDH1 Gliomas. Cancer Res. 2018, 78, 2290–2304. [Google Scholar] [CrossRef]
- Zhang, Y.; Pusch, S.; Innes, J.; Sidlauskas, K.; Ellis, M.; Lau, J.; El-Hassan, T.; Aley, N.; Launchbury, F.; Richard-Loendt, A.; et al. Mutant IDH sensitizes gliomas to endoplasmic reticulum stress and triggers apoptosis via miR-183-mediated inhibition of semaphorin 3E. Cancer Res. 2019, 79, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Yalaza, C.; Ak, H.; Cagli, M.S.; Ozgiray, E.; Atay, S.; Aydin, H.H. R132H mutation in IDH1 gene is associated with increased tumor HIF1-alpha and serum VEGF levels in primary glioblastoma multiforme. Ann. Clin. Lab. Sci. 2017, 47, 362–364. [Google Scholar]
- Jensen, R.L. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg. Focus 2006, 20, E24. [Google Scholar] [CrossRef]
- Gillespie, D.L.; Whang, K.; Ragel, B.T.; Flynn, J.R.; Kelly, D.A.; Jensen, R.L. Silencing of hypoxia inducible factor-1α by RNA interference attenuates human glioma cell growth in vivo. Clin. Cancer Res. 2007, 13, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Nakagawa, K.; Harada, H.; Nagato, S.; Furukawa, K.; Teraoka, M.; Seno, T.; Oka, K.; Iwata, S.; Ohnishi, T. Si-lencing hypoxia-inducible factor-1α inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int. J. Oncol. 2007, 30, 793–802. [Google Scholar] [PubMed]
- Tang, J.H.; Ma, Z.X.; Huang, G.H.; Xu, Q.F.; Xiang, Y.; Li, N.; Sidlauskas, K.; Zhang, E.E.; Lv, S.Q. Downregulation of HIF-1a sensitizes U251 glioma cells to the temozolomide (TMZ) treatment. Exp. Cell Res. 2016, 343, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pi-etsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblas-tomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; DeMatteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Lai-gle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade glio-mas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Dubbink, H.J.; Marie, Y.; Brandes, A.A.; Taphoorn, M.J.B.; Wesseling, P.; Frenay, M.; Tijssen, C.C.; Lacombe, D.; Idbaih, A.; et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oli-godendroglial tumors: A report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin. Cancer Res. 2010, 16, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chou, A.P.; Chen, W.; Chen, R.; Deng, Y.; Phillips, H.S.; Selfridge, J.; Zurayk, M.; Lou, J.J.; Everson, R.G.; et al. Overex-pression of isocitrate dehydrogenase 1/2 (IDH1/2) mutant protein renders glioma cells more sensitive to radiation. Neuro-Oncology 2013, 15, 57–68. [Google Scholar] [CrossRef]
- Tran, A.N.; Lai, A.; Li, S.; Pope, W.B.; Teixeira, S.; Harris, R.J.; Woodworth, D.C.; Nghiemphu, P.L.; Cloughesy, T.F.; El-lingson, B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantita-tive MR volumetry. Neuro-Oncology 2014, 16, 414–420. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; el Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus. 2015, 38, E6. [Google Scholar] [CrossRef]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Target-ing therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vac-cine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Muta-tional burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint im-munotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vac-cine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017, 19, 1460–1468. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Núñez, F.J.; Liu, Y.; Yu, M.; Taher, A.W.; et al. Inhibition of 2-Hydroxyglutarate Elicits Metabolic-reprograming and Mutant IDH1 Glioma Immunity in Mice. J. Clin. Invest. 2020. [Google Scholar] [CrossRef]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patanè, M.; Zucca, I.; Sirtori, F.R.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef]
- Platten, M.; Schilling, D.; Bunse, L.; Wick, A.; Bunse, T.; Riehl, D.; Green, E.; Sanghvi, K.; Karapanagiotou-Schenkel, I.; Hart-ing, I.; et al. ATIM-33. NOA-16: A first-in-man multicenter phase i clinical trial of the german neurooncology working group evaluating a mutation-specific peptide vaccine targeting idh1r132h in patients with newly diagnosed malignant as-trocytomas. Neuro-Oncology 2018, 20, vi8–vi9. [Google Scholar] [CrossRef]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Senhaji, N.; Louati, S.; Chbani, L.; el Fatemi, H.; Hammas, N.; Mikou, K.; Maaroufi, M.; Benzagmout, M.; Boujraf, S.; el Bardai, S.; et al. EGFR Amplification and IDH Mutations in Glioblastoma Patients of the Northeast of Morocco. Biomed Res. Int. 2017. [Google Scholar] [CrossRef]
- Taher, M.M.; Dairi, G.; Butt, E.M.; Al-Quthami, K.; Al-Khalidi, H.; Jastania, R.A.; Nageeti, T.H.; Bogari, N.M.; Athar, M.; Al-Allaf, F.A.; et al. EGFRvIII expression and isocitrate dehydrogenase mutations in patients with glioma. Oncol. Lett. 2020, 20, 384. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, J.; Wen, X.; Xu, B.; Que, Y.; Yu, K.; Xu, L.; Zhao, J.; Pan, Q.; Zhou, P.; et al. Chimeric antigen recep-tor-modified T-cell therapy for platelet-derived growth factor receptor α-positive rhabdomyosarcoma. Cancer 2020, 126, 2093–2100. [Google Scholar] [CrossRef]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.M.; Baia, G.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a pa-tient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.S.; Rosa, M.d.; Borodovsky, A.; Festuccia, W.T.; Chan, T.; Riggins, G.J. Demethylation and epigenetic modifica-tion with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro-Oncology 2019, 21, 189–200. [Google Scholar] [CrossRef]
- Oganesian, A.; Redkar, S.; Taverna, P.; Joshi-Hangal, R.; Azab, M. Preclinical data in cynomolgus (cyn) monkeys of ASTX727, a novel oral hypomethylating agent (HMA) composed of low-dose oral decitabine combined with a novel cytidine deami-nase inhibitor (CDAi) E7727. Blood 2013, 122, 2526. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic random-ized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef]
- Bai, H.; Harmancı, A.S.; Erson-Omay, E.Z.; Li, J.; Coşkun, S.; Simon, M.; Krischek, B.; Özduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mutant IDH Inhibitors | |||||
|---|---|---|---|---|---|
| ClinicalTrials.gov Identifier | Drug | Mechanism of Action | Phase | Status | Announced Results |
| NCT02073994 | Ivosidenib (AG-120) | mIDH1 inhibitor | Phase I | Active, not recruiting | Favorable safety profile. Reduction in volume and growth rate of only non-enhancing gliomas [50] |
| NCT03030066 | DS-1001b | mIDH1 inhibitor | Phase I | Active, not recruiting | Favorable safety profile under 100 mg/day. One partial response and one minor response in 14 patients [51] |
| NCT02381886 | IDH305 | mIDH1 inhibitor | Phase I | Active, not recruiting | Favorable safety profile, therapeutic responses were announced only for AML [52] |
| NCT02746081 | BAY1436032 | mIDH1 inhibitor | Phase I | Active, not recruiting | |
| NCT04458272 | DS-1001b | mIDH1 inhibitor | Phase II | Active, not recruiting | |
| NCT02977689 | IDH305 | mIDH1 inhibitor | Phase II | Withdrawn | Novartis paused all study start-up activities due to the safety evaluation of the IDH305 compound |
| NCT02987010 | IDH305 | mIDH1 inhibitor | Phase II | Withdrawn | The sponsor did not want to move forward with the protocol; the study was never opened |
| NCT04521686 | LY3410738 | mIDH1 inhibitor | Phase I | Recruiting | |
| NCT04195555 | Ivosidenib (AG-120) | mIDH1 inhibitor | Phase II | Recruiting | |
| NCT02273739 | Enasidenib (AG-221) | mIDH2 inhibitor | Phase I/II | Completed | Closed earlier than planned. High ratio of adverse events |
| NCT02481154 | Vorasidenib (AG-881) | mIDH1/2 inhibitor | Phase I | Active, not recruiting | Favorable safety profile under 100 mg/day. Efficacy data not announced yet |
| NCT04762602 | HMPL-306 | mIDH1/2 inhibitor | Phase I | Recruiting | |
| NCT04164901 | Vorasidenib (AG-881) | mIDH1/2 inhibitor | Phase III | Recruiting | |
| NCT03343197 | Ivosidenib (AG-120) or vorasidenib (AG-881) | mIDH1 or mIDH1/2 inhibitor | Phase I | Active, not recruiting | |
| Combinatorial Approaches | |||||
|---|---|---|---|---|---|
| ClinicalTrials.gov Identifier | Drug | Mechanism of Action | Phase | Status | Announced Results |
| NCT03893903 | IDH1R132H peptide vaccine and avelumab | Combination of an IDH1R132H-specific vaccine and an immune checkpoint inhibitor | Phase I | Recruiting | |
| NCT03749187 | BGB-290 and temozolomide | Combination of the PARP1/2 inhibitor and an alkylating agent | Phase I | Recruiting | |
| NCT02193347 | IDH1 peptide vaccine and temozolomide | Combination of a PEPIDH1M vaccine and an alkylating agent | Phase I | Active, not recruiting | |
| NCT04056910 | Ivosidenib (AG-120) with nivolumab | Combination of an mIDH1 inhibitor and an immune checkpoint inhibitor | Phase II | Recruiting | |
| NCT03991832 | Olaparib and durvalumab | Combination of a PARP inhibitor and an immune checkpoint inhibitor | Phase II | Recruiting | |
| NCT03180502 | Proton beam or intensity-modulated radiation therapy and temozolomide | Combination of radiation and an alkylating agent | Phase II | Recruiting | |
| NCT02968940 | Avelumab and hypofractionated radiation therapy | Combination of an immune checkpoint inhibitor and radiation | Phase II | Completed | |
| NCT03914742 | BGB-290 and temozolomide | Combination of a PARP1/2 inhibitor and an alkylating agent | Phase I/II | Recruiting | |
| NCT03684811 | Olutasidenib (FT-2102) and azacitidine | Single-agent treatment with an mIDH1 inhibitor and a combination with a DNA methyltransferase 1 inhibitor | Phase I/II | Active, not recruiting | Favorable safety profile, low efficacy with a single agent, evaluation of the combination is ongoing [53] |
| NCT02496741 | Metformin and chloroquine | Combination of antidiabetic and antimalarial agents | Phase I/II | Completed | |
| NCT03528642 | CB-839 with radiation therapy and temozolomide | Combination of radiation and an alkylating agent with a glutaminase inhibitor | Phase I | Recruiting | |
| NCT00626990 | Radiation and temozolomide | Combination of radiation and an alkylating agent | Phase III | Active, not recruiting | Increased overall survival with adjuvant TMZ in IDH-mutant gliomas [54] |
| DNA Methyltransferase (DNMT) Inhibitors | |||||
| ClinicalTrials.gov Identifier | Drug | Mechanism of Action | Phase | Status | Announced Results |
| NCT03922555 | ASTX727 | DNMT and cytidine deaminase inhibitor | Phase I | Recruiting | |
| NCT03666559 | Azacitidine | DNMT1 inhibitor | Phase II | Recruiting | |
| Poly ADP Ribose Polymerase (PARP) Inhibitors | |||||
| ClinicalTrials.gov Identifier | Drug | Mechanism of Action | Phase | Status | |
| NCT03212274 | Olaparib (AZD2281) | PARP1/2 inhibitor | Phase II | Recruiting | |
| NCT03561870 | Olaparib | PARP1/2 inhibitor | Phase II | Active, not recruiting | |
| Immunotherapy | |||||
| ClinicalTrials.gov Identifier | Drug | Mechanism of Action | Phase | Status | |
| NCT02454634 | IDH1 peptide vaccine (NOA-16) | IDH1 peptide vaccine | Phase I | Completed | Favorable safety profile, high percentage of IDH peptide-specific immune response [107] |
| NCT02771301 | IDH1R132H-DC vaccine | IDH1R132H-dendritic cell vaccine | Phase I | Unknown | |
| NCT03557359 | Nivolumab | Immune checkpoint inhibitor | Phase II | Recruiting | |
| NCT03718767 | Nivolumab | Immune checkpoint inhibitor | Phase II | Recruiting | |
| NCT03925246 | Nivolumab | Immune checkpoint inhibitor | Phase II | Active, not recruiting | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. https://doi.org/10.3390/biomedicines9070799
Kayabolen A, Yilmaz E, Bagci-Onder T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines. 2021; 9(7):799. https://doi.org/10.3390/biomedicines9070799
Chicago/Turabian StyleKayabolen, Alisan, Ebru Yilmaz, and Tugba Bagci-Onder. 2021. "IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications?" Biomedicines 9, no. 7: 799. https://doi.org/10.3390/biomedicines9070799
APA StyleKayabolen, A., Yilmaz, E., & Bagci-Onder, T. (2021). IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines, 9(7), 799. https://doi.org/10.3390/biomedicines9070799