
Merkel Cell Carcinoma

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Epidemiology
3. Clinical Features
4. Histopathological Features
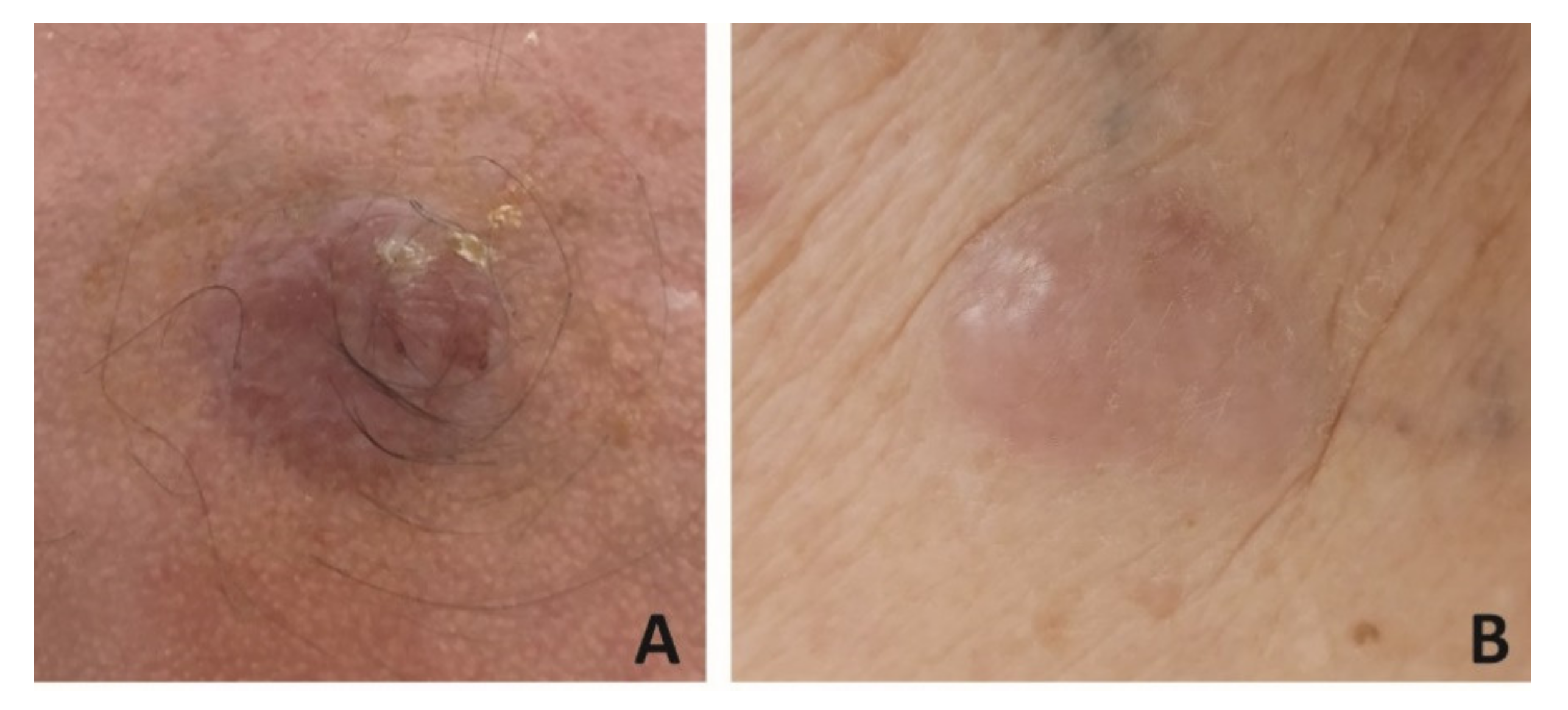
5. Dermoscopy
6. Reflectance Confocal Microscopy (RCM)
7. Pathogenesis
7.1. MCCP Tumors
7.2. MCCN Tumors
7.3. Pathways Deregulated in MCCP and MCCN
7.4. Cell of Origin for MCC
8. Immunology
9. Angiogenesis
10. Current Treatments
10.1. Surgery
10.2. SLNB
10.3. Complete Lymph Node Dissection (CLND)
10.4. Adjuvant RT
10.5. Chemotherapy
10.6. Target Therapy
10.7. Immunotherapy Clinical Trials
11. Future Perspectives
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Xavier-Junior, J.C.C.; Ocanha-Xavier, J.P. WHO (2018) Classification of Skin Tumors. Am. J. Dermatopathol. 2019, 41, 699–700. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Rywlin, A.M. Malignant Merkel-cell tumor is a more accurate description than trabecular carcinoma. Am. J. Dermatopathol. 1982, 4, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.M.; Cerroni, L. Merkel cell carcinoma: A review. J. Cutan. Pathol. 2021, 48, 411–421. [Google Scholar] [CrossRef]
- Zwijnenburg, E.M.; Lubeek, S.F.K.; Werner, J.E.M.; Amir, A.L.; Weijs, W.L.J.; Takes, R.P.; Pegge, S.A.H.; van Herpen, C.M.L.; Adema, G.J.; Kaanders, J.H.A.M. Merkel cell carcinoma: New trends. Cancers 2021, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Hussain, K. Merkel cell carcinoma. Clin. Exp. Dermatol. 2020, 1–6. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Decaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Rabinowits, G.; Lezcano, C.; Catalano, P.J.; McHugh, P.; Becker, H.; Reilly, M.M.; Huang, J.; Tyagi, A.; Thakuria, M.; Bresler, S.C.; et al. Cabozantinib in Patients with Advanced Merkel Cell Carcinoma. Oncologist 2018, 23, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The genomic landscape of Merkel cell carcinom and clinicogenomic biomarkers of response to immune checkpoint inhibitor therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Lebbé, C.; zur Hausen, A.; Avril, M.F.; Hariharan, S.; Bharmal, M.; Becker, J.C. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Orphanet: Cutaneous Neuroendocrine Carcinoma. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11166&Disease_Disease_Search_diseaseGroup=Merkel-cell-carcinoma&Disease_Disease_Search_diseaseType=Pat&Disease (accessed on 17 May 2021).
- Key Statistics for Merkel Cell Carcinoma. Available online: http://www.cancer.org/cancer/skincancer-Merkelcell/detailedguide/skin-cancer-Merkel-cell-carcinoma-key-statistics (accessed on 17 May 2021).
- Youlden, D.R.; Soyer, H.P.; Youl, P.H.; Fritschi, L.; Baade, P.D. Incidence and survival for merkel cell carcinoma in Queensland, Australia, 1993–2010. JAMA Dermatol. 2014, 150, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.P.; Liang, E.S.; Martin, R.C.W. Epidemiology of Merkel cell carcinoma in New Zealand: A population-based study. Br. J. Dermatol. 2015, 173, 835–837. [Google Scholar] [CrossRef]
- Girschik, J.; Thorn, K.; Beer, T.W.; Heenan, P.J.; Fritschi, L. Merkel cell carcinoma in Western Australia: A population-based study of incidence and survival. Br. J. Dermatol. 2011, 165, 1051–1057. [Google Scholar] [CrossRef]
- Chun, S.M.; Yun, S.J.; Lee, S.C.; Won, Y.H.; Lee, J.B. Merkel cell polyomavirus is frequently detected in korean patients with merkel cell carcinoma. Ann. Dermatol. 2013, 25, 203–207. [Google Scholar] [CrossRef]
- Song, P.I.; Liang, H.; Wei, W.Q.; Jiang, Y.Q.; Smith, J.S.; Qiao, Y.L. The clinical profile of Merkel cell carcinoma in mainland China. Int. J. Dermatol. 2012, 51, 1054–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.C.; Stang, A.; Decaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3. [Google Scholar] [CrossRef]
- Hodgson, N.C. Merkel cell carcinoma: Changing incidence trends. J. Surg. Oncol. 2005, 89, 1–4. [Google Scholar] [CrossRef]
- Lyhne, D.; Lock-Andersen, J.; Dahlstrøm, K.; Drzewiecki, K.T.; Balslev, E.; Muhic, A.; Krarup-Hansen, A. Rising incidence of Merkel cell carcinoma. J. Plast. Surg. Hand Surg. 2011, 45, 274–280. [Google Scholar] [CrossRef]
- Eisemann, N.; Waldmann, A.; Geller, A.C.; Weinstock, M.A.; Volkmer, B.; Greinert, R.; Breitbart, E.W.; Katalinic, A. Non-melanoma skin cancer incidence and impact of skin cancer screening on incidence. J. Investig. Dermatol. 2014, 134, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Reichgelt, B.A.; Visser, O. Epidemiology and survival of Merkel cell carcinoma in the Netherlands. A population-based study of 808 cases in 1993-2007. Eur. J. Cancer 2011, 47, 579–585. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population based study. J. Cutan. Pathol. 2010, 37, 20–27. [Google Scholar] [CrossRef]
- Agbai, O.N.; Buster, K.; Sanchez, M.; Hernandez, C.; Kundu, R.V.; Chiu, M.; Roberts, W.E.; Draelos, Z.D.; Bhushan, R.; Taylor, S.C.; et al. Skin cancer and photoprotection in people of color: A review and recommendations for physicians and the public. J. Am. Acad. Dermatol. 2014, 70, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Kaae, J.; Hansen, A.V.; Biggar, R.J.; Boyd, H.A.; Moore, P.S.; Wohlfahrt, J.; Melbye, M. Merkel cell carcinoma: Incidence, mortality, and risk of other cancers. J. Natl. Cancer Inst. 2010, 102, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Van Der Zwan, J.M.; Trama, A.; Otter, R.; Larrañaga, N.; Tavilla, A.; Marcos-Gragera, R.; Dei Tos, A.P.; Baudin, E.; Poston, G.; Links, T.; et al. Rare neuroendocrine tumours: Results of the surveillance of rare cancers in Europe project. Eur. J. Cancer 2013, 49, 2565–2578. [Google Scholar] [CrossRef] [Green Version]
- Lemos, B.D.; Storer, B.E.; Iyer, J.G.; Phillips, J.L.; Bichakjian, C.K.; Fang, L.C.; Johnson, T.M.; Liegeois-Kwon, N.J.; Otley, C.C.; Paulson, K.G.; et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5823 cases as the basis of the first consensus staging system. J. Am. Acad. Dermatol. 2010, 63, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Kieny, A.; Cribier, B.; Meyer, N.; Velten, M.; Jégu, J.; Lipsker, D. Epidemiology of Merkel cell carcinoma. A population-based study from 1985 to 2013, in northeastern of France. Int. J. Cancer 2019, 144, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Kauczok, C.S.; Ugurel, S.; Eib, S.; Bröcker, E.B.; Houben, R. Merkel cell carcinoma: Molecular pathogenesis, clinical features and therapy. JDDG J. Ger. Soc. Dermatology 2008, 6, 709–719. [Google Scholar] [CrossRef]
- Llombart, B.; Monteagudo, C.; López-Guerrero, J.A.; Carda, C.; Jorda, E.; Sanmartín, O.; Almenar, S.; Molina, I.; Martín, J.M.; Llombart-Bosch, A. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005, 46, 622–634. [Google Scholar] [CrossRef]
- Sparks, J.; Sparks, M.; Malone, J.C. Cutaneous Merkel cell carcinoma: Multiple asynchronous primary lesions in a patient on immunosuppressive therapy. J. Cutan. Pathol. 2017, 44, 309–312. [Google Scholar] [CrossRef]
- Gupta, S.G.; Wang, L.C.; Peñas, P.F.; Gellenthin, M.; Lee, S.J.; Nghiem, P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch. Dermatol. 2006, 142, 685–690. [Google Scholar] [CrossRef]
- Rizzo, J.M.; Harms, P.W.; Harms, K.L.; Plaska, A.; Brenner, C.; Durham, A.B. Unknown primary Merkel cell carcinoma in the immunosuppressed patient: Case series. JAAD Case Reports 2021, 8, 19–22. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network NCCN clinical practice guidelines in oncology: Merkel cell carcinoma (version 1.2021). NCCN.org 2021, 1, 1–69.
- Keohane, S.G.; Proby, C.M.; Newlands, C.; Motley, R.J.; Nasr, I.; Mohd Mustapa, M.F.; Slater, D.N. The new 8th edition of TNM staging and its implications for skin cancer: A review by the British Association of Dermatologists and the Royal College of Pathologists, U.K. Br. J. Dermatol. 2018, 179, 824–828. [Google Scholar] [CrossRef]
- Walsh, N.M. Complete spontaneous regression of Merkel cell carcinoma (1986–2016): A 30 year perspective. J. Cutan. Pathol. 2016, 43, 1150–1154. [Google Scholar] [CrossRef]
- Harms, K.L.; Zhao, L.; Johnson, B.; Wang, X.; Carskadon, S.; Palanisamy, N.; Rhodes, D.R.; Mannan, R.; Vo, J.N.; Choi, J.E.; et al. Virus-positive Merkel Cell Carcinoma Is an Independent Prognostic Group with Distinct Predictive Biomarkers. Clin. Cancer Res. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e2. [Google Scholar] [CrossRef]
- Tope, W.D.; Sangüeza, O.P. Merkel Cell Carcinoma: Histopathology, Immunohistochemistry, and Cytogenetic Analysis. J. Dermatol. Surg. Oncol. 1994, 20, 648–652. [Google Scholar] [CrossRef]
- Gaitskell, K.; Nassar, S.; Ibrahim, H. Merkel cell carcinoma with divergent differentiation. Clin. Exp. Dermatol. 2020, 45, 327–332. [Google Scholar] [CrossRef]
- Pasternak, S.; Carter, M.D.; Ly, T.Y.; Doucette, S.; Walsh, N.M. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum. Pathol. 2018, 82, 232–238. [Google Scholar] [CrossRef]
- Jaeger, T.; Ring, J.; Andres, C. Histological, Immunohistological, and Clinical Features of Merkel Cell Carcinoma in Correlation to Merkel Cell Polyomavirus Status. J. Skin Cancer 2012, 2012, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argenziano, G.; Zalaudek, I.; Corona, R.; Sera, F.; Cicale, L.; Petrillo, G.; Ruocco, E.; Hofmann-Wellenhof, R.; Soyer, H.P. Vascular structures in skin tumors: A dermoscopy study. Arch. Dermatol. 2004, 140, 1485–1489. [Google Scholar] [CrossRef]
- Harting, M.S.; Ludgate, M.W.; Fullen, D.R.; Johnson, T.M.; Bichakjian, C.K. Dermatoscopic vascular patterns in cutaneous Merkel cell carcinoma. J. Am. Acad. Dermatol. 2012, 66, 923–927. [Google Scholar] [CrossRef]
- Dalle, S.; Parmentier, L.; Moscarella, E.; Phan, A.; Argenziano, G.; Thomas, L. Dermoscopy of merkel cell carcinoma. Dermatology 2012, 224, 140–144. [Google Scholar] [CrossRef]
- Kato, J.; Horimoto, K.; Sato, S.; Minowa, T.; Uhara, H. Dermoscopy of Melanoma and Non-melanoma Skin Cancers. Front. Med. 2019, 6, 180. [Google Scholar] [CrossRef] [Green Version]
- Lallas, A.; Moscarella, E.; Argenziano, G.; Longo, C.; Apalla, Z.; Ferrara, G.; Piana, S.; Rosato, S.; Zalaudek, I. Dermoscopy of uncommon skin tumours. Australas. J. Dermatol. 2014, 55, 53–62. [Google Scholar] [CrossRef]
- Jalilian, C.; Chamberlain, A.J.; Haskett, M.; Rosendahl, C.; Goh, M.; Beck, H.; Keir, J.; Varghese, P.; Mar, A.; Hosking, S.; et al. Clinical and dermoscopic characteristics of Merkel cell carcinoma. Br. J. Dermatol. 2013, 169, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Benati, E.; Borsari, S.; Bombonato, C.; Pampena, R.; Moscarella, E.; Piana, S.; Pellacani, G. Merkel cell carcinoma: Morphologic aspects on reflectance confocal microscopy. J. Eur. Acad. Dermatology Venereol. 2017, 31, e480–e481. [Google Scholar] [CrossRef]
- Cinotti, E.; Provvidenziale, L.; Habougit, C.; Fimiani, M.; Tognetti, L.; Perrot, J.L.; Rubegni, P. Dermoscopic and reflectance microscopy features of primary and metastatic Merkel cell carcinoma: Ten cases. Ski. Res. Technol. 2019, 25, 407–409. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel cell polyomavirus and merkel cell carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef] [Green Version]
- Coursaget, P.; Samimi, M.; Nicol, J.T.J.; Gardair, C.; Touzé, A. Human Merkel cell polyomavirus: Virological background and clinical implications. APMIS 2013, 121, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Shuda, M.; Camacho, C.J.; Gamper, A.M.; Thant, M.; Chang, Y.; Moore, P.S. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J. Virol. 2015, 89, 4191–4200. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; McGrath, J.P.; Lim, M.Y.; Cushman, C.; Swanson, S.K.; Tillgren, M.L.; Paulo, J.A.; Gokhale, P.C.; Florens, L.; et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 2020, 22, 603–615. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Bardot, E.S.; Valdes, V.J.; Zhang, J.; Perdigoto, C.N.; Nicolis, S.; Hearn, S.A.; Silva, J.M.; Ezhkova, E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 2013, 32, 1990–2000. [Google Scholar] [CrossRef] [Green Version]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic Regulation of Epidermal Differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T Antigen-Induced Atonal Homolog 1 Is a Lineage-Dependency Oncogene in Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 56–65.e3. [Google Scholar] [CrossRef] [Green Version]
- Schrama, D.; Sarosi, E.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Horny, K.; Gerhardt, P.; Hebel-Cherouny, A.; Wülbeck, C.; Utikal, J.; Becker, J.C. Mutational landscape of virus-and uv-associated merkel cell carcinoma cell lines is comparable to tumor tissue. Cancers 2021, 13, 649. [Google Scholar] [CrossRef]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Toberer, F.; Haenssle, H.A.; Heinzel-Gutenbrunner, M.; Enk, A.; Hartschuh, W.; Helmbold, P.; Kutzner, H. Metabolic reprogramming and angiogenesis in primary cutaneous Merkel cell carcinoma: Expression of hypoxia-inducible factor-1α and its central downstream factors. J. Eur. Acad. Dermatology Venereol. 2021, 35, 88–94. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.M.; Berthon, P.; et al. Polyomavirus-Positive Merkel Cell Carcinoma Derived from a Trichoblastoma Suggests an Epithelial Origin of this Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef]
- Chan, I.S.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for Merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Afanasiev, O.; Nghiem, P. Immunobiology of Merkel cell carcinoma: Implications for immunotherapy of a polyomavirus-associated cancer. Curr. Oncol. Rep. 2011, 13, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel cell carcinoma in the age of immunotherapy: Facts and hopes. Clin. Cancer Res. 2018, 24, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Nghiem, P.; Bhatia, S.; Hauschild, A.; Saiag, P.; Mahnke, L.; Hariharan, S.; Kaufman, H.L. Immune evasion mechanisms and immune checkpoint inhibition in advanced merkel cell carcinoma. Oncoimmunology 2017, 6, e1338237. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Hudgens, C.W.; Ray-Lyons, G.; Nagarajan, P.; Aung, P.P.; Curry, J.L.; Torres-Cabala, C.A.; Mino, B.; Rodriguez-Canales, J.; Reuben, A.; et al. Density, distribution, and composition of immune infiltrates correlate with survival in Merkel cell carcinoma. Clin. Cancer Res. 2016, 22, 5553–5563. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Gaboriaud, P.; Berthon, P.; Zaragoza, J.; Schrama, D.; Houben, R.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Aubin, F.; Bens, G.; et al. Merkel cell carcinomas infiltrated with CD33+ myeloid cells and CD8+ T cells are associated with improved outcome. J. Am. Acad. Dermatol. 2018, 78, 973–982.e8. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Iyer, J.G.; Tegeder, A.R.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.T.; Lemos, B.D.; Byrd, D.R.; et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral cd8+ lymphocyte invasion as an independent predictor of survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farah, M.; Reuben, A.; Spassova, I.; Yang, R.K.; Kubat, L.; Nagarajan, P.; Ning, J.; Li, W.; Aung, P.P.; Curry, J.L.; et al. T-Cell Repertoire in Combination with T-Cell Density Predicts Clinical Outcomes in Patients with Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 2146–2156.e4. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitteldorf, C.; Berisha, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. PD-1 and PD-L1 in neoplastic cells and the tumor microenvironment of Merkel cell carcinoma. J. Cutan. Pathol. 2017, 44, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Figueras, M.T.; Puig, L.; Musulén, E.; Gilaberte, M.; Lerma, E.; Serrano, S.; Ferrándiz, C.; Ariza, A. Expression profiles associated with aggressive behavior in Merkel cell carcinoma. Mod. Pathol. 2007, 20, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Brunner, M.; Thurnher, D.; Pammer, J.; Geleff, S.; Heiduschka, G.; Reinisch, C.M.; Petzelbauer, P.; Erovic, B.M. Expression of VEGF-A/C, VEGF-R2, PDGF-α/β, c-kit, EGFR, Her-2/Neu, Mcl-1 and Bmi-1 in Merkel cell carcinoma. Mod. Pathol. 2008, 21, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Kukko, H.; Koljonen, V.; Lassus, P.; Tukiainen, E.; Haglund, C.; Bohling, T. Expression of Vascular Endothelial Growth Factor Receptor-2 in Merkel Cell Carcinoma. Anticancer Res. 2007, 27, 2587–2589. [Google Scholar]
- Bob, A.; Nielen, F.; Krediet, J.; Schmitter, J.; Freundt, D.; Terhorst, D.; Röwert-Huber, J.; Kanitakis, J.; Stockfleth, E.; Ulrich, C.; et al. Tumor vascularization and clinicopathologic parameters as prognostic factors in merkel cell carcinoma. J. Cancer Res. Clin. Oncol. 2017, 143, 1999–2010. [Google Scholar] [CrossRef]
- Ng, L.; Beer, T.W.; Murray, K. Vascular density has prognostic value in merkel cell carcinoma. Am. J. Dermatopathol. 2008, 30, 442–445. [Google Scholar] [CrossRef]
- Werchau, S.; Toberer, F.; Enk, A.; Dammann, R.; Helmbold, P. Merkel cell carcinoma induces lymphatic microvessel formation. J. Am. Acad. Dermatol. 2012, 67, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Gaboriaud, P.; Tallet, A.; Leblond, V.; Arnold, F.; Berthon, P.; Schweinitzer, S.; Larcher, T.; Guyétant, S.; Schrama, D.; et al. VEGF-A Inhibition as a Potential Therapeutic Approach in Merkel Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 736–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidelines Detail. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1444 (accessed on 17 May 2021).
- Perez, M.C.; de Pinho, F.R.; Holstein, A.; Oliver, D.E.; Naqvi, S.M.H.; Kim, Y.; Messina, J.L.; Burke, E.; Gonzalez, R.J.; Sarnaik, A.A.; et al. Resection Margins in Merkel Cell Carcinoma: Is a 1-cm Margin Wide Enough? Ann. Surg. Oncol. 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Storer, B.E.; Iyer, J.G.; Moshiri, A.; Parvathaneni, U.; Byrd, D.; Sober, A.J.; Sondak, V.K.; Gershenwald, J.E.; Nghiem, P. Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases from the National Cancer Data Base. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.P.; Holtzman, M.P. Surgical resection improves median overall survival with marginal improvement in long-term survival when compared with definitive radiotherapy in Merkel cell carcinoma: A propensity score matched analysis of the National Cancer Database. Am. J. Surg. 2018, 215, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.L.; Bichakjian, C.K.; Lowe, L.; Griffith, K.A.; Frohm, M.L.; Fullen, D.R.; Hayman, J.A.; Lao, C.D.; Shah, K.S.; McLean, S.A.; et al. Clinicopathologic features of primary merkel cell carcinoma: A detailed descriptive analysis of a large contemporary cohort. Dermatologic Surg. 2013, 39, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.S.; Whalley, D.; Haydu, L.E.; Murali, R.; Tippett, J.; Thompson, J.F.; Hruby, G.; Scolyer, R.A. Increasing tumor thickness is associated with recurrence and poorer survival in patients with merkel cell carcinoma. Ann. Surg. Oncol. 2012, 19, 3325–3334. [Google Scholar] [CrossRef] [PubMed]
- Vargo, J.A.; Ghareeb, E.R.; Balasubramani, G.K.; Beriwal, S. RE: Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases from the National Cancer Data Base. J. Natl. Cancer Inst. 2017, 109, 108. [Google Scholar] [CrossRef] [PubMed]
- Luryi, A.L.; Chen, M.M.; Mehra, S.; Roman, S.A.; Sosa, J.A.; Judson, B.L. Treatment factors associated with survival in early-stage oral cavity cancer: Analysis of 6830 cases from the National Cancer Data Base. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Qureshi, M.M.; Truong, M.T.; Sahni, D. Demographics and outcomes of stage I and II Merkel cell carcinoma treated with Mohs micrographic surgery compared with wide local excision in the National Cancer Database. J. Am. Acad. Dermatol. 2018, 79, 126–134.e3. [Google Scholar] [CrossRef] [PubMed]
- Grotz, T.E.; Joseph, R.W.; Pockaj, B.A.; Foote, R.L.; Otley, C.C.; Bagaria, S.P.; Weaver, A.L.; Jakub, J.W. Negative Sentinel Lymph Node Biopsy in Merkel Cell Carcinoma is Associated with a Low Risk of Same-Nodal-Basin Recurrences. Ann. Surg. Oncol. 2015, 22, 4060–4066. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Eigentler, T.; Frerich, B.; Gambichler, T.; Grabbe, S.; Höller, U.; Klumpp, B.; Loquai, C.; Krause-Bergmann, A.; Müller-Richter, U.; et al. S2k guidelines for Merkel cell carcinoma (MCC, neuroendocrine carcinoma of the skin)–update 2018. JDDG J. Ger. Soc. Dermatology 2019, 17, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Durham, A.B.; Bichakjian, C.K.; Harms, P.W.; Hayman, J.A.; McLean, S.A.; Harms, K.L.; Burns, W.R. Completion Lymph Node Dissection or Radiation Therapy for Sentinel Node Metastasis in Merkel Cell Carcinoma. Ann. Surg. Oncol. 2019, 26, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.C.; Oliver, D.E.; Weitman, E.S.; Boulware, D.; Messina, J.L.; Torres-Roca, J.; Cruse, C.W.; Gonzalez, R.J.; Sarnaik, A.A.; Sondak, V.K.; et al. Management of Sentinel Lymph Node Metastasis in Merkel Cell Carcinoma: Completion Lymphadenectomy, Radiation, or Both? Ann. Surg. Oncol. 2019, 26, 379–385. [Google Scholar] [CrossRef]
- Fang, L.C.; Lemos, B.; Douglas, J.; Iyer, J.; Nghiem, P. Radiation monotherapy as regional treatment for lymph node-positive merkel cell carcinoma. Cancer 2010, 116, 1783–1790. [Google Scholar] [CrossRef]
- Strom, T.; Carr, M.; Zager, J.S.; Naghavi, A.; Smith, F.O.; Cruse, C.W.; Messina, J.L.; Russell, J.; Rao, N.G.; Fulp, W.; et al. Radiation Therapy is Associated with Improved Outcomes in Merkel Cell Carcinoma. Ann. Surg. Oncol. 2016, 23, 3572–3578. [Google Scholar] [CrossRef]
- Fields, R.C.; Busam, K.J.; Chou, J.F.; Panageas, K.S.; Pulitzer, M.P.; Allen, P.J.; Kraus, D.H.; Brady, M.S.; Coit, D.G. Recurrence after complete resection and selective use of adjuvant therapy for stage i through III Merkel cell carcinoma. Cancer 2012, 118, 3311–3320. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, A.; Torchio, M.; Prinzi, N.; Trevisan, F.; Dallera, P.; De Stefani, A.; Russo, A.; Vitali, E.; Bruschieri, L.; et al. Adjuvant radiotherapy for Merkel cell carcinoma: A systematic review and meta-analysis. Radiother. Oncol. 2019, 134, 211–219. [Google Scholar] [CrossRef]
- Mojica, P.; Smith, D.; Ellenhorn, J.D.I. Adjuvant radiation therapy is associated with improved survival in merkel cell carcinoma of the skin. J. Clin. Oncol. 2007, 25, 1043–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, M.; Gaskins, J.; Wall, W.; Tennant, P.; Bumpous, J.; Dunlap, N. Optimal adjuvant radiotherapy dose for stage I, II or III Merkel cell carcinoma: An analysis of the National Cancer Database. Jpn. J. Clin. Oncol. 2019, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Bichakjian, C.K.; Olencki, T.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Blitzblau, R.; Bowen, G.M.; Contreras, C.M.; Daniels, G.A.; Decker, R.; et al. Merkel Cell Carcinoma, Version 1.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 742–774. [Google Scholar] [CrossRef] [PubMed]
- Frohm, M.L.; Griffith, K.A.; Harms, K.L.; Hayman, J.A.; Fullen, D.R.; Nelson, C.C.; Won, S.L.; Schwartz, J.L.; Bichakjian, C.K. Recurrence and survival in patients with merkel cell carcinoma undergoing surgery without adjuvant radiation therapy to the primary site. JAMA Dermatology 2016, 152, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Voog, E.; Biron, P.; Martin, J.P.; Blay, J.Y. Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer 1999, 85, 2589–2595. [Google Scholar] [CrossRef]
- Tai, P.T.H.; Yu, E.; Winquist, E.; Hammond, A.; Stitt, L.; Tonita, J.; Gilchrist, J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: Case series and review of 204 cases. J. Clin. Oncol. 2000, 18, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.G.; Rischin, D.; Porter, I.; Walpole, E.; Harvey, J.; Hamilton, C.; Keller, J.; Tripcony, L. Does Chemotherapy Improve Survival in High-Risk Stage I and II Merkel cell Carcinoma of the Skin? Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.D.; Gaunt, P.; Wheatley, K.; Bowden, S.J.; Savage, J.; Faust, G.; Nobes, J.; Goodman, A.; Ritchie, D.; Kumar, S.; et al. UKMCC-01: A phase II study of pazopanib (PAZ) in metastatic Merkel cell carcinoma. J. Clin. Oncol. 2016, 34, 9542. [Google Scholar] [CrossRef]
- Samlowski, W.E.; Moon, J.; Tuthill, R.J.; Heinrich, M.C.; Balzer-Haas, N.S.; Merl, S.A.; DeConti, R.C.; Thompson, J.A.; Witter, M.T.; Flaherty, L.E.; et al. A phase II trial of imatinib mesylate in merkel cell carcinoma (Neuroendocrine carcinoma of the skin): A Southwest oncology group study (S0331). Am. J. Clin. Oncol. Cancer Clin. Trials 2010, 33, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarotto, R.; Cardnell, R.; Su, S.; Diao, L.; Eterovic, A.K.; Prieto, V.; Morrisson, W.H.; Wang, J.; Kies, M.S.; Glisson, B.S.; et al. Poly ADP-ribose polymerase-1 as a potential therapeutic target in Merkel cell carcinoma. Head Neck 2018, 40, 1676–1684. [Google Scholar] [CrossRef]
- Park, S.Y.; Doolittle-Amieva, C.; Moshiri, Y.; Akaike, T.; Parvathaneni, U.; Bhatia, S.; Zaba, L.C.; Nghiem, P. How we treat Merkel cell carcinoma: Within and beyond current guidelines. Futur. Oncol. 2021, 17, 1363–1377. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. New Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable tumor regression and overall survival in patients with advanced Merkel cell carcinoma receiving pembrolizumab as first-line therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Lee, A.Y.; Berman, R.S. The Landmark Series: Non-melanoma Skin Cancers. Ann. Surg. Oncol. 2020, 27, 22–27. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.K.; Dimitrakopoulou-Strauss, A.; Sachpekidis, C.; Enk, A.; Hassel, J.C. Ipilimumab has efficacy in metastatic Merkel cell carcinoma: A case series of five patients. J. Eur. Acad. Dermatology Venereol. 2017, 31, e389–e391. [Google Scholar] [CrossRef] [PubMed]
- A Study of INCMGA00012 in Metastatic Merkel Cell Carcinoma (POD1UM-201)-Full Text View-ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT03599713 (accessed on 17 May 2021).
- Glutsch, V.; Kneitz, H.; Gesierich, A.; Goebeler, M.; Haferkamp, S.; Becker, J.C.; Ugurel, S.; Schilling, B. Activity of ipilimumab plus nivolumab in avelumab-refractory Merkel cell carcinoma. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Khushalani, N.I.; Eroglu, Z.; Russell, J.; Wuthrick, E.; Caudell, J.; Harrison, L.; Aoki, M.; Shah, H.; Blakaj, D.; et al. A phase II, randomized study of nivolumab (NIVO) and Ipilimumab (IPI) versus NIVO, IPI and stereotactic body radiation therapy (SBRT) for metastatic Merkel cell carcinoma (MCC, NCT03071406): A preliminary report. Ann. Oncol. 2019, 30, v538–v539. [Google Scholar] [CrossRef]
- Domatinostat in Combination with Avelumab in Patients with Advanced Merkel Cell Carcinoma Progressing on Anti-PD-(L)1-Full Text View-ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT04393753?term=NCT04393753&cond=merkel&draw=2&rank=1 (accessed on 6 May 2021).
- Topalian, S.L.; Bhatia, S.; Amin, A.; Kudchadkar, R.R.; Sharfman, W.H.; Lebbé, C.; Delord, J.P.; Dunn, L.A.; Shinohara, M.M.; Kulikauskas, R.; et al. Neoadjuvant nivolumab for patients with resectable merkel cell carcinoma in the CheckMate 358 trial. J. Clin. Oncol. 2020, 38, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Angeles, C.V.; Sabel, M.S. Immunotherapy for Merkel cell carcinoma. J. Surg. Oncol. 2021, 123, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Hassel, J.C.; Menzer, C.; Kähler, K.C.; Eigentler, T.K.; Meier, F.E.; Berking, C.; Gutzmer, R.; Mohr, P.; Kiecker, F.; et al. Adjuvant ipilimumab compared with observation in completely resected Merkel cell carcinoma (ADMEC): A randomized, multicenter DeCOG/ADO study. J. Clin. Oncol. 2018, 36, 9527. [Google Scholar] [CrossRef]
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021. [Google Scholar] [CrossRef]
- Mucaj, V.; Shay, J.E.S.; Simon, M.C. Effects of hypoxia and HIFs on cancer metabolism. Int. J. Hematol. 2012, 95, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Barroil, M.; Fasquelle, F.; Foulongne, V.; Ramos, J.; Delfour, C.; Guillot, B.; Du Thanh, A. Divergent differentiation of Merkel cell carcinoma between primary and metastatic lesions. Ann. Dermatol. Venereol. 2021, 148, 51–54. [Google Scholar] [CrossRef]
- Akaike, T.; Qazi, J.; Anderson, A.; Behnia, F.S.; Shinohara, M.M.; Akaike, G.; Hippe, D.S.; Thomas, H.; Takagishi, S.R.; Lachance, K.; et al. High somatostatin receptor expression and efficacy of somatostatin analogues in patients with metastatic Merkel cell carcinoma*. Br. J. Dermatol. 2021, 184, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Manegold, C.; Dingemans, A.M.C.; Gray, J.E.; Nakagawa, K.; Nicolson, M.; Peters, S.; Reck, M.; Wu, Y.L.; Brustugun, O.T.; Crinò, L.; et al. The Potential of Combined Immunotherapy and Antiangiogenesis for the Synergistic Treatment of Advanced NSCLC. J. Thorac. Oncol. 2017, 12, 194–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzozowska, M.; Wierzba, W.; Szafraniec-Buryło, S.; Czech, M.; Połowinczak-Przybyłek, J.; Potemski, P.; Śliwczyński, A. Real-world evidence of patient outcome following treatment of advanced gastrointestinal stromal tumor (GIST) with imatinib, sunitinib, and sorafenib in publicly funded health care in Poland. Med. Sci. Monit. 2019, 25, 3846–3853. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.R.; Grimm, D.; Bauer, J.; Wehland, M.; Magnusson, N.E. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 461. [Google Scholar] [CrossRef] [PubMed]
- Manzo, A.; Montanino, A.; Carillio, G.; Costanzo, R.; Sandomenico, C.; Normanno, N.; Piccirillo, M.C.; Daniele, G.; Perrone, F.; Rocco, G.; et al. Angiogenesis inhibitors in NSCLC. Int. J. Mol. Sci. 2017, 18, 2021. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trail | Study Phase | Line of Treatment | Agent | Primary Endpoint |
|---|---|---|---|---|
| Advanced MCC | ||||
| Keynote-017 | 2 | First line | Pembro | ORR: 56% |
| JAVELIN Merkel 200 | 2 | ≥1 prior line | Avelumab | ORR: 33% |
| POD1UM-201 | 2 | First line | INCMGA00012 | ORR: ongoing |
| NCT03071406 | 2 | ≥1 prior line | IPI + Nivo + SBRT | ORR a |
| MERKELIN2 trial | 2 | After anti-PD-1/PD-L1 | Domat + Avelumab | ORR: ongoing |
| Neoadjuvant MCC | ||||
| NCT04869137 | 2 | Neoadjuvant | Lenvatinib + Pembro | pCR |
| Adjuvant MCC | ||||
| ADMEC-O | 2 | Adjuvant, stage I–III | Nivo | PFS at 12 months |
| NCT03798639 | 1 | Adjuvant, stage III | Nivo + IPI or + RT | % of patients b |
| I-MAT | 2 | Adjuvant, stage I–III | Avelumab | RFS |
| ADAM | 3 | Adjuvant, stage III | Avelumab | RFS |
| STAMP | 3 | Adjuvant, stage I–III | Pembro or RT | RFS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellambra, E.; Carbone, M.L.; Ricci, F.; Ricci, F.; Di Pietro, F.R.; Moretta, G.; Verkoskaia, S.; Feudi, E.; Failla, C.M.; Abeni, D.; et al. Merkel Cell Carcinoma. Biomedicines 2021, 9, 718. https://doi.org/10.3390/biomedicines9070718
Dellambra E, Carbone ML, Ricci F, Ricci F, Di Pietro FR, Moretta G, Verkoskaia S, Feudi E, Failla CM, Abeni D, et al. Merkel Cell Carcinoma. Biomedicines. 2021; 9(7):718. https://doi.org/10.3390/biomedicines9070718
Chicago/Turabian StyleDellambra, Elena, Maria Luigia Carbone, Francesca Ricci, Francesco Ricci, Francesca Romana Di Pietro, Gaia Moretta, Sofia Verkoskaia, Elisa Feudi, Cristina M. Failla, Damiano Abeni, and et al. 2021. "Merkel Cell Carcinoma" Biomedicines 9, no. 7: 718. https://doi.org/10.3390/biomedicines9070718
APA StyleDellambra, E., Carbone, M. L., Ricci, F., Ricci, F., Di Pietro, F. R., Moretta, G., Verkoskaia, S., Feudi, E., Failla, C. M., Abeni, D., & Fania, L. (2021). Merkel Cell Carcinoma. Biomedicines, 9(7), 718. https://doi.org/10.3390/biomedicines9070718