
Neuropsychiatric and Cognitive Deficits in Parkinson’s Disease and Their Modeling in Rodents



Abstract
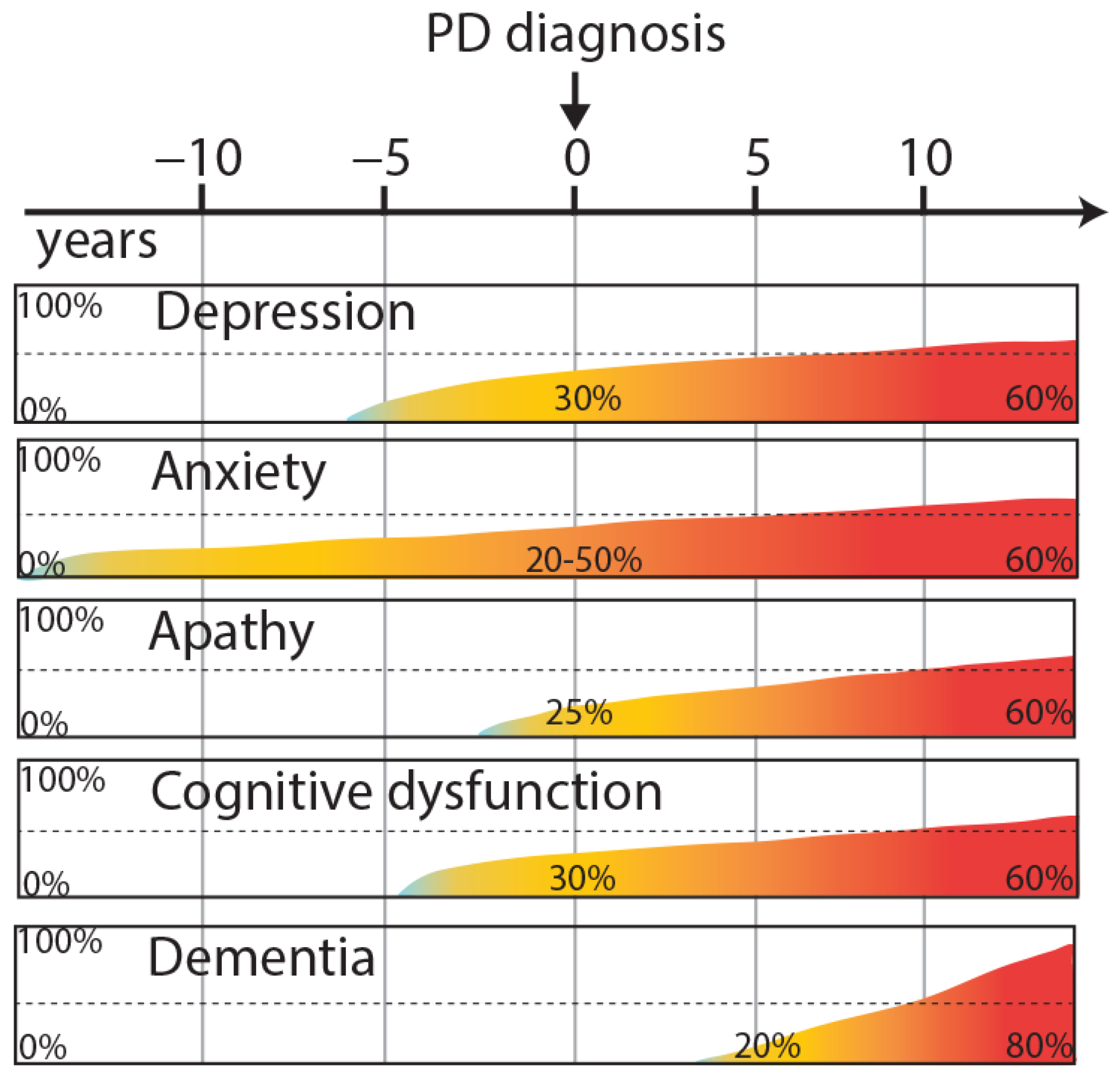
1. Introduction
2. Overview of Neuropsychiatric and Cognitive Deficits in PD
2.1. Neuropsychiatric Disorders
2.1.1. Depression
2.1.2. Anxiety
2.1.3. Apathy
2.2. Cognitive Dysfunction
2.2.1. Working Memory and Attention
2.2.2. Long-Term Memory and Learning
Declarative Episodic Memory
Declarative Emotional Memory and Reward-Learning
Non-Declarative Memory
2.3. Inhibitory Control
2.4. Behavioral Flexibility
2.5. Dementia
3. Overview of Neuropsychiatric and Cognitive Deficits in Rodent Models of PD
3.1. Rodent Models of Neuropsychiatric Disorders
3.1.1. Depression and Anxiety
3.1.2. Apathy
3.2. Rodent Models of Cognitive Dysfunctions
3.2.1. Attention
3.2.2. Memory Impairment and Learning Deficits
3.2.3. Inhibitory Control
3.2.4. Behavioral Flexibility

{kind=link}
| Model | Flexibility Deficits | Inhibitory Control Deficits | Attention Deficits | Learning Deficits | Short-Term Memory Impairment | Long-Term Memory Impairment | Ref |
|---|---|---|---|---|---|---|---|
| 6-OHDA (Bilateral, STR) | +/− | Discrepancies | + + (if attentional load increased) | n.r. | + + | + + | [154,156,165,188,189,190,191] |
| 6-OHDA (Bilateral, SNc) | n.r | n.r. | n.r. | n.r. | + + | + + | [160] |
| 6-OHDA (Bilateral, SNc + VTA) | n.r. | Intact | n.r. | n.r. | n.r. | n.r. | [185] |
| 6-OHDA (Bilateral, MFB) | n.r. | n.r. | n.r. | n.r. | + + | + + | [151,159] |
| 6-OHDA (Unilateral, SNc) | n.r. | n.r. | n.r. | + + | + + | Intact | [115] |
| 6-OHDA (Unilateral, MFB) | + + | Reduced tendency to binge-eating | + + (particularly ipsilateral to the lesion) | n.r. | n.r. | + + | [153,155,158,193] |
| MPTP (bilateral, systemic administration) | n.r. | n.r. | n.r. | + + (only for learning new rules) | + + | Intact | [168] |
| MPTP (unilateral, SNc) | n.r. | n.r. | n.r. | + + | + + | Intact | [158] |
| MPTP (bilateral, SNc) | n.r. | n.r. | n.r. | + + | + + | + + | [158,160,167,169] |
| hA53T alpha-synuclein mice (mouse prion promoter) | n.r. | n.r. | n.r. | + + (older mice) | n.r. | + + (older mice) | [137,177,178] |
| TH Knock-Out mice (Cre recombinase) | + + | n.r. | n.r. | n.r. | n.r. | n.r. | [198,199] |
| WT alpha-synuclein mice (mouse Thy-1 promoter) line 61 | + + | n.r. | n.r. | + + | + + | + + | [171,173] |
| WT alpha-synuclein mice (PDGF-β promoter) | n.r. | n.r. | n.r. | n.r. | n.r. | + + | [174] |
| A30P alpha-synuclein mice (mouse Thy-1 promoter) | n.r. | n.r. | n.r. | + + (older mice) | n.r. | + + (older mice) | [175,176] |
| Y39C alpha-synuclein mice (mouse Thy-1 promoter) | n.r. | n.r. | n.r. | n.r. | n.r. | + + (older mice) | [179] |
| WT alpha-synuclein mice (conditional expression in the midbrain and forebrain) | n.r. | n.r. | n.r. | n.r. | n.r. | + + | [180] |
| Truncated 1-120 alpha-synuclein mice | n.r. | n.r. | n.r. | + + | n.r. | + + | [182] |
| MitoPark mice | n.r. | n.r. | n.r. | + + | n.r. | + + | [181] |
| BAC LRRK2 transgenic rats (G2019S and R1441C mutations) | n.r. | n.r. | n.r. | n.r. | + + | n.r. | [183] |
| BAC WT alpha-synuclein transgenic rats | n.r. | n.r. | n.r. | + + | n.r. | n.r. | [140] |
| AAV alpha-syn (VTA) | n.r. | n.r. | n.r. | + + | n.r. | + + | [184] |
| AAV A53T alpha-syn (SNc) | n.r. | + + | + + | n.r. | n.r. | n.r. | [156,187] |
3.2.5. Dementia
4. Clinical Relevance and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savica, R.; Rocca, W.A.; Ahlskog, J.E. When Does Parkinson Disease Start? Arch. Neurol. 2010, 67, 798–801. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathology of Nonmotor Symptoms of Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 13–62. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Barone, P.; Antonini, A.; Colosimo, C.; Marconi, R.; Morgante, L.; Avarello, T.P.; Bottacchi, E.; Cannas, A.; Ceravolo, G.; Ceravolo, R.; et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov. Disord. 2009, 24, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Jellinger, K.A. Synuclein deposition and non-motor symptoms in Parkinson disease. J. Neurol. Sci. 2011, 310, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Agarwal, P. Depression and Anxiety in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 93–104. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Publishing: Washington, DC, USA; London, UK, 2013. [Google Scholar] [CrossRef]
- Gonera, E.G.; Hof, M.V.; Berger, H.J.C.; van Weel, C.; Horstink, M.W.I.M. Symptoms and duration of the prodromal phase in parkinson’s disease. Mov. Disord. 1997, 12, 871–876. [Google Scholar] [CrossRef]
- Shiba, M.; Bower, J.H.; Maraganore, D.M.; Ms, S.K.M.; Bs, B.J.P.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Anxiety disorders and depressive disorders preceding Parkinson’s disease: A case-control study. Mov. Disord. 2000, 15, 669–677. [Google Scholar] [CrossRef]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brücke, T.; Bayés, A.; et al. The Onset of Nonmotor Symptoms in Parkinson’s disease (The ONSET PDStudy). Mov. Disord. 2014, 30, 229–237. [Google Scholar] [CrossRef]
- Remy, P.; Doder, M.; Lees, A.; Turjanski, N.; Brooks, D. Depression in Parkinson’s disease: Loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005, 128, 1314–1322. [Google Scholar] [CrossRef]
- Aarsland, D.; Påhlhagen, S.; Ballard, C.; Ehrt, U.; Svenningsson, P. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat. Rev. Neurol. 2011, 8, 35–47. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2015, 16, 22–34. [Google Scholar] [CrossRef]
- Numakawa, T.; Richards, M.; Nakajima, S.; Adachi, N.; Furuta, M.; Odaka, H.; Kunugi, H. The Role of Brain-Derived Neurotrophic Factor in Comorbid Depression: Possible Linkage with Steroid Hormones, Cytokines, and Nutrition. Front. Psychiatry 2014, 5. [Google Scholar] [CrossRef]
- Liu, J.; Dong, J.; Wang, L.; Su, Y.; Yan, P.; Sun, S. Comparative Efficacy and Acceptability of Antidepressants in Parkinson’s Disease: A Network Meta-Analysis. PLoS ONE 2013, 8, e76651. [Google Scholar] [CrossRef]
- Barone, P.; Poewe, W.; Albrecht, S.; Debieuvre, C.; Massey, D.; Rascol, O.; Tolosa, E.; Weintraub, D. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010, 9, 573–580. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Martinez-Martin, P.; Antonini, A.; Brown, R.G.; Friedman, J.H.; Onofrj, M.; Surmann, E.; Ghys, L.; Trenkwalder, C. Rotigotine and specific non-motor symptoms of Parkinson’s disease: Post hoc analysis of RECOVER. Park. Relat. Disord. 2013, 19, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Nègre-Pagès, L.; Grandjean, H.; Lapeyre-Mestre, M.; Montastruc, J.L.; Fourrier, A.; Lépine, J.P.; Rascol, O.; on behalf of the DoPaMiP Study Group. Anxious and depressive symptoms in Parkinson’s disease: The French cross-sectionnal DoPaMiP study. Mov. Disord. 2009, 25, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Mamikonyan, E. The Neuropsychiatry of Parkinson Disease: A Perfect Storm. Am. J. Geriatr. Psychiatry 2019, 27, 998–1018. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; Chen, H.; Schwarzschild, M.A.; Kawachi, I.; Ascherio, A. Prospective study of phobic anxiety and risk of Parkinson’s disease. Mov. Disord. 2003, 18, 646–651. [Google Scholar] [CrossRef]
- Pontone, G.M.; Williams, J.; Anderson, K.E.; Chase, G.; Goldstein, S.A.; Grill, S.; Bs, E.S.H.; Lehmann, S.; Little, J.T.; Margolis, R.L.; et al. Prevalence of anxiety disorders and anxiety subtypes in patients with Parkinson’s disease. Mov. Disord. 2009, 24, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Carey, G.; Görmezoğlu, M.; de Jong, J.J.; Hofman, P.A.; Backes, W.H.; Dujardin, K.; Leentjens, A. Neuroimaging of Anxiety in Parkinson’s Disease: A Systematic Review. Mov. Disord. 2020, 36, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Rektorova, I.; Balaz, M.; Svatova, J.; Zarubova, K.; Honig, I.; Dostal, V.; Sedlackova, S.; Nestrasil, I.; Mastik, J.; Bares, M.; et al. Effects of Ropinirole on Nonmotor Symptoms of Parkinson Disease. Clin. Neuropharmacol. 2008, 31, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Sauerbier, A.; Cova, I.; Rosa-Grilo, M.; Taddei, R.N.; Mischley, L.K.; Chaudhuri, K.R. Treatment of Nonmotor Symptoms in Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 132, 361–337. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Zimmermann, R.; Gschwandtner, U.; Hatz, F.; Bousleiman, H.; Schwarz, N.; Fuhr, P. Apathy in Parkinson’s disease is related to executive function, gender and age but not to depression. Front. Aging Neurosci. 2015, 6, 350–350. [Google Scholar] [CrossRef] [PubMed]
- Radakovic, R.; Gray, D.; Dudley, K.; Mioshi, E.; Dick, D.; Melchiorre, G.; Gordon, H.; Newton, J.; Colville, S.; Pal, S.; et al. Reliability and validity of the brief dimensional apathy scale. Arch. Clin. Neuropsychol. 2020, 35, 539–544. [Google Scholar] [CrossRef]
- Radakovic, R.; Davenport, R.; Starr, J.M.; Abrahams, S. Apathy dimensions in Parkinson’s disease. Int. J. Geriatr. Psychiatry 2017, 33, 151–158. [Google Scholar] [CrossRef]
- Dujardin, K.; Sockeel, P.; Devos, D.; Delliaux, M.; Krystkowiak, P.; Destée, A.; Defebvre, L. Characteristics of apathy in Parkinson’s disease. Mov. Disord. 2007, 22, 778–784. [Google Scholar] [CrossRef]
- Aarsland, D.; Brønnick, K.; Ehrt, U.; De Deyn, P.; Tekin, S.; Emre, M.; Cummings, J. Neuropsychiatric symptoms in patients with Parkinson’s disease and dementia: Frequency, profile and associated care giver stress. J. Neurol. Neurosurg. Psychiatry 2007, 78, 36–42. [Google Scholar] [CrossRef]
- Brown, D.S.; Barrett, M.J.; Flanigan, J.L.; Sperling, S. Clinical and demographic correlates of apathy in Parkinson’s disease. J. Neurol. 2019, 266, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Krack, P.; Lhommée, E.; Metereau, E.; Klinger, H.; Favre, E.; Le Bars, D.; Schmitt, E.; Bichon, A.; Pelissier, P.; et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression inde novoParkinson’s disease. Brain 2016, 139, 2486–2502. [Google Scholar] [CrossRef] [PubMed]
- Prange, S.; Metereau, E.; Maillet, A.; Lhommée, E.; Klinger, H.; Pelissier, P.; Ibarrola, D.; Heckemann, R.A.; Castrioto, A.; Tremblay, L.; et al. Early limbic microstructural alterations in apathy and depression in de novo Parkinson’s disease. Mov. Disord. 2019, 34, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Thobois, S.; Lhommée, E.; Klinger, H.; Ardouin, C.; Schmitt, E.; Bichon, A.; Kistner, A.; Castrioto, A.; Xie, J.; Fraix, V.; et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain 2013, 136, 1568–1577. [Google Scholar] [CrossRef] [PubMed]
- Leentjens, A.F.; Koester, J.; Fruh, B.; Shephard, D.T.S.; Barone, P.; Houben, J.J. The effect of pramipexole on mood and motivational symptoms in parkinson’s disease: A meta-analysis of placebo-controlled studies. Clin. Ther. 2009, 31, 89–98. [Google Scholar] [CrossRef]
- Czernecki, V. Motivation, reward, and Parkinson’s disease: Influence of dopatherapy. Neuropsychologia 2002, 40, 2257–2267. [Google Scholar] [CrossRef]
- Biundo, R.; Weis, L.; Facchini, S.; Formento-Dojot, P.; Vallelunga, A.; Pilleri, M.; Antonini, A. Cognitive profiling of Parkinson disease patients with mild cognitive impairment and dementia. Park. Relat. Disord. 2014, 20, 394–399. [Google Scholar] [CrossRef]
- Kehagia, A.A.; Barker, R.A.; Robbins, T.W. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 2010, 9, 1200–1213. [Google Scholar] [CrossRef]
- Ott, T.; Nieder, A. Dopamine and Cognitive Control in Prefrontal Cortex. Trends Cogn. Sci. 2019, 23, 213–234. [Google Scholar] [CrossRef]
- Baggio, H.C.; Junqué, C. Functional MRI in Parkinson’s Disease Cognitive Impairment. Int. Rev. Neurobiol. 2019, 144, 29–58. [Google Scholar] [CrossRef]
- Santangelo, G.; Vitale, C.; Picillo, M.; Moccia, M.; Cuoco, S.; Longo, K.; Pezzella, D.; di Grazia, A.; Erro, R.; Pellecchia, M.T.; et al. Mild Cognitive Impairment in newly diagnosed Parkinson’s disease: A longitudinal prospective study. Park. Relat. Disord. 2015, 21, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Czernecki, V.; Benchetrit, E.; Houot, M.; Pineau, F.; Mangone, G.; Corvol, J.-C.; Vidailhet, M.; Levy, R. Social cognitive impairment in early Parkinson’s disease: A novel “mild impairment”? Park. Relat. Disord. 2021, 85, 117–121. [Google Scholar] [CrossRef]
- Santangelo, G.; Vitale, C.; Trojano, L.; Errico, D.; Amboni, M.; Barbarulo, A.M.; Grossi, D.; Barone, P. Neuropsychological correlates of theory of mind in patients with early Parkinson’s disease. Mov. Disord. 2011, 27, 98–105. [Google Scholar] [CrossRef]
- Oberauer, K. Working Memory and Attention—A Conceptual Analysis and Review. J. Cognit. 2019, 2, 36. [Google Scholar] [CrossRef]
- Muslimović, D.; Post, B.; Speelman, J.D.; Schmand, B. Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 2005, 65, 1239–1245. [Google Scholar] [CrossRef]
- Wright, M.; Burns, R.; Geffen, G.; Geffen, L. Covert orientation of visual attention in Parkinson’s disease: An impairment in the maintenance of attention. Neuropsychologia 1990, 28, 151–159. [Google Scholar] [CrossRef]
- Cools, R.; D’Esposito, M. Inverted-U–Shaped Dopamine Actions on Human Working Memory and Cognitive Control. Biol. Psychiatry 2011, 69, e113–e125. [Google Scholar] [CrossRef]
- Alexander, G.A.; Delong, M.R.; Strick, P.L. Parallel Organization of Functionally Segregated Circuits Linking Basal Ganglia and Cortex. Annu. Rev. Neurosci. 1986, 9, 357–381. [Google Scholar] [CrossRef] [PubMed]
- Polito, C.; Berti, V.; Ramat, S.; Vanzi, E.; De Cristofaro, M.T.R.; Pellicanò, G.; Mungai, F.; Marini, P.; Formiconi, A.R.; Sorbi, S.; et al. Interaction of caudate dopamine depletion and brain metabolic changes with cognitive dysfunction in early Parkinson’s disease. Neurobiol. Aging 2010, 33, 206–e29. [Google Scholar] [CrossRef]
- Pillon, B.; Deweer, B.; Vidailhet, M.; Bonnet, A.-M.; Hahn-Barma, V.; Dubois, B. Is impaired memory for spatial location in Parkinson’s disease domain specific or dependent on ‘strategic’ processes? Neuropsychologia 1998, 36, 1–9. [Google Scholar] [CrossRef]
- Chahine, L.; Weintraub, D.; Hawkins, K.; Siderowf, A.; Ms, S.E.; Oakes, D.; Seibyl, J.P.; Stern, M.B.; Marek, K.; Jennings, D.; et al. Cognition in individuals at risk for Parkinson’s: Parkinson associated risk syndrome (PARS) study findings. Mov. Disord. 2015, 31, 86–94. [Google Scholar] [CrossRef]
- Mattay, V.S.; Tessitore, A.; Callicott, J.; Bertolino, A.; Goldberg, T.E.; Chase, T.N.; Hyde, T.M.; Weinberger, D.R. Dopaminergic modulation of cortical function in patients with Parkinson’s disease. Ann. Neurol. 2002, 51, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Serences, J.T.; Shomstein, S.; Leber, A.; Golay, X.; Egeth, H.E.; Yantis, S. Coordination of Voluntary and Stimulus-Driven Attentional Control in Human Cortex. Psychol. Sci. 2005, 16, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.; Stephan, K.E.; Friston, K.; Frackowiak, R.; Lees, A.; Passingham, R. Attention to action in Parkinson’s disease. Brain 2002, 125, 276–289. [Google Scholar] [CrossRef] [PubMed]
- González-Redondo, R.; García-García, D.; Clavero-Ibarra, P.; Gasca-Salas, C.; Garcia-Eulate, R.; Zubieta, J.L.; Arbizu, J.; Obeso, J.A.; Rodríguez-Oroz, M.C. Grey matter hypometabolism and atrophy in Parkinson’s disease with cognitive impairment: A two-step process. Brain 2014, 137, 2356–2367. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Del Tredici, K. Cognitive decline correlates with neuropathological stage in Parkinson’s disease. J. Neurol. Sci. 2006, 248, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Sarter, M.; Gehring, W.J.; Kozak, R. More attention must be paid: The neurobiology of attentional effort. Brain Res. Rev. 2006, 51, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Gratwicke, J.; Jahanshahi, M.; Foltynie, T. Parkinson’s disease dementia: A neural networks perspective. Brain 2015, 138, 1454–1476. [Google Scholar] [CrossRef]
- Pinto, L.; Goard, M.; Estandian, D.; Xu, M.; Kwan, A.; Lee, S.-H.; Harrison, T.C.; Feng, G.; Dan, Y. Fast modulation of visual perception by basal forebrain cholinergic neurons. Nat. Neurosci. 2013, 16, 1857–1863. [Google Scholar] [CrossRef]
- Wesnes, K.A.; McKeith, I.; Edgar, C.; Emre, M.; Lane, R. Benefits of rivastigmine on attention in dementia associated with Parkinson disease. Neurology 2005, 65, 1654–1656. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Rajkowski, J.; Cohen, J. Role of locus coeruleus in attention and behavioral flexibility. Biol. Psychiatry 1999, 46, 1309–1320. [Google Scholar] [CrossRef]
- Bezdicek, O.; Ballarini, T.; Buschke, H.; Růžička, F.; Roth, J.; Albrecht, F.; Růžička, E.; Mueller, K.; Schroeter, M.L.; Jech, R. Memory impairment in Parkinson’s disease: The retrieval versus associative deficit hypothesis revisited and reconciled. Neuropsychology 2019, 33, 391–405. [Google Scholar] [CrossRef]
- Watson, G.S.; Leverenz, J.B. Profile of Cognitive Impairment in Parkinson’s Disease. Brain Pathol. 2010, 20, 640–645. [Google Scholar] [CrossRef]
- Camina, E.; Güell-Pelayo, F. The Neuroanatomical, Neurophysiological and Psychological Basis of Memory: Current Models and Their Origins. Front. Pharmacol. 2017, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Yarnall, A.J.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Coleman, S.Y.; Firbank, M.J.; Nombela, C.; Winder-Rhodes, S.; Evans, J.R.; Rowe, J.B.; et al. Characterizing mild cognitive impairment in incident Parkinson disease: The ICICLE-PD Study. Neurology 2013, 82, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Monaco, M.; Zabberoni, S.; Peppe, A.; Perri, R.; Fadda, L.; Iannarelli, F.; Caltagirone, C.; Carlesimo, G.A. Free and Cued Recall Memory in Parkinson’s Disease Associated with Amnestic Mild Cognitive Impairment. PLoS ONE 2014, 9, e86233. [Google Scholar] [CrossRef]
- Chiaravalloti, N.; Ibarretxe-Bilbao, N.; DeLuca, J.; Rusu, O.; Pena, J.; García-Gorostiaga, I.; Ojeda, N. The source of the memory impairment in Parkinson’s disease: Acquisition versus retrieval. Mov. Disord. 2014, 29, 765–771. [Google Scholar] [CrossRef]
- Higginson, C.I.; Wheelock, V.L.; Carroll, K.E.; Sigvardt, K.A. Recognition Memory in Parkinson’s Disease With and Without Dementia: Evidence Inconsistent with the Retrieval Deficit Hypothesis. J. Clin. Exp. Neuropsychol. 2005, 27, 516–528. [Google Scholar] [CrossRef]
- Nombela, C.; Rowe, J.; Winder-Rhodes, S.E.; Hampshire, A.; Owen, A.M.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Yarnall, A.; Firbank, M.J.; et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain 2014, 137, 2743–2758. [Google Scholar] [CrossRef]
- La, C.; Linortner, P.; Bernstein, J.D.; Ua Cruadhlaoich, M.A.; Fenesy, M.; Deutsch, G.K.; Rutt, B.K.; Tian, L.; Wagner, A.D.; Zeineh, M.; et al. Hippocampal CA1 subfield predicts episodic memory impairment in Parkinson’s disease. NeuroImage Clin. 2019, 23, 101824. [Google Scholar] [CrossRef]
- Das, T.; Hwang, J.J.; Poston, K.L. Episodic recognition memory and the hippocampus in Parkinson’s disease: A review. Cortex 2018, 113, 191–209. [Google Scholar] [CrossRef]
- Pan, P.L.; Shi, H.C.; Zhong, J.G.; Xiao, P.R.; Shen, Y.; Wu, L.J.; Song, Y.Y.; He, G.X.; Li, H.L. Gray matter atrophy in Parkinson’s disease with dementia: Evidence from meta-analysis of voxel-based morphometry studies. Neurol. Sci. 2012, 34, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.A.; Aarsland, D.; Brønnick, K.S.; Meng, X.; Tekin, S.; Olin, J.T. Evaluating Rivastigmine in Mild-to-Moderate Parkinson’s Disease Dementia Using ADAS-Cog Items. Am. J. Alzheimer’s Dis. Other Dement. 2010, 25, 407–413. [Google Scholar] [CrossRef]
- Rolls, E.T. Limbic systems for emotion and for memory, but no single limbic system. Cortex 2015, 62, 119–157. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.H.; Ostlund, S.B.; Balleine, B. Reward-guided learning beyond dopamine in the nucleus accumbens: The integrative functions of cortico-basal ganglia networks. Eur. J. Neurosci. 2008, 28, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.D. Learning and memory mechanisms involved in compulsive drug use and relapse. Methods Mol. Med. 2003, 79. [Google Scholar]
- Bódi, N.; Kéri, S.; Nagy, H.; Moustafa, A.; Myers, C.E.; Daw, N.; Dibó, G.; Takáts, A.; Bereczki, D.; Gluck, M.A. Reward-learning and the novelty-seeking personality: A between- and within-subjects study of the effects of dopamine agonists on young Parkinson’s patients. Brain 2009, 132, 2385–2395. [Google Scholar] [CrossRef]
- Palminteri, S.; Lebreton, M.; Worbe, Y.; Grabli, D.; Hartmann, A.; Pessiglione, M. Pharmacological modulation of subliminal learning in Parkinson’s and Tourette’s syndromes. Proc. Natl. Acad. Sci. USA 2009, 106, 19179–19184. [Google Scholar] [CrossRef]
- Freedberg, M.; Schacherer, J.; Chen, K.-H.; Uc, E.; Narayanan, N.S.; Hazeltine, E. Separating the effect of reward from corrective feedback during learning in patients with Parkinson’s disease. Cogn. Affect. Behav. Neurosci. 2017, 21, 557–695. [Google Scholar] [CrossRef]
- Van Nuland, A.J.; Helmich, R.C.; Dirkx, M.F.; Zach, H.; Toni, I.; Cools, R.; Ouden, H.E.M.D. Effects of dopamine on reinforcement learning in Parkinson’s disease depend on motor phenotype. Brain 2020, 143, 3422–3434. [Google Scholar] [CrossRef]
- Wojtala, J.; Heber, I.A.; Neuser, P.; Heller, J.; Kalbe, E.; Rehberg, S.P.; Storch, A.; Linse, K.; Schneider, C.; Gräber, S.; et al. Cognitive decline in Parkinson’s disease: The impact of the motor phenotype on cognition. J. Neurol. Neurosurg. Psychiatry 2018, 90, 171–179. [Google Scholar] [CrossRef]
- Appollonio, I.; Grafman, J.; Clark, K.; Nichelli, P.; Zeffiro, T.; Hallett, M. Implicit and Explicit Memory in Patients With Parkinson’s Disease With and Without Dementia. Arch. Neurol. 1994, 51, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Witt, K.; Daniels, C.; Daniel, V.; Schmitt-Eliassen, J.; Volkmann, J.; Deuschl, G. Patients with Parkinson’s disease learn to control complex systems—an indication for intact implicit cognitive skill learning. Neuropsychologia 2006, 44, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- van Asselen, M.; Almeida, I.; Andre, R.; Januário, C.; Gonçalves, A.F.; Castelo-Branco, M. The role of the basal ganglia in implicit contextual learning: A study of Parkinson’s disease. Neuropsychologia 2009, 47, 1269–1273. [Google Scholar] [CrossRef]
- Nicastro, N.; Manuel, A.L.; Garibotto, V.; Burkhard, P.R.; Schnider, A. Consolidation of a Learned Skill Correlates with Dopamine SPECT Uptake in Early Parkinson’s Disease. J. Clin. Neurol. 2018, 14, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.S.; Dibble, L.E.; Olivier, G.N.; Walter, C.; Duff, K.; Schaefer, S.Y. Dopamine replacement improves motor learning of an upper extremity task in people with Parkinson disease. Behav. Brain Res. 2019, 377, 112213. [Google Scholar] [CrossRef]
- Horiba, M.; Ueki, Y.; Nojima, I.; Shimizu, Y.; Sahashi, K.; Itamoto, S.; Suzuki, A.; Yamada, G.; Matsukawa, N.; Wada, I. Impaired Motor Skill Acquisition Using Mirror Visual Feedback Improved by Transcranial Direct Current Stimulation (tDCS) in Patients With Parkinson’s Disease. Front. Neurosci. 2019, 13, 602. [Google Scholar] [CrossRef]
- Bari, A.; Robbins, T.W. Inhibition and impulsivity: Behavioral and neural basis of response control. Prog. Neurobiol. 2013, 108, 44–79. [Google Scholar] [CrossRef]
- Dalley, J.; Everitt, B.; Robbins, T. Impulsivity, Compulsivity, and Top-Down Cognitive Control. Neuron 2011, 69, 680–694. [Google Scholar] [CrossRef]
- Jiménez-Urbieta, H.; Gago, B.; de la Riva, P.; Delgado-Alvarado, M.; Marin, C.; Oroz, M.C.R. Dyskinesias and impulse control disorders in Parkinson’s disease: From pathogenesis to potential therapeutic approaches. Neurosci. Biobehav. Rev. 2015, 56, 294–314. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.A.; Claassen, D.O.; Huizenga, H.M.; Schewel, K.D.; Ridderinkhof, K.R.; Bashore, T.R.; Wildenberg, W.P.M.V.D. Dopamine Agonists and the Suppression of Impulsive Motor Actions in Parkinson Disease. J. Cogn. Neurosci. 2012, 24, 1709–1724. [Google Scholar] [CrossRef] [PubMed]
- Manza, P.; Amandola, M.; Tatineni, V.; Li, C.-S.R.; Leung, H.-C. Response inhibition in Parkinson’s disease: A meta-analysis of dopaminergic medication and disease duration effects. NPJ Park. Dis. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Borchert, R.J.; Rittman, T.; Passamonti, L.; Ye, Z.; Sami, S.; Jones, S.P.; Nombela, C.; Rodríguez, P.V.; Vatansever, D.; Rae, C.L.; et al. Atomoxetine Enhances Connectivity of Prefrontal Networks in Parkinson’s Disease. Neuropsychopharmacology 2016, 41, 2171–7. [Google Scholar] [CrossRef]
- Al-Khaled, M.; Heldmann, M.; Bolstorff, I.; Hagenah, J.; Münte, T.F. Intertemporal choice in Parkinson’s disease and restless legs syndrome. Park. Relat. Disord. 2015, 21, 1330–1335. [Google Scholar] [CrossRef]
- Milenkova, M.; Mohammadi, B.; Kollewe, K.; Schrader, C.; Fellbrich, A.; Wittfoth, M.; Dengler, R.; Münte, T.F. Intertemporal choice in Parkinson’s disease. Mov. Disord. 2011, 26, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.H.D.R.; Averbeck, B.; O’Sullivan, S.S.; Vincent, M.B.; Rosso, A.L.; Lees, A.J.; Djamshidian, A. Jumping to conclusions in untreated patients with Parkinson’s disease. Neuropsychologia 2016, 85, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.A. Cognitive Complexity and Cognitive Flexibility. Sociometry 1962, 25, 405. [Google Scholar] [CrossRef]
- Tyrer, P.J.; Slifstein, M.; Verster, J.C.; Fromme, K.; Patel, A.B.; Hahn, B.; Allgulander, C.; Cuello, A.C.; Hernandez, G.; Shizgal, P.; et al. Behavioral Flexibility: Attentional Shifting, Rule Switching and Response Reversal. In Encyclopedia of Psychopharmacology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 209–213. [Google Scholar] [CrossRef]
- Eklanker, M.; Efeenstra, M.; Edenys, D. Dopaminergic control of cognitive flexibility in humans and animals. Front. Neurosci. 2013, 7, 201. [Google Scholar] [CrossRef]
- Monchi, O.; Petrides, M.; Doyon, J.; Postuma, R.B.; Worsley, K.; Dagher, A. Neural Bases of Set-Shifting Deficits in Parkinson’s Disease. J. Neurosci. 2004, 24, 702–710. [Google Scholar] [CrossRef]
- Scheffels, J.F.; Engels, J.E.; Kalbe, E.; Kessler, J. Screening exekutiver Funktionen bei Parkinson-Patienten durch den neuen Schnelltest PAL-5. Fortschritte der Neurol.·Psychiatr. 2017, 86, 219–225. [Google Scholar] [CrossRef]
- Costa, A.; Peppe, A.; Mazzù, I.; Longarzo, M.; Caltagirone, C.; Carlesimo, G.A. Dopamine Treatment and Cognitive Functioning in Individuals with Parkinson’s Disease: The “Cognitive Flexibility” Hypothesis Seems to Work. Behav. Neurol. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Lange, F.; Seer, C.; Loens, S.; Wegner, F.; Schrader, C.; Dressler, D.; Dengler, R.; Kopp, B. Neural mechanisms underlying cognitive inflexibility in Parkinson’s disease. Neuropsychologia 2016, 93, 142–150. [Google Scholar] [CrossRef]
- Cools, R.; Barker, R.A.; Sahakian, B.; Robbins, T. l-Dopa medication remediates cognitive inflexibility, but increases impulsivity in patients with Parkinson’s disease. Neuropsychologia 2003, 41, 1431–1441. [Google Scholar] [CrossRef]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 2007, 22, 1689–1707. [Google Scholar] [CrossRef]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A. Prevalence and Characteristics of Dementia in Parkinson Disease. Arch. Neurol. 2003, 60, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney multicenter study of Parkinson’s disease: Non- L -dopa–responsive problems dominate at 15 years. Mov. Disord. 2004, 20, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Schupf, N.; Tang, M.-X.; Cote, L.J.; Louis, E.D.; Mejia, H.; Stern, Y.; Marder, K. Combined effect of age and severity on the risk of dementia in Parkinson’s disease. Ann. Neurol. 2002, 51, 722–729. [Google Scholar] [CrossRef]
- Dubois, B.; Pillon, B. Cognitive deficits in Parkinson’s disease. J. Neurol. 1996, 244, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D. Donepezil for cognitive impairment in Parkinson’s disease: A randomised controlled study. J. Neurol. Neurosurg. Psychiatry 2002, 72, 708–712. [Google Scholar] [CrossRef]
- Poewe, W.; Wolters, E.; Emre, M.; Onofrj, M.; Hsu, C.; Tekin, S.; Lane, R. Long-term benefits of rivastigmine in dementia associated with Parkinson’s disease: An active treatment extension study. Mov. Disord. 2005, 21, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Ballard, C.; Walker, Z.; Bostrom, F.; Alves, G.; Kossakowski, K.; Leroi, I.; Pozo-Rodriguez, F.; Minthon, L.; Londos, E. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: A double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009, 8, 613–618. [Google Scholar] [CrossRef]
- Campos, F.L.; Carvalho, M.M.; Cristovão, A.C.; Eje, G.; Ebaltazar, G.; Salgado, A.J.; Ekim, Y.-S.; Esousa, N. Rodent models of Parkinson’s disease: Beyond the motor symptomatology. Front. Behav. Neurosci. 2013, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Tadaiesky, M.; Dombrowski, P.; Figueiredo, C.; Cargnin-Ferreira, E.; Da Cunha, C.; Takahashi, R. Emotional, cognitive and neurochemical alterations in a premotor stage model of Parkinson’s disease. Neuroscience 2008, 156, 830–840. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.A.; Feustel, P.; Ramirez-Zamora, A.; Molho, E.; Pilitsis, J.G.; Shin, D.S. Investigation of diazepam efficacy on anxiety-like behavior in hemiparkinsonian rats. Behav. Brain Res. 2016, 301, 226–237. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Mokhemer, E.; Abuirmeileh, A.N. Beneficial effect of etazolate on depression-like behavior and, learning, and memory impairment in a model of Parkinson’s disease. Behav. Brain Res. 2018, 350, 109–115. [Google Scholar] [CrossRef]
- Liu, K.-C.; Li, J.-Y.; Tan, H.-H.; Du, C.-X.; Xie, W.; Zhang, Y.-M.; Ma, W.-L.; Zhang, L. Serotonin6 receptors in the dorsal hippocampus regulate depressive-like behaviors in unilateral 6-hydroxydopamine-lesioned Parkinson’s rats. Neuropharmacology 2015, 95, 290–298. [Google Scholar] [CrossRef]
- Antunes, M.S.; Souza, L.C.; Ladd, F.V.L.; Ladd, A.A.B.L.; Moreira, A.L.; Bortolotto, V.C.; Silva, M.R.P.; Araújo, S.M.; Prigol, M.; Nogueira, C.W.; et al. Hesperidin Ameliorates Anxiety-Depressive-Like Behavior in 6-OHDA Model of Parkinson’s Disease by Regulating Striatal Cytokine and Neurotrophic Factors Levels and Dopaminergic Innervation Loss in the Striatum of Mice. Mol. Neurobiol. 2020, 57, 3027–3041. [Google Scholar] [CrossRef]
- Loiodice, S.; Young, H.W.; Rion, B.; Méot, B.; Montagne, P.; Denibaud, A.-S.; Viel, R.; La Rochelle, C.D. Implication of nigral dopaminergic lesion and repeated L-dopa exposure in neuropsychiatric symptoms of Parkinson’s disease. Behav. Brain Res. 2018, 360, 120–127. [Google Scholar] [CrossRef]
- Jaunarajs, K.E.; George, J.; Bishop, C. L-DOPA-induced dysregulation of extrastriatal dopamine and serotonin and affective symptoms in a bilateral rat model of Parkinson’s disease. Neuroscience 2012, 218, 243–256. [Google Scholar] [CrossRef]
- Berghauzen-Maciejewska, K.; Kuter, K.; Kolasiewicz, W.; Głowacka, U.; Dziubina, A.; Ossowska, K.; Wardas, J. Pramipexole but not imipramine or fluoxetine reverses the “depressive-like” behaviour in a rat model of preclinical stages of Parkinson’s disease. Behav. Brain Res. 2014, 271, 343–353. [Google Scholar] [CrossRef]
- Oliva, A.E.; Masini, D.; Efisone, G. A mouse model of non-motor symptoms in Parkinson’s disease: Focus on pharmacological interventions targeting affective dysfunctions. Front. Behav. Neurosci. 2014, 8, 290. [Google Scholar] [CrossRef]
- Carnicella, S.; Drui, G.; Boulet, S.; Carcenac, C.; Favier, M.; Duran, T.; Savasta, M. Implication of dopamine D3 receptor activation in the reversion of Parkinson’s disease-related motivational deficits. Transl. Psychiatry 2014, 4, e401–e401. [Google Scholar] [CrossRef]
- Chiu, W.-H.; Depboylu, C.; Hermanns, G.; Maurer, L.; Windolph, A.; Oertel, W.; Ries, V.; Höglinger, G. Long-term treatment with l-DOPA or pramipexole affects adult neurogenesis and corresponding non-motor behavior in a mouse model of Parkinson’s disease. Neuropharmacology 2015, 95, 367–376. [Google Scholar] [CrossRef]
- Han, N.-R.; Kim, Y.-K.; Ahn, S.; Hwang, T.-Y.; Lee, H.; Park, H.-J. A Comprehensive Phenotype of Non-motor Impairments and Distribution of Alpha-Synuclein Deposition in Parkinsonism-Induced Mice by a Combination Injection of MPTP and Probenecid. Front. Aging Neurosci. 2021, 12. [Google Scholar] [CrossRef]
- Cunha, M.P.; Pazini, F.L.; Lieberknecht, V.; Budni, J.; Oliveira-Giacomelli, Á.; Rosa, J.; Mancini, G.; Mazzardo, L.; Colla, A.R.; Leite, M.C.; et al. MPP+-Lesioned Mice: An Experimental Model of Motor, Emotional, Memory/Learning, and Striatal Neurochemical Dysfunctions. Mol. Neurobiol. 2016, 54, 6356–6377. [Google Scholar] [CrossRef]
- Moretti, M.T.; Neis, V.B.; Matheus, F.C.; Cunha, M.; Rosa, P.B.; Ribeiro, C.M.; Rodrigues, A.L.; Prediger, R. Effects of Agmatine on Depressive-Like Behavior Induced by Intracerebroventricular Administration of 1-Methyl-4-phenylpyridinium (MPP+). Neurotox. Res. 2015, 28, 222–231. [Google Scholar] [CrossRef]
- Vučković, M.G.; Wood, R.I.; Holschneider, D.P.; Abernathy, A.; Togasaki, D.M.; Smith, A.; Petzinger, G.M.; Jakowec, M.W. Memory, mood, dopamine, and serotonin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. Neurobiol. Dis. 2008, 32, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.N.; Caudle, W.M.; Shepherd, K.R.; Noorian, A.; Jackson, C.R.; Iuvone, P.M.; Weinshenker, D.; Greene, J.G.; Miller, G.W. Nonmotor Symptoms of Parkinson’s Disease Revealed in an Animal Model with Reduced Monoamine Storage Capacity. J. Neurosci. 2009, 29, 8103–8113. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Moreira, C.G.; Morawska, M.; Masneuf, S.; Baumann, C.; Noain, D. Preliminary Evidence of Apathetic-Like Behavior in Aged Vesicular Monoamine Transporter 2 Deficient Mice. Front. Hum. Neurosci. 2016, 10, 587. [Google Scholar] [CrossRef]
- Fukui, M.; Rodriguiz, R.M.; Zhou, J.; Jiang, X.; Phillips, L.E.; Caron, M.G.; Wetsel, W.C. Vmat2 Heterozygous Mutant Mice Display a Depressive-Like Phenotype. J. Neurosci. 2007, 27, 10520–10529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-R.; Maskri, L.; Herold, C.; Bader, V.; Stichel, C.C.; Güntürkün, O.; L\ufcbbert, H. Non-motor behavioural impairments in parkin-deficient mice. Eur. J. Neurosci. 2007, 26, 1902–1911. [Google Scholar] [CrossRef]
- Hennis, M.R.; Seamans, K.W.; Marvin, M.A.; Casey, B.H.; Goldberg, M.S. Behavioral and Neurotransmitter Abnormalities in Mice Deficient for Parkin, DJ-1 and Superoxide Dismutase. PLoS ONE 2013, 8, e84894. [Google Scholar] [CrossRef]
- Rial, D.; Castro, A.A.; Machado, N.; Garção, P.; Gonçalves, F.Q.; Silva, H.; Tome, A.; Köfalvi, A.; Corti, O.; Raisman-Vozari, R.; et al. Behavioral Phenotyping of Parkin-Deficient Mice: Looking for Early Preclinical Features of Parkinson’s Disease. PLoS ONE 2014, 9, e114216. [Google Scholar] [CrossRef]
- Wang, W.; Song, N.; Jia, F.; Tang, T.; Bao, W.; Zuo, C.; Xie, J.; Jiang, H. Genomic DNA levels of mutant alpha-synuclein correlate with non-motor symptoms in an A53T Parkinson’s disease mouse model. Neurochem. Int. 2018, 114, 71–79. [Google Scholar] [CrossRef]
- Paumier, K.L.; Rizzo, S.J.S.; Berger, Z.; Chen, Y.; Gonzales, C.; Kaftan, E.; Li, L.; Lotarski, S.; Monaghan, M.; Shen, W.; et al. Behavioral Characterization of A53T Mice Reveals Early and Late Stage Deficits Related to Parkinson’s Disease. PLoS ONE 2013, 8, e70274. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.R.; Sidhu, A. Mice expressing the A53T mutant form of human alpha-synuclein exhibit hyperactivity and reduced anxiety-like behavior. J. Neurosci. Res. 2010, 88, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Oaks, A.W.; Frankfurt, M.; Finkelstein, D.I.; Sidhu, A. Age-Dependent Effects of A53T Alpha-Synuclein on Behavior and Dopaminergic Function. PLoS ONE 2013, 8, e60378. [Google Scholar] [CrossRef]
- Nuber, S.; Harmuth, F.; Kohl, Z.; Adame, A.; Trejo, M.; Schönig, K.; Zimmermann, F.; Bauer, C.; Casadei, N.; Giel, C.; et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain 2013, 136, 412–432. [Google Scholar] [CrossRef]
- Caudal, D.; Alvarsson, A.; Björklund, A.; Svenningsson, P. Depressive-like phenotype induced by AAV-mediated overexpression of human α-synuclein in midbrain dopaminergic neurons. Exp. Neurol. 2015, 273, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Cinar, E.; Yalcin-Cakmakli, G.; Saka, E.; Ulusoy, A.; Yuruker, S.; Elibol, B.; Tel, B.C. Modelling cognitive deficits in Parkinson’s disease: Is CA2 a gateway for hippocampal synucleinopathy? Exp. Neurol. 2020, 330, 113357. [Google Scholar] [CrossRef]
- Willner, P.; Towell, A.; Sampson, D.; Sophokleous, S.; Muscat, R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 1987, 93, 358–364. [Google Scholar] [CrossRef]
- Santiago, R.M.; Barbiero, J.; Gradowski, R.W.; Bochen, S.; Lima, M.M.S.; Da Cunha, C.; Andreatini, R.; Vital, M.A. Induction of depressive-like behavior by intranigral 6-OHDA is directly correlated with deficits in striatal dopamine and hippocampal serotonin. Behav. Brain Res. 2014, 259, 70–77. [Google Scholar] [CrossRef]
- Vecchia, D.D.; Kanazawa, L.K.S.; Wendler, E.; Hocayen, P.D.A.S.; Bruginski, E.; Campos, F.R.; Stern, C.A.J.; Vital, M.A.B.F.; Miyoshi, E.; Wöhr, M.; et al. Effects of ketamine on vocal impairment, gait changes, and anhedonia induced by bilateral 6-OHDA infusion into the substantia nigra pars compacta in rats: Therapeutic implications for Parkinson’s disease. Behav. Brain Res. 2018, 342, 1–10. [Google Scholar] [CrossRef]
- Chen, L.; Deltheil, T.; Turle-Lorenzo, N.; Liberge, M.; Rosier, C.; Watabe, I.; Sreng, L.; Amalric, M.; Mourre, C. SK channel blockade reverses cognitive and motor deficits induced by nigrostriatal dopamine lesions in rats. Int. J. Neuropsychopharmacol. 2014, 17, 1295–1306. [Google Scholar] [CrossRef]
- Kamińska, K.; Lenda, T.; Konieczny, J.; Czarnecka, A.M.; Lorenc-Koci, E. Depressive-like neurochemical and behavioral markers of Parkinson’s disease after 6-OHDA administered unilaterally to the rat medial forebrain bundle. Pharmacol. Rep. 2017, 69, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.M.; Campos, F.L.; Coimbra, B.; Pêgo, J.M.; Rodrigues, C.; Lima, R.; Rodrigues, A.J.; Sousa, N.; Salgado, A.J. Behavioral characterization of the 6-hydroxidopamine model of Parkinson’s disease and pharmacological rescuing of non-motor deficits. Mol. Neurodegener. 2013, 8, 14–14. [Google Scholar] [CrossRef] [PubMed]
- Gorton, L.M.; Vuckovic, M.G.; Vertelkina, N.; Petzinger, G.M.; Jakowec, M.W.; Wood, R.I. Exercise effects on motor and affective behavior and catecholamine neurochemistry in the MPTP-lesioned mouse. Behav. Brain Res. 2010, 213, 253–262. [Google Scholar] [CrossRef]
- Engeln, M.; Fasano, S.; Ahmed, S.H.; Cador, M.; Baekelandt, V.; Bezard, E.; Fernagut, P.-O. Levodopa gains psychostimulant-like properties after nigral dopaminergic loss. Ann. Neurol. 2013, 74, 140–144. [Google Scholar] [CrossRef]
- Dowd, E.; Monville, C.; Torres, E.M.; Dunnett, S. The Corridor Task: A simple test of lateralised response selection sensitive to unilateral dopamine deafferentation and graft-derived dopamine replacement in the striatum. Brain Res. Bull. 2005, 68, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Grealish, S.; Xie, L.; Kelly, M.; Dowd, E. Unilateral axonal or terminal injection of 6-hydroxydopamine causes rapid-onset nigrostriatal degeneration and contralateral motor impairments in the rat. Brain Res. Bull. 2008, 77, 312–319. [Google Scholar] [CrossRef]
- Lindgren, H.S.; Klein, A.; Dunnett, S. Nigral 6-hydroxydopamine lesion impairs performance in a lateralised choice reaction time task—Impact of training and task parameters. Behav. Brain Res. 2014, 266, 207–215. [Google Scholar] [CrossRef]
- Baunez, C.; Robbins, T. Effects of dopamine depletion of the dorsal striatum and further interaction with subthalamic nucleus lesions in an attentional task in the rat. Neuroscience 1999, 92, 1343–1356. [Google Scholar] [CrossRef]
- Smith, E.S.; Hardy, G.A.; Schallert, T.; Lee, H.J. The impact of l-dopa on attentional impairments in a rat model of Parkinson’s disease. Neuroscience 2016, 337, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Urbieta, H.; Gago, B.; Quiroga-Varela, A.; Rodríguez-Chinchilla, T.; Galán, L.M.; Oregi, A.; Belloso-Iguerategui, A.; Delgado-Alvarado, M.; Navalpotro-Gómez, I.; Marin, C.; et al. Pramipexole-induced impulsivity in mildparkinsonian rats: A model of impulse control disorders in Parkinson’s disease. Neurobiol. Aging 2018, 75, 126–135. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.J.; White, N. Parallel information processing in the water maze: Evidence for independent memory systems involving dorsal striatum and hippocampus. Behav. Neural Biol. 1994, 61, 260–270. [Google Scholar] [CrossRef]
- Costa, C.; Sgobio, C.; Siliquini, S.; Tozzi, A.; Tantucci, M.; Ghiglieri, V.; Di Filippo, M.; Pendolino, V.; de Iure, A.; Marti, M.; et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 2012, 135, 1884–1899. [Google Scholar] [CrossRef]
- Mura, A.; Feldon, J. Spatial learning in rats is impaired after degeneration of the nigrostriatal dopaminergic system. Mov. Disord. 2003, 18, 860–871. [Google Scholar] [CrossRef]
- Ferro, M.M.; Bellissimo, M.I.; Anselmo-Franci, J.A.; Angellucci, M.E.M.; Canteras, N.S.; Da Cunha, C. Comparison of bilaterally 6-OHDA- and MPTP-lesioned rats as models of the early phase of Parkinson’s disease: Histological, neurochemical, motor and memory alterations. J. Neurosci. Methods 2005, 148, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, e55718. [Google Scholar] [CrossRef]
- Sy, H.-N.; Wu, S.-L.; Wang, W.-F.; Chen, C.-H.; Huang, Y.-T.; Liou, Y.-M.; Chiou, C.-S.; Pawlak, C.R.; Ho, Y.-J. MPTP-induced dopaminergic degeneration and deficits in object recognition in rats are accompanied by neuroinflammation in the hippocampus. Pharmacol. Biochem. Behav. 2010, 95, 158–165. [Google Scholar] [CrossRef]
- Hsieh, M.-H.; Gu, S.-L.; Ho, S.-C.; Pawlak, C.R.; Lin, C.-L.; Ho, Y.-J.; Lai, T.-J.; Wu, F.-Y. Effects of MK-801 on recognition and neurodegeneration in an MPTP-induced Parkinson’s rat model. Behav. Brain Res. 2012, 229, 41–47. [Google Scholar] [CrossRef]
- Moriguchi, S.; Yabuki, Y.; Fukunaga, K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J. Neurochem. 2012, 120, 541–551. [Google Scholar] [CrossRef]
- Bonito-Oliva, A.; Pignatelli, M.; Spigolon, G.; Yoshitake, T.; Seiler, S.; Longo, F.; Piccinin, S.; Kehr, J.; Mercuri, N.B.; Nistico, R.; et al. Cognitive Impairment and Dentate Gyrus Synaptic Dysfunction in Experimental Parkinsonism. Biol. Psychiatry 2014, 75, 701–710. [Google Scholar] [CrossRef]
- De Leonibus, E.; Managò, F.; Giordani, F.; Petrosino, F.; Lopez, S.; Oliverio, A.; Amalric, M.; Mele, A.; Manag, F. Metabotropic Glutamate Receptors 5 Blockade Reverses Spatial Memory Deficits in a Mouse Model of Parkinson’s Disease. Neuropsychopharmacology 2008, 34, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.; Kouzmine, I.; Canteras, N.S.; Da Cunha, C. Lesion of the substantia nigra, pars compacta impairs delayed alternation in a Y-maze in rats. Exp. Neurol. 2005, 192, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Tanila, H.; Björklund, M.; Riekkinen, P. Cognitive Changes in Mice Following Moderate MPTP Exposure. Brain Res. Bull. 1998, 45, 577–582. [Google Scholar] [CrossRef]
- Da Cunha, C. Memory disruption in rats with nigral lesions induced by MPTP: A model for early Parkinson’s disease amnesia. Behav. Brain Res. 2001, 124, 9–18. [Google Scholar] [CrossRef]
- Dehay, B.; Fernagut, P.-O. Alpha-synuclein-based models of Parkinson’s disease. Rev. Neurol. 2016, 172, 371–378. [Google Scholar] [CrossRef]
- Magen, I.; Fleming, S.M.; Zhu, C.; Garcia, E.C.; Cardiff, K.M.; Dinh, D.; De La Rosa, K.; Sanchez, M.; Torres, E.R.; Masliah, E.; et al. Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease. Eur. J. Neurosci. 2012, 35, 870–882. [Google Scholar] [CrossRef]
- Magen, I.; Torres, E.R.; Dinh, D.; Chung, A.; Masliah, E.; Chesselet, M.-F. Social Cognition Impairments in Mice Overexpressing Alpha-Synuclein Under the Thy1 Promoter, a Model of Pre-manifest Parkinson’s Disease. J. Park. Dis. 2015, 5, 669–680. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Magen, I.; Bove, N.; Zhu, C.; Lemesre, V.; Dutta, G.; Elias, C.J.; Lester, H.A.; Chesselet, M.-F. Chronic nicotine improves cognitive and social impairment in mice overexpressing wild type α-synuclein. Neurobiol. Dis. 2018, 117, 170–180. [Google Scholar] [CrossRef]
- Hatami, A.; Chesselet, M.-F. Transgenic Rodent Models to Study Alpha-Synuclein Pathogenesis, with a Focus on Cognitive Deficits. In Behavioral Neurobiology of Huntington’s Disease and Parkinson’s Disease. Current Topics in Behavioral Neurosciences,; Springer: Berlin/Heidelberg, Germany, 2014; Volume 22, pp. 303–330. [Google Scholar] [CrossRef]
- Freichel, C.; Neumann, M.; Ballard, T.; Müller, V.; Woolley, M.; Ozmen, L.; Borroni, E.; Kretzschmar, H.A.; Haass, C.; Spooren, W.; et al. Age-dependent cognitive decline and amygdala pathology in α-synuclein transgenic mice. Neurobiol. Aging 2007, 28, 1421–1435. [Google Scholar] [CrossRef] [PubMed]
- Schell, H.; Boden, C.; Chagas, A.M.; Kahle, P.J. Impaired c-Fos and Polo-Like Kinase 2 Induction in the Limbic System of Fear-conditioned α-Synuclein Transgenic Mice. PLoS ONE 2012, 7, e50245. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.I.; Hare, D.J.; Billings, J.L.; Sedjahtera, A.; Nurjono, M.; Arthofer, E.; George, S.; Culvenor, J.G.; Bush, A.I.; Adlard, P.A. Clioquinol Improves Cognitive, Motor Function, and Microanatomy of the Alpha-Synuclein hA53T Transgenic Mice. ACS Chem. Neurosci. 2015, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Covelo, A.; Martell-Martínez, H.; Nanclares, C.; Sherman, M.A.; Okematti, E.; Meints, J.; Teravskis, P.; Gallardo, C.; Savonenko, A.V.; et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy. Acta Neuropathol. 2019, 138, 551–574. [Google Scholar] [CrossRef]
- Zhou, W.; Milder, J.B.; Freed, C.R. Transgenic Mice Overexpressing Tyrosine-to-Cysteine Mutant Human α-Synuclein. J. Biol. Chem. 2008, 283, 9863–9870. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Petrasch-Parwez, E.; Winner, B.; Winkler, J.; von Hörsten, S.; Schmidt, T.; Boy, J.; Kuhn, M.; Nguyen, H.P.; Teismann, P.; et al. Neurodegeneration and Motor Dysfunction in a Conditional Model of Parkinson’s Disease. J. Neurosci. 2008, 28, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Redus, L.; Chen, C.; Martinez, P.A.; Strong, R.; Li, S.; O’Connor, J.C. Cognitive Dysfunction Precedes the Onset of Motor Symptoms in the MitoPark Mouse Model of Parkinson’s Disease. PLoS ONE 2013, 8, e71341. [Google Scholar] [CrossRef]
- Hall, K.; Yang, S.; Sauchanka, O.; Spillantini, M.G.; Anichtchik, O. Behavioural deficits in transgenic mice expressing human truncated (1–120 amino acid) alpha-synuclein. Exp. Neurol. 2015, 264, 8–13. [Google Scholar] [CrossRef]
- Sloan, M.; Alegre-Abarrategui, J.; Potgieter, D.; Kaufmann, A.-K.; Exley, R.; Deltheil, T.; Threlfell, S.; Connor-Robson, N.; Brimblecombe, K.; Wallings, R.; et al. LRRK2BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function. Hum. Mol. Genet. 2016, 25, 951–963. [Google Scholar] [CrossRef]
- Hall, H.; Jewett, M.; Landeck, N.; Nilsson, N.; Schagerlöf, U.; Leanza, G.; Kirik, D. Characterization of Cognitive Deficits in Rats Overexpressing Human Alpha-Synuclein in the Ventral Tegmental Area and Medial Septum Using Recombinant Adeno-Associated Viral Vectors. PLoS ONE 2013, 8, e64844. [Google Scholar] [CrossRef]
- Carvalho, M.M.; Campos, F.L.; Marques, M.; Soares-Cunha, C.; Kokras, N.; Dalla, C.; Leite-Almeida, H.; Sousa, N.; Salgado, A.J. Effect of Levodopa on Reward and Impulsivity in a Rat Model of Parkinson’s Disease. Front. Behav. Neurosci. 2017, 11, 145–145. [Google Scholar] [CrossRef]
- Leite-Almeida, H.; Melo, A.; Pêgo, J.M.; Bernardo, S.; Milhazes, N.; Borges, F.; Sousa, N.; Almeida, A.; Cerqueira, J.J. Variable delay-to-signal: A fast paradigm for assessment of aspects of impulsivity in rats. Front. Behav. Neurosci. 2013, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Engeln, M.; Ansquer, S.; Dugast, E.; Bezard, E.; Belin, D.; Fernagut, P.-O. Multi-facetted impulsivity following nigral degeneration and dopamine replacement therapy. Neuropharmacology 2016, 109, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Magnard, R.; Vachez, Y.; Carcenac, C.; Boulet, S.; Houeto, J.-L.; Savasta, M.; Belin, D.; Carnicella, S. Nigrostriatal Dopaminergic Denervation Does Not Promote Impulsive Choice in the Rat: Implication for Impulse Control Disorders in Parkinson’s Disease. Front. Behav. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Tedford, S.E.; Persons, A.L.; Napier, T.C. Dopaminergic Lesions of the Dorsolateral Striatum in Rats Increase Delay Discounting in an Impulsive Choice Task. PLoS ONE 2015, 10, e0122063–e0122063. [Google Scholar] [CrossRef]
- Rokosik, S.L.; Napier, T.C. Pramipexole-Induced Increased Probabilistic Discounting: Comparison Between a Rodent Model of Parkinson’s Disease and Controls. Neuropsychopharmacology 2012, 37, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Urbieta, H.; Gago, B.; Quiroga-Varela, A.; Rodríguez-Chinchilla, T.; Galán, L.M.; Delgado-Alvarado, M.; Navalpotro-Gómez, I.; Belloso-Iguerategui, A.; Marin, C.; Rodríguez-Oroz, M.C. Motor impulsivity and delay intolerance are elicited in a dose-dependent manner with a dopaminergic agonist in parkinsonian rats. Psychopharmacology 2020, 237, 2419–2431. [Google Scholar] [CrossRef]
- Dardou, D.; Reyrolle, L.; Chassain, C.; Durif, F. Chronic pramipexole treatment induces compulsive behavior in rats with 6-OHDA lesions of the substantia nigra and ventral tegmental area. Behav. Brain Res. 2017, 332, 327–336. [Google Scholar] [CrossRef]
- Mineo, D.; Cacace, F.; Mancini, M.; Vannelli, A.; Campanelli, F.; Natale, G.; Marino, G.; Cardinale, A.; Calabresi, P.; Picconi, B.; et al. Dopamine drives binge-like consumption of a palatable food in experimental Parkinsonism. Mov. Disord. 2019, 34, 821–831. [Google Scholar] [CrossRef]
- Grospe, G.M.; Baker, P.M.; Ragozzino, M.E. Cognitive Flexibility Deficits Following 6-OHDA Lesions of the Rat Dorsomedial Striatum. Neuroscience 2018, 374, 80–90. [Google Scholar] [CrossRef]
- Kwak, C.; Lim, C.-S.; Kaang, B.-K. Assessments of cognitive abilities in a mouse model of Parkinson’s disease with a touch screen test. Behav. Brain Res. 2016, 301, 63–71. [Google Scholar] [CrossRef]
- Seip-Cammack, K.M.; Young, J.J.; Young, M.E.; Shapiro, M.L. Partial lesion of the nigrostriatal dopamine pathway in rats impairs egocentric learning but not spatial learning or behavioral flexibility. Behav. Neurosci. 2017, 131, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.A.; King, K.M.; Kortagere, S. Limitations of the rat medial forebrain lesion model to study prefrontal cortex mediated cognitive tasks in Parkinson’s disease. Brain Res. 2019, 1702, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Darvas, M.; Palmiter, R.D. Contributions of Striatal Dopamine Signaling to the Modulation of Cognitive Flexibility. Biol. Psychiatry 2011, 69, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Darvas, M.; Henschen, C.W.; Palmiter, R.D. Contributions of signaling by dopamine neurons in dorsal striatum to cognitive behaviors corresponding to those observed in Parkinson’s disease. Neurobiol. Dis. 2014, 65, 112–123. [Google Scholar] [CrossRef] [PubMed]

| Model | Pathophysology | Depressive Symptoms | Anxiety | Apathy | Ref |
|---|---|---|---|---|---|
| Paraquat | ↓ motor performance | + + | + + | n.r. | [113] |
| 6-OHDA (Bilateral, STR) | ↓ motor performance ↓ number of TH neurons (SNc) ↓ dopamine level in STR | + + | + + | + + | [115,119,146] |
| 6-OHDA (Bilateral, SNc) | ↓ TH + neurons in SNc | + + | + + | + + | [120,144,145] |
| 6-OHDA (Unilateral, MFB) | 96% loss of TH (STR); 87% loss of TH neurons (SNc) | + + | + + | + + | [117,118,147] |
| MPTP (bilateral, systemic administration) | Motor impairment & ↓ TH + neurons in SNc | + + | + + | − − | [126,127,128,129,149] |
| VMAT-2 deficient mice | ↓ striatal dopamine level↓ dopaminergic neurons of SNc alpha-synuclein agregation | + + (12–15 mo) − − (12 mo) | − − (3–5 mo) + + (6 mo) | + + (12 mo) | [130,131,132] |
| Parkin deficient mice (deletion of exon 3) | No significant motor impairments (6–21 months) | n.r. | +/− (6 and 15 mo) | n.r. | [133,134,135] |
| hA53T alpha-synuclein mice (mouse prion promoter) | Inclusions in several brain areas altered neuronal morphology, ↓ TH+ in SNc at 12 months | + + (6 mo) | + + (3 and 6 mo) − − (12 mo) | n.r. | [136,137,138,139,140] |
| BAC WT alpha-synuclein transgenic rats | Age-dependent accumulation of alpha-synuclein aggregates (mainly striatum) | n.r. | + + | n.r. | [140] |
| AAV alpha-syn (SNc) | Progressive motor impairment, alpha-synuclein agregation, ↓ DA striatum and ↓ TH+ neurons in SNc | +/− | − − | − − | [114,141,142,150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decourt, M.; Jiménez-Urbieta, H.; Benoit-Marand, M.; Fernagut, P.-O. Neuropsychiatric and Cognitive Deficits in Parkinson’s Disease and Their Modeling in Rodents. Biomedicines 2021, 9, 684. https://doi.org/10.3390/biomedicines9060684
Decourt M, Jiménez-Urbieta H, Benoit-Marand M, Fernagut P-O. Neuropsychiatric and Cognitive Deficits in Parkinson’s Disease and Their Modeling in Rodents. Biomedicines. 2021; 9(6):684. https://doi.org/10.3390/biomedicines9060684
Chicago/Turabian StyleDecourt, Mélina, Haritz Jiménez-Urbieta, Marianne Benoit-Marand, and Pierre-Olivier Fernagut. 2021. "Neuropsychiatric and Cognitive Deficits in Parkinson’s Disease and Their Modeling in Rodents" Biomedicines 9, no. 6: 684. https://doi.org/10.3390/biomedicines9060684
APA StyleDecourt, M., Jiménez-Urbieta, H., Benoit-Marand, M., & Fernagut, P.-O. (2021). Neuropsychiatric and Cognitive Deficits in Parkinson’s Disease and Their Modeling in Rodents. Biomedicines, 9(6), 684. https://doi.org/10.3390/biomedicines9060684