
An Integrative Transcriptomic and Methylation Approach for Identifying Differentially Expressed Circular RNAs Associated with DNA Methylation Change


Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Identification of Dysregulated mRNAs and circRNAs
2.3. Small RNA Data Analysis
2.4. Differential Methylation Analysis
2.5. Prediction of miRNAs Related to circRNAs
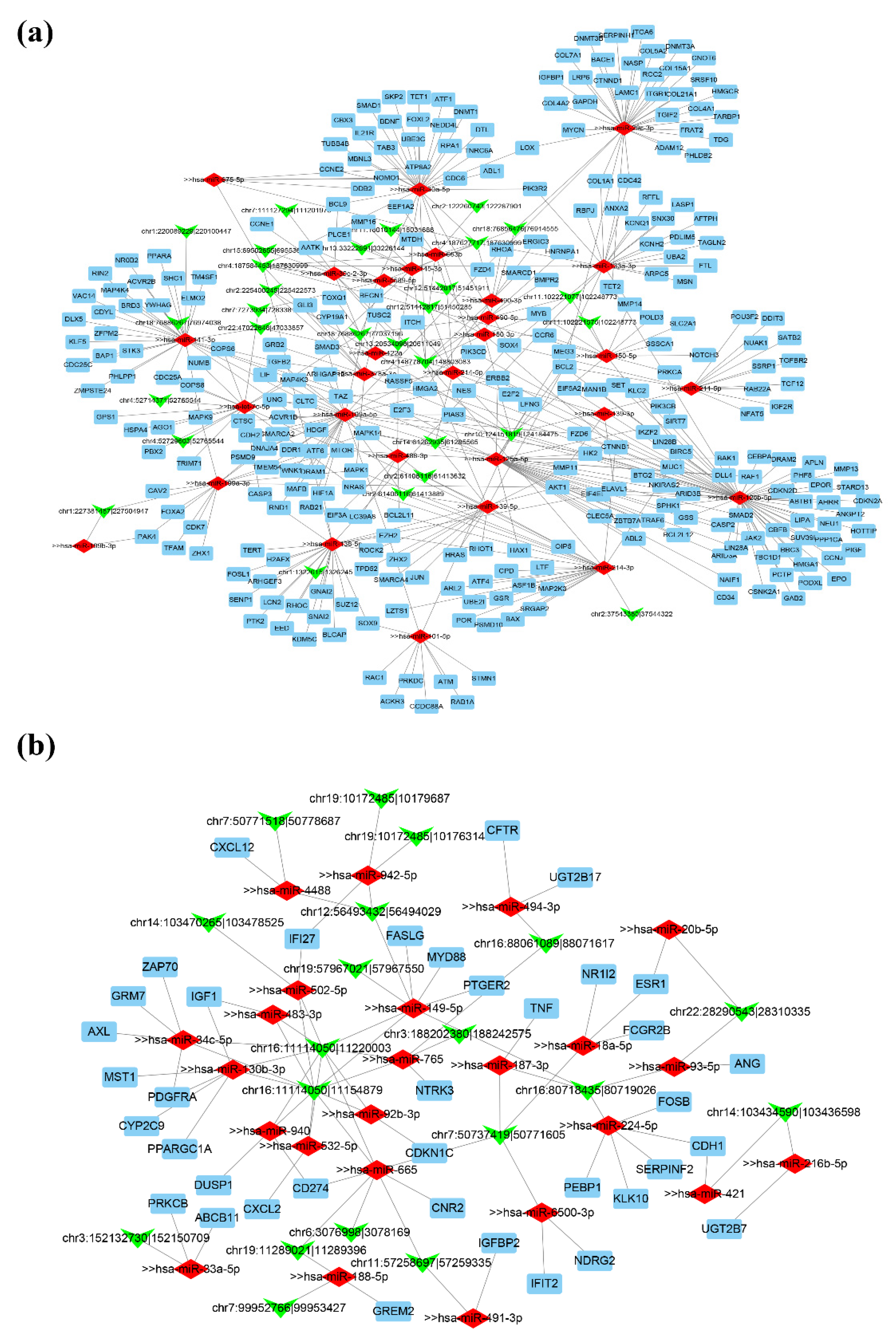
2.6. Construction of the circRNA–miRNA–mRNA Network
2.7. Integrated Functional Enrichment Analysis
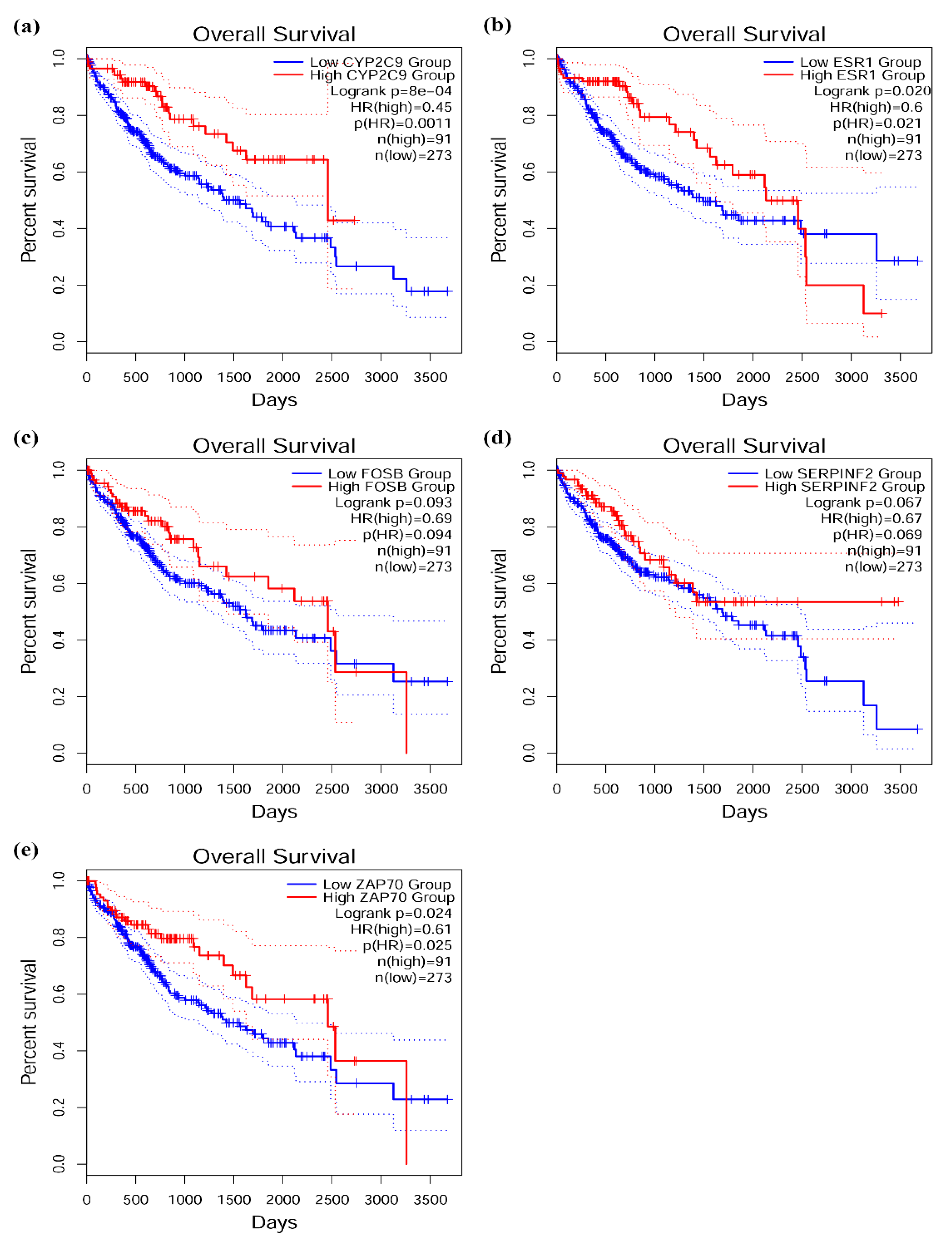
2.8. Survival Analysis of Target Genes
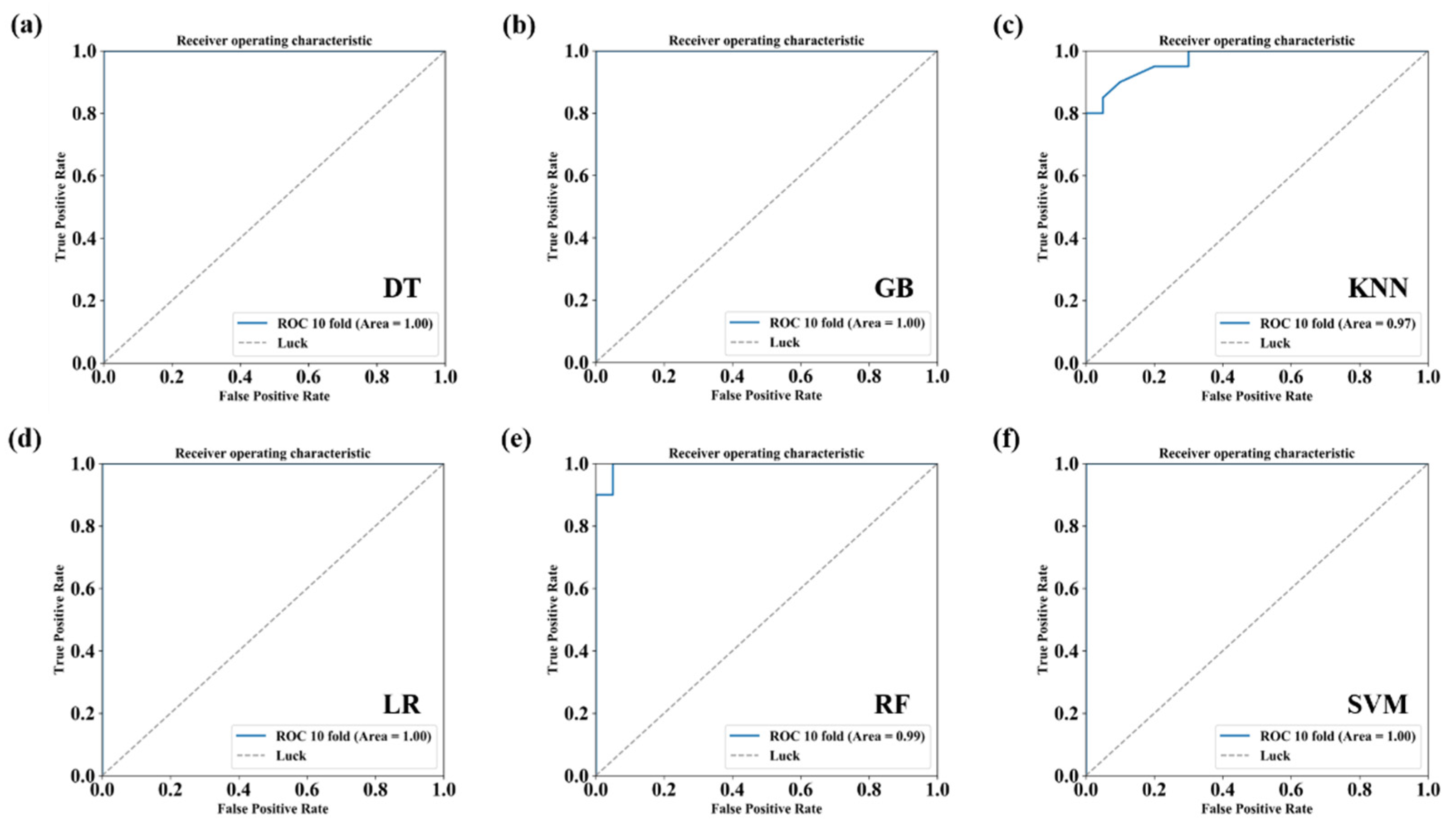
2.9. Conventional Classification Algorithms
3. Results
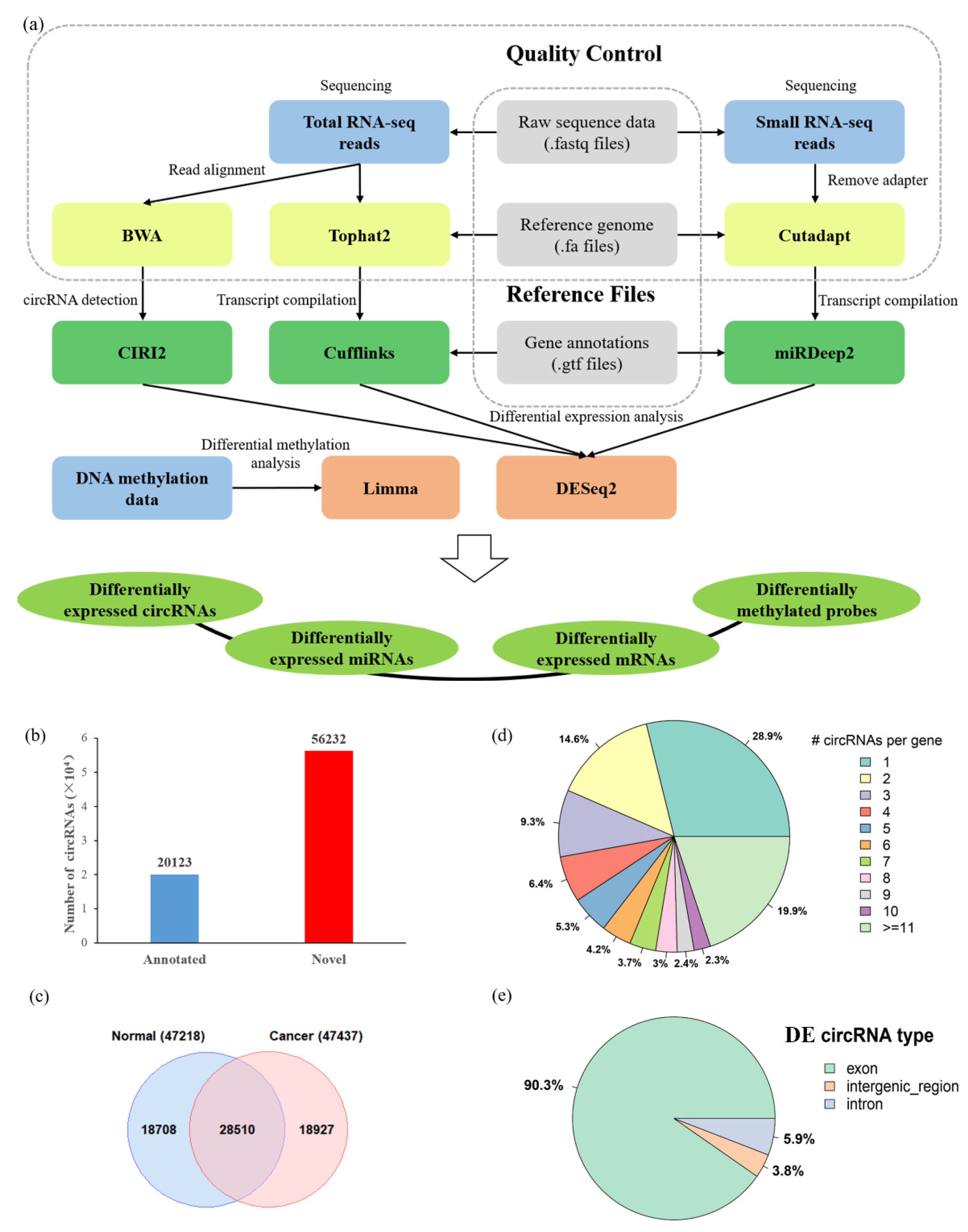
3.1. The Landscape of Differentially Expressed circRNAs in HCC
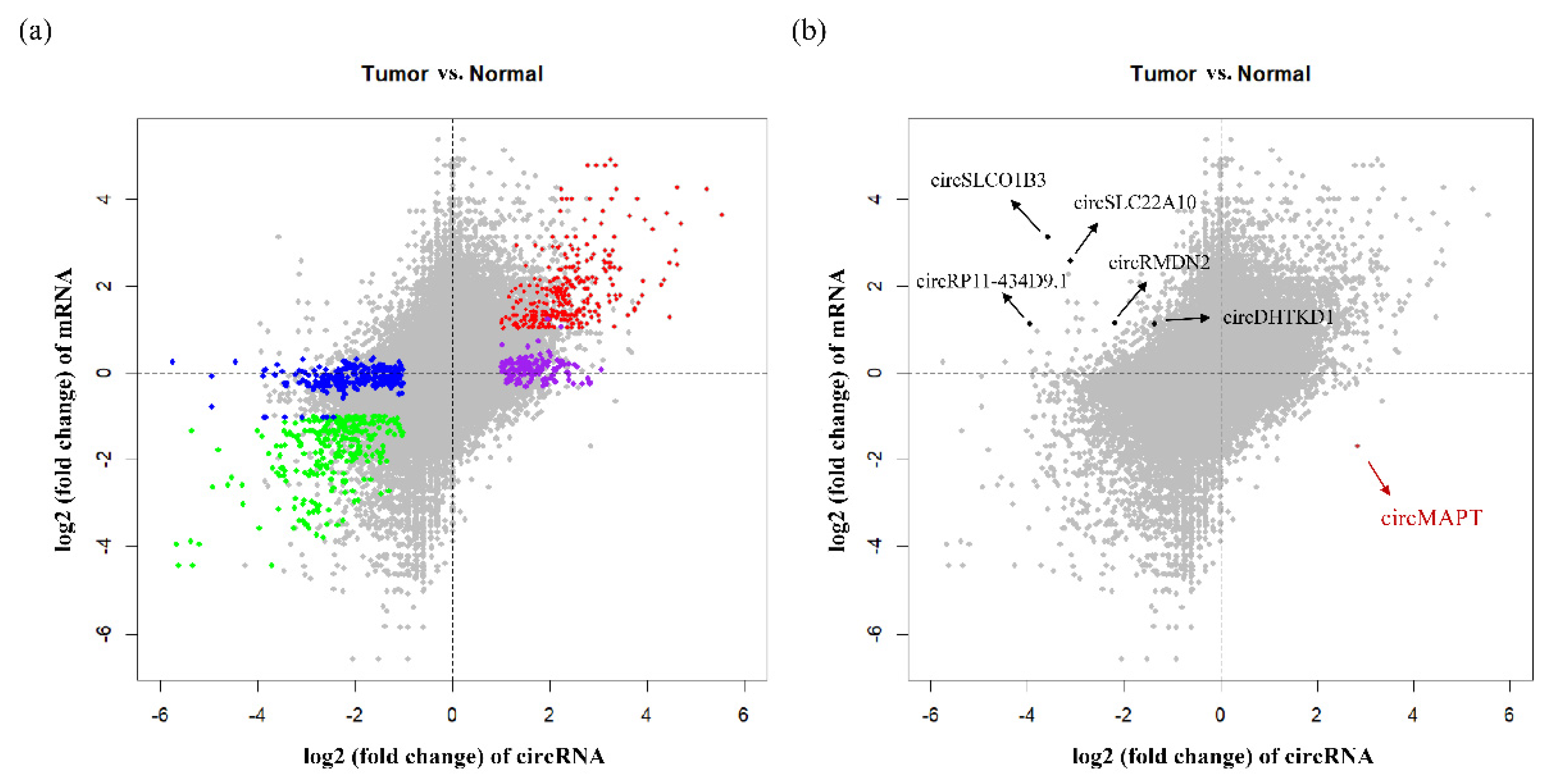
3.2. The Differential Expression Pattern of circRNAs and Genes in HCC
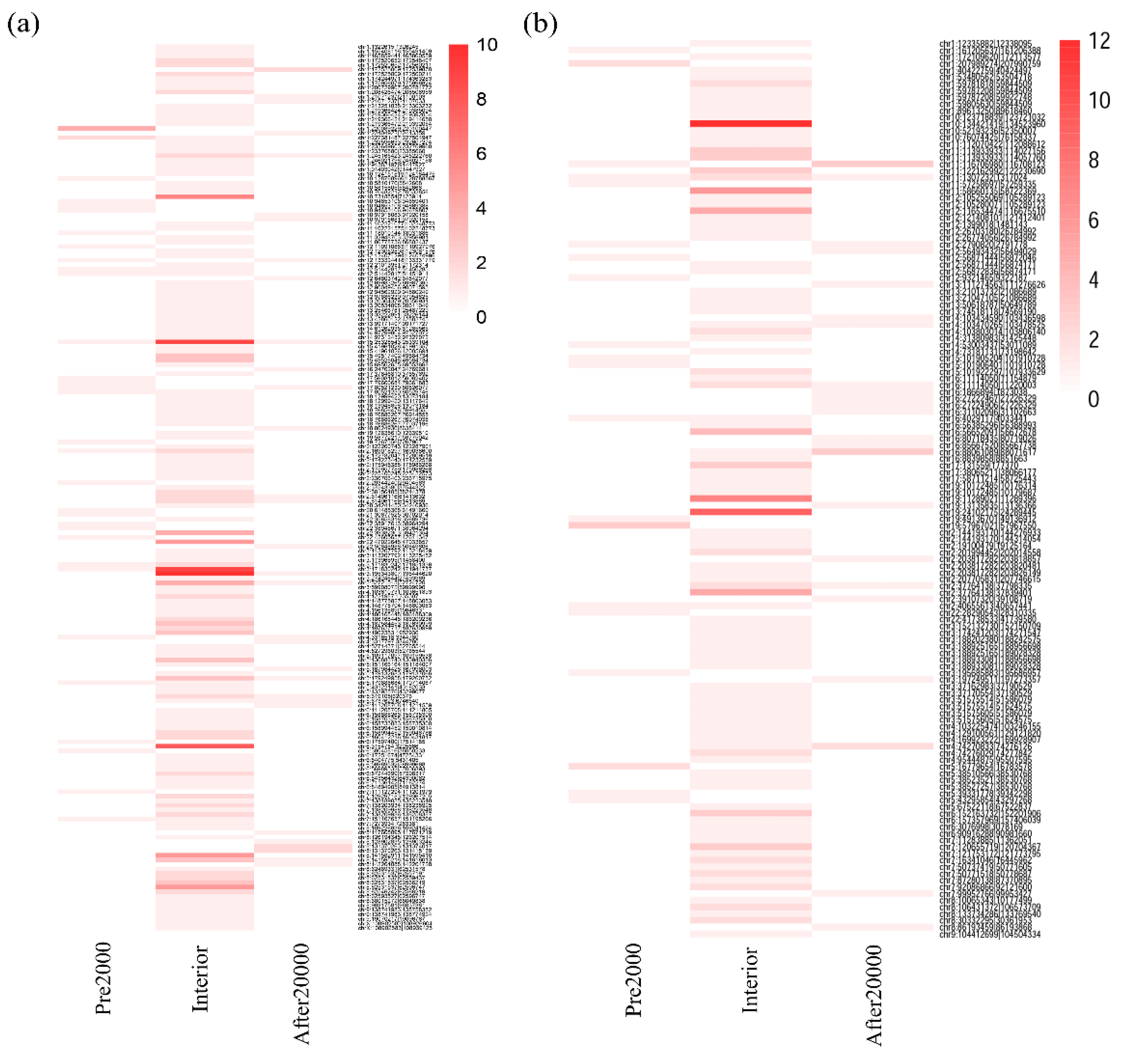
3.3. Altered DNA Methylation Events Associated with circRNAs in HCC
3.4. Aberrant DNA Methylation Profiles May have Specific Effect on Circular RNAs That Are Not Observed on Linear RNAs in HCC
3.5. Construction of ceRNA Regulatory Network in HCC
3.6. Survival Analysis of Target Genes in Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Zoratto, F.; Papa, A.; Iodice, F.; Minozzi, M.; Frati, L.; Tomao, S. Current approach in the treatment of hepatocellular carcinoma. World J. Gastrointest. Oncol. 2010, 2, 348–359. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Zhong, J.; Berhane, S.; Toyoda, H.; Cucchetti, A.; Shi, K.; Tada, T.; Chong, C.C.; Xiang, B.-D.; Li, L.-Q.; et al. Development of pre and post-operative models to predict early recurrence of hepatocellular carcinoma after surgical resection. J. Hepatol. 2018, 69, 1284–1293. [Google Scholar] [CrossRef]
- Jia, P.; Zhao, Z. Characterization of Tumor-Suppressor Gene Inactivation Events in 33 Cancer Types. Cell Rep. 2019, 26, 496–506.e3. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nat. Cell Biol. 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nat. Cell Biol. 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302. [Google Scholar] [CrossRef]
- Song, X.; Zhang, N.; Han, P.; Moon, B.-S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wu, J.; Han, P.; Zhao, Z.; Song, X. Circular RNA expression profiles and features in human tissues: A study using RNA-seq data. BMC Genom. 2017, 18 (Suppl. 6), 680. [Google Scholar] [CrossRef]
- Chen, S.; Huang, V.; Xu, X.; Livingstone, J.; Soares, F.; Jeon, J.; Zeng, Y.; Hua, J.T.; Petricca, J.; Guo, H.; et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 2019, 176, 831–843.e822. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e813. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Wei, C.-Y.; Huang, X.-Y.; Peng, R.; Yang, X.; Lu, J.-C.; Zhang, C.; Gao, C.; Cai, J.-B.; Gao, P.-T.; et al. Circular RNA circTRIM33–12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol. Cancer 2019, 18, 105. [Google Scholar] [CrossRef]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-C.; Wong, C.-W.; Liang, P.-P.; Shi, M.; Cao, Y.; Rao, S.-T.; Tsui, S.K.-W.; Waye, M.M.-Y.; Zhang, Q.; Fu, W.-M.; et al. Translation of the circular RNA circβ-catenin promotes liver cancer cell growth through activation of the Wnt pathway. Genome Biol. 2019, 20, 84. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Zhou, Y.; Wang, Q.; Zheng, Z.; Xu, B.; Wu, C.; Zhou, Q.; Hu, W.; Wu, C.; et al. A novel protein encoded by a circular RNA circPPP1R12A promotes tumor pathogenesis and metastasis of colon cancer via Hippo-YAP signaling. Mol. Cancer 2019, 18, 47. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, G.; Yan, X.; Lv, Z.; Yin, H.; Zhang, S.; Song, W.; Li, X.; Li, L.; Du, Z.; et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018, 19, 218. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Chen, Y.; Wu, Z.; Zhang, C.; Shi, W. The down-regulation of hsa_circ_0012919, the sponge for miR-125a-3p, contributes to DNA methylation of CD11a and CD70 in CD4+ T cells of systemic lupus erythematous. Clin. Sci. 2018, 132, 2285–2298. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Y.; Huang, Z.; Kong, Y.; Hu, X.; Xiao, W.; Quan, J.; Fan, X. CircRNA-5692 inhibits the progression of hepatocellular carcinoma by sponging miR-328-5p to enhance DAB2IP expression. Cell Death Dis. 2019, 10, 900. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Davalos, V.; de Moura, M.C.; Soler, M.; Perez-Salvia, M.; Bueno-Costa, A.; Setien, F.; Moran, S.; Villanueva, A.; Esteller, M. Circular RNA CpG island hypermethylation-associated silencing in human cancer. Oncotarget 2018, 9, 29208–29219. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L.; Gu, J.; Zhang, H.; Yuan, J.; Lian, Q.; Lv, G.; Wang, S.; Wu, Y.; Yang, Y.-C.T.; et al. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat. Commun. 2017, 8, 14421. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- H, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1301. [Google Scholar]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Chang, N.-W.; Shrestha, S.; Hsu, S.-D.; Lin, Y.-L.; Lee, W.-H.; Yang, C.-D.; Hong, H.-C.; Wei-Hsiang, L.; Tu, S.-J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Miuma, S.; Ichikawa, T.; Arima, K.; Takeshita, S.; Muraoka, T.; Matsuzaki, T.; Ootani, M.; Shibata, H.; Akiyama, M.; Ozawa, E.; et al. Branched-chain amino acid deficiency stabilizes insulin-induced vascular endothelial growth factor mRNA in hepatocellular carcinoma cells. J. Cell. Biochem. 2012, 113, 3113–3121. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Wang, L.; Zhang, L.; Yan, R.; Li, K.; Xing, S.; Wu, G.; Hu, L.; Jia, W.; et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016, 26, 1112–1130. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Bian, X.-L.; Chen, H.-Z.; Yang, P.-B.; Li, Y.-P.; Zhang, F.-N.; Zhang, J.-Y.; Wang, W.-J.; Zhao, W.-X.; Zhang, S.; Chen, Q.-T.; et al. Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat. Commun. 2017, 8, 14420. [Google Scholar] [CrossRef]
- Guo, P.; Ma, X.; Zhao, W.; Huai, W.; Li, T.; Qiu, Y.; Zhang, Y.; Han, L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1–TSC2 complex. Oncogene 2017, 37, 478–488. [Google Scholar] [CrossRef]
- Ji, P.; Liang, S.; Li, P.; Xie, C.; Li, J.; Zhang, K.; Zheng, X.; Feng, M.; Li, Q.; Jiao, H.; et al. Speckle-type POZ protein suppresses hepatocellular carcinoma cell migration and invasion via ubiquitin-dependent proteolysis of SUMO1/sentrin specific peptidase 7. Biochem. Biophys. Res. Commun. 2018, 502, 30–42. [Google Scholar] [CrossRef]
- Massafra, V.; Milona, A.; Vos, H.; Ramos, R.J.; Gerrits, J.; Willemsen, E.C.; Pittol, J.M.R.; Ijssennagger, N.; Houweling, M.; Prinsen, H.C.; et al. Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice. Gastroenterol. 2017, 152, 1462–1476.e10. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Mok, M.T.S.; Kang, W.; Yang, W.; Tang, W.; Wu, F.; Xu, L.; Yan, M.; Yu, Z.; Lee, S.-D.; et al. Loss of tumor suppressor IGFBP4 drives epigenetic reprogramming in hepatic carcinogenesis. Nucleic Acids Res. 2018, 46, 8832–8847. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, F. Sequence context analysis of 8.2 million single nucleotide polymorphisms in the human genome. Gene 2006, 366, 316–324. [Google Scholar] [CrossRef]
- Zhou, X.-J.; Klionsky, D.J.; Zhang, H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy 2019, 15, 908–912. [Google Scholar] [CrossRef]
- Pandey, R.; Bakay, M.; Hain, H.S.; Strenkowski, B.; Yermakova, A.; Kushner, J.A.; Orange, J.S.; Hakonarson, H. The Autoimmune Disorder Susceptibility Gene CLEC16A Restrains NK Cell Function in YTS NK Cell Line and Clec16a Knockout Mice. Front. Immunol. 2019, 10, 68. [Google Scholar] [CrossRef]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Ádány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Cui, H.; Banerjee, S.; Guo, S.; Xie, N.; Ge, J.; Jiang, D.; Zörnig, M.; Thannickal, V.J.; Liu, G. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Gerold, K.D.; Schober, K.; Probst, L.; Boerner, K.; Kim, M.-J.; Ruckdeschel, A.; Serwold, T.; Kissler, S. The Autoimmunity-Associated Gene CLEC16A Modulates Thymic Epithelial Cell Autophagy and Alters T Cell Selection. Immunity 2015, 42, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, N.; Wang, C.; Yu, Y.; Yuan, L.; Zhang, M.; Cao, X. Cyclin L2, a Novel RNA Polymerase II-associated Cyclin, Is Involved in Pre-mRNA Splicing and Induces Apoptosis of Human Hepatocellular Carcinoma Cells. J. Biol. Chem. 2004, 279, 11639–11648. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Tansey, W.P.; Hiebert, S.W.; Zhao, Z. Integrative network analysis identifies key genes and pathways in the progression of hepatitis C virus induced hepatocellular carcinoma. BMC Med. Genom. 2011, 4, 62. [Google Scholar] [CrossRef]
- Yu, H.; Lin, C.-C.; Li, Y.-Y.; Zhao, Z. Dynamic protein interaction modules in human hepatocellular carcinoma progression. BMC Syst. Biol. 2013, 7 (Suppl. 5), S2. [Google Scholar] [CrossRef]
- Hishida, M.; Nomoto, S.; Inokawa, Y.; Hayashi, M.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Tanaka, C.; Kobayashi, D.; Yamada, S.; et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int. J. Oncol. 2013, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-T.; Ye, J.; Xia, S.-L.; Zhang, Y.-F.; Su, Q.; Zhang, Z.-H.; Li, X. Polymorphism of estrogen receptor alpha (ESR1) is associated with virological response to entecavir (ETV) in nucleoside-naïve adult patients with chronic hepatitis B. Infection 2013, 41, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Nano, J.; Ghanbari, M.; Wang, W.; de Vries, P.; Dhana, K.; Muka, T.; Uitterlinden, A.G.; van Meurs, J.B.; Hofman, A.; Franco, O.H.; et al. Epigenome-Wide Association Study Identifies Methylation Sites Associated with Liver Enzymes and Hepatic Steatosis. Gastroenterology 2017, 153, 1096–1106.e2. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Age (Years) | Tumor Size (cm) | Hbv Infection |
|---|---|---|---|---|
| #3 | female | 46 | 6 | Yes |
| #6 | male | 35 | 14.1 | Yes |
| #7 | male | 42 | 38 | Yes |
| #8 | male | 61 | 8 | No |
| #10 | male | 66 | 12 | Yes |
| #11 | male | 53 | 8 | Yes |
| #12 | female | 49 | 10 | Yes |
| #13 | male | 52 | 17 | Yes |
| #14 | female | 51 | 5.5 | Yes |
| #15 | male | 47 | 5 | Yes |
| #16 | male | 43 | 10 | Yes |
| #17 | male | 60 | 3 | Yes |
| #18 | male | 61 | 10 | Yes |
| #19 | male | 43 | 7 | Yes |
| #20 | male | 64 | 10 | Yes |
| #21 | male | 40 | 7 | NA |
| #22 | male | 53 | 19 | Yes |
| #24 | male | 62 | 2.4 | Yes |
| #25 | male | 48 | 6.7 | Yes |
| #26 | male | 49 | NA | Yes |
| circRNA ID | circRNA Type | Gene ID | Gene Symbol | log2 (Fold Change) | p_adj |
|---|---|---|---|---|---|
| chr12:96381971-96384310 | exon | ENSG00000084110.6 | HAL | −3.63 | 1.96 × 10−27 |
| chr10:96701615-96732002 | exon | ENSG00000138109.9 | CYP2C9 | −4.61 | 5.57 × 10−24 |
| chr19:10183600-10184111 | exon | ENSG00000167798.12 | C3P1 | −3.97 | 6.33 × 10−24 |
| chr3:51575514-51586079 | intron | ENSG00000164080.9 | RAD54L2 | −2.73 | 2.43 × 10−22 |
| chr16:56385296-56388993 | exon | ENSG00000087258.9 | GNAO1 | −5.55 | 1.09 × 10−20 |
| chr10:96818092-96827448 | exon | ENSG00000138115.9 | CYP2C8 | −3.81 | 3.32 × 10−19 |
| chr8:62593527-62596747 | exon | ENSG00000198363.11 | ASPH | 2.73 | 7.18 × 10−17 |
| chr7:87068983-87069718 | exon | ENSG00000005471.11 | ABCB4 | −2.65 | 2.34 × 10−16 |
| chr12:56871444-56872046 | exon | ENSG00000135423.8 | GLS2 | −5.25 | 3.61 × 10−16 |
| chr5:113740135-113740553 | exon | ENSG00000080709.10 | KCNN2 | −4.42 | 4.26 × 10−16 |
| chr6:161157919-161162449 | exon | ENSG00000122194.14 | PLG | −2.94 | 6.63 × 10−16 |
| chr4:128995615-128999117 | exon | ENSG00000138709.13 | LARP1B | −1.74 | 2.69 × 10−15 |
| chr1:225140372-225156576 | exon | ENSG00000185842.10 | DNAH14 | 5.36 | 4.75 × 10−15 |
| chr16:87935518-87936126 | exon | ENSG00000174990.3 | CA5A | −3.30 | 5.00 × 10−15 |
| chr1:225140372-225161855 | exon | ENSG00000185842.10 | DNAH14 | 2.67 | 2.57 × 10−14 |
| chr7:87031149-87032597 | exon | ENSG00000005471.11 | ABCB4 | −4.37 | 2.99 × 10−14 |
| chr11:122162992-122165713 | intron | ENSG00000255090.1 | RP11-820L6.1 | −4.48 | 5.70 × 10−14 |
| chr4:1902353-1936989 | exon | ENSG00000109685.13 | WHSC1 | 3.52 | 7.04 × 10−14 |
| chr13:46577274-46619651 | exon | ENSG00000123200.12 | ZC3H13 | −2.56 | 5.98 × 10−13 |
| chr8:131370263-131374017 | exon | ENSG00000153317.10 | ASAP1 | 2.11 | 9.06 × 10−13 |
| Dysregulated circRNAs | Number (by Region) | Hypomethylated Sites | Hypermethylated Sites | |
|---|---|---|---|---|
| Upregulated | 195 | 31 (After2000) | 5 | 23 |
| 155 (Interior) | 17 | 179 | ||
| 32 (Pre2000) | 3 | 27 | ||
| Downregulated | 134 | 23 (After2000) | 8 | 19 |
| 102 (Interior) | 30 | 113 | ||
| 19 (Pre2000) | 3 | 20 | ||
| Genes | Expression | Number | Methylation | Promoter | Gene body | Number |
|---|---|---|---|---|---|---|
| Parental genes of upregulated circRNAs | With DE | 436 | With DM | 121 | 345 | 367 |
| Without DM | 237 | 88 | 69 | |||
| Without probe | NA | NA | NA | |||
| Without DE | 187 | With DM | 54 | 125 | 146 | |
| Without DM | 86 | 59 | 38 | |||
| Without probe | NA | NA | 3 | |||
| Parental genes of downregulated circRNAs | With DE | 337 | With DM | 84 | 266 | 283 |
| Without DM | 180 | 66 | 49 | |||
| Without probe | NA | NA | NA | |||
| Without DE | 147 | With DM | 32 | 107 | 112 | |
| Without DM | 84 | 32 | 29 | |||
| Without probe | NA | NA | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, T.; Wang, L.; Jia, P.; Song, X.; Zhao, Z. An Integrative Transcriptomic and Methylation Approach for Identifying Differentially Expressed Circular RNAs Associated with DNA Methylation Change. Biomedicines 2021, 9, 657. https://doi.org/10.3390/biomedicines9060657
Xu T, Wang L, Jia P, Song X, Zhao Z. An Integrative Transcriptomic and Methylation Approach for Identifying Differentially Expressed Circular RNAs Associated with DNA Methylation Change. Biomedicines. 2021; 9(6):657. https://doi.org/10.3390/biomedicines9060657
Chicago/Turabian StyleXu, Tianyi, LiPing Wang, Peilin Jia, Xiaofeng Song, and Zhongming Zhao. 2021. "An Integrative Transcriptomic and Methylation Approach for Identifying Differentially Expressed Circular RNAs Associated with DNA Methylation Change" Biomedicines 9, no. 6: 657. https://doi.org/10.3390/biomedicines9060657
APA StyleXu, T., Wang, L., Jia, P., Song, X., & Zhao, Z. (2021). An Integrative Transcriptomic and Methylation Approach for Identifying Differentially Expressed Circular RNAs Associated with DNA Methylation Change. Biomedicines, 9(6), 657. https://doi.org/10.3390/biomedicines9060657