
Neural Plasticity in the Brain during Neuropathic Pain


Abstract
1. Introduction
2. The Primary Somatosensory Cortex
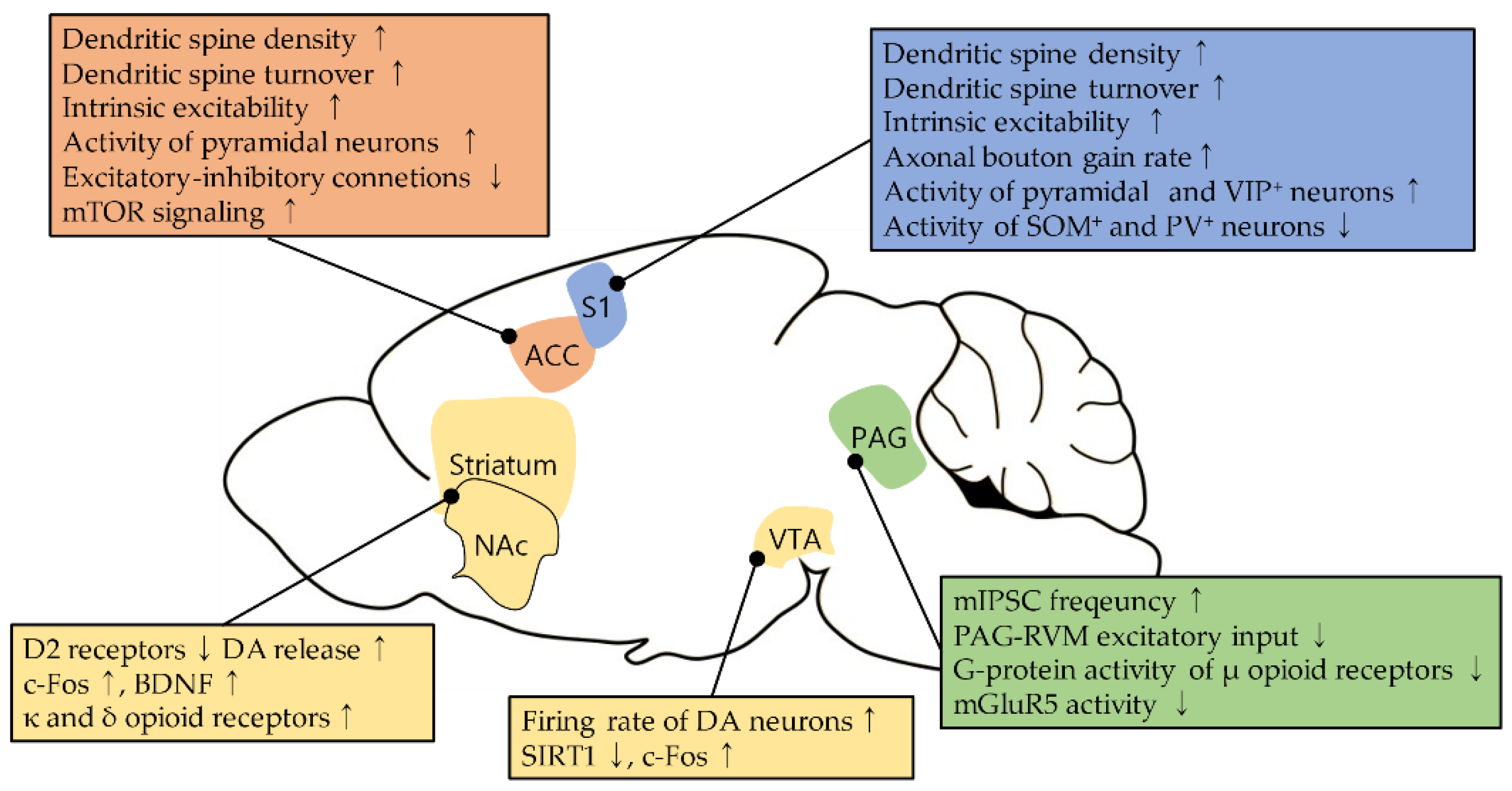
2.1. Neural Plasticity in the S1 during Neuropathic Pain
2.2. Experimental Manipulations to Reverse the Neural Plasticity
3. The Anterior Cingulate Cortex
3.1. Neural Plasticity in the ACC during Neuropathic Pain
3.2. Modulation of Neural Plasticity in the ACC
4. The Periaqueductal Gray
4.1. Changes in Opioid Sensitivity in the PAG during Neuropathic Pain
4.2. Plasticity in Glutamatergic Pathway in the PAG during Neuropathic Pain
5. The Basal Ganglia
Neural Plasticity in the Basal Ganglia during Neuropathic Pain and Its Modulation
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.C.; Treede, R.-D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Asbury, A.K. Diabetic neuropathy. Annal. Neurol. 1984, 15, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef]
- Huang, J.; Gadotti, V.M.; Chen, L.; Souza, I.A.; Huang, S.; Wang, D.; Ramakrishnan, C.; Deisseroth, K.; Zhang, Z.; Zamponi, G.W. A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci. 2019, 22, 1659–1668. [Google Scholar] [CrossRef]
- Bonnet, U.; Scherbaum, N. How addictive are gabapentin and pregabalin? A systematic review. Eur. Neuropsychopharmacol. 2017, 27, 1185–1215. [Google Scholar] [CrossRef]
- Wiffen, P.J.; Derry, S.; Bell, R.F.; Rice, A.S.; Tölle, T.R.; Phillips, T.; Moore, R.A. Gabapentin for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 6, Cd007938. [Google Scholar] [CrossRef]
- Martínez-Navarro, M.; Maldonado, R.; Baños, J.-E. Why mu-opioid agonists have less analgesic efficacy in neuropathic pain? Eur. J. Pain 2019, 23, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Singh, N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res. 2011, 1381, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2017, 18, 20–30. [Google Scholar] [CrossRef]
- Kim, W.; Kim, S.K.; Nabekura, J. Functional and structural plasticity in the primary somatosensory cortex associated with chronic pain. J. Neurochem. 2017, 141, 499–506. [Google Scholar] [CrossRef]
- Zhuo, M. Long-term cortical synaptic changes contribute to chronic pain and emotional disorders. Neurosci. Lett. 2019, 702, 66–70. [Google Scholar] [CrossRef]
- Sarnthein, J.; Stern, J.; Aufenberg, C.; Rousson, V.; Jeanmonod, D. Increased EEG power and slowed dominant frequency in patients with neurogenic pain. Brain A J. Neurol. 2005, 129, 55–64. [Google Scholar] [CrossRef]
- Min, Z. A Synaptic model for pain: Long-term potentiation in the anterior cingulate cortex. Mol. Cells 2007, 23, 259–271. [Google Scholar]
- Ossipov, M.H.; Morimura, K.; Porreca, F. Descending pain modulation and chronification of pain. Curr. Opin. Support Palliat. Care 2014, 8, 143–151. [Google Scholar] [CrossRef]
- Torta, R.; Ieraci, V.; Zizzi, F. A Review of the Emotional Aspects of Neuropathic Pain: From Comorbidity to Co-Pathogenesis. Pain Ther. 2017, 6, 11–17. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, C.-E.; Yoon, H.; Kim, S.K.; Kim, S.J. S1 Employs Feature-Dependent Differential Selectivity of Single Cells and Distributed Patterns of Populations to Encode Mechanosensations. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.L.; Minoshima, S.; Morrow, T.J.; Koeppe, R.A. Comparison of human cerebral activation pattern during cutaneous warmth, heat pain, and deep cold pain. J. Neurophysiol. 1996, 76, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ploner, M.; Schmitz, F.; Freund, H.J.; Schnitzler, A. Differential organization of touch and pain in human primary somatosensory cortex. J. Neurophysiol. 2000, 83, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Coghill, R.C.; Sang, C.N.; Maisog, J.M.; Iadarola, M.J. Pain Intensity Processing Within the Human Brain: A Bilateral, Distributed Mechanism. J. Neurophysiol. 1999, 82, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Kenshalo, D.R., Jr.; Anton, F.; Dubner, R. The detection and perceived intensity of noxious thermal stimuli in monkey and in human. J. Neurophysiol. 1989, 62, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Uhelski, M.L.; Davis, M.A.; Fuchs, P.N. Pain affect in the absence of pain sensation: Evidence of asomaesthesia after somatosensory cortex lesions in the rat. Pain 2012, 153, 885–892. [Google Scholar] [CrossRef]
- Jin, Y.; Meng, Q.; Mei, L.; Zhou, W.; Zhu, X.; Mao, Y.; Xie, W.; Zhang, X.; Luo, M.-H.; Tao, W.; et al. A somatosensory cortex input to the caudal dorsolateral striatum controls comorbid anxiety in persistent pain. Pain 2020, 161, 416–428. [Google Scholar] [CrossRef]
- Singh, A.; Patel, D.; Li, A.; Hu, L.; Zhang, Q.; Liu, Y.; Guo, X.; Robinson, E.; Martinez, E.; Doan, L.; et al. Mapping Cortical Integration of Sensory and Affective Pain Pathways. Curr. Biol. 2020, 30, 1703–1715. [Google Scholar] [CrossRef]
- Bornhövd, K.; Quante, M.; Glauche, V.; Bromm, B.; Weiller, C.; Büchel, C. Painful stimuli evoke different stimulus–response functions in the amygdala, prefrontal, insula and somatosensory cortex: A single-trial fMRI study. Brain A J. Neurol. 2002, 125, 1326–1336. [Google Scholar] [CrossRef]
- Chen, L.M.; Friedman, R.M.; Roe, A.W. Area-specific representation of mechanical nociceptive stimuli within SI cortex of squirrel monkeys. Pain 2009, 141, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kenshalo, D.R., Jr.; Chudler, E.H.; Anton, F.; Dubner, R. SI nociceptive neurons participate in the encoding process by which monkeys perceive the intensity of noxious thermal stimulation. Brain Res. 1988, 454, 378–382. [Google Scholar] [CrossRef]
- Krout, K.E.; Loewy, A.D. Parabrachial nucleus projections to midline and intralaminar thalamic nuclei of the rat. J. Comp. Neurol. 2000, 428, 475–494. [Google Scholar] [CrossRef]
- Deng, J.; Zhou, H.; Lin, J.-K.; Shen, Z.-X.; Chen, W.-Z.; Wang, L.-H.; Li, Q.; Mu, D.; Wei, Y.-C.; Xu, X.-H.; et al. The Parabrachial nucleus directly channels spinal nociceptive signals to the intralaminar thalamic nuclei, but not the amygdala. Neuron 2020, 107, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Gustin, S.M.; Peck, C.C.; Cheney, L.B.; Macey, P.M.; Murray, G.M.; Henderson, L.A. Pain and plasticity: Is chronic pain always associated with somatosensory cortex activity and reorganization? J. Neurosci. 2012, 32, 14874–14884. [Google Scholar] [CrossRef] [PubMed]
- Peyron, R.; Schneider, F.; Faillenot, I.; Convers, P.; Barral, F.-G.; Garcia-Larrea, L.; Laurent, B. An fMRI study of cortical representation of mechanical allodynia in patients with neuropathic pain. Neurology 2004, 63, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef]
- Ishikawa, T.; Eto, K.; Kim, S.K.; Wake, H.; Takeda, I.; Horiuchi, H.; Moorhouse, A.J.; Ishibashi, H.; Nabekura, J. Cortical astrocytes prime the induction of spine plasticity and mirror image pain. Pain 2018, 159, 1592–1606. [Google Scholar] [CrossRef]
- Kim, S.K.; Kato, G.; Ishikawa, T.; Nabekura, J. Phase-specific plasticity of synaptic structures in the somatosensory cortex of living mice during neuropathic pain. Mol. Pain 2011, 7, 87. [Google Scholar] [CrossRef]
- Xiong, W.; Ping, X.; Ripsch, M.S.; Chavez, G.S.C.; Hannon, H.E.; Jiang, K.; Bao, C.; Jadhav, V.; Chen, L.; Chai, Z.; et al. Enhancing excitatory activity of somatosensory cortex alleviates neuropathic pain through regulating homeostatic plasticity. Sci. Rep. 2017, 7, 12743. [Google Scholar] [CrossRef]
- Cichon, J.; Blanck, T.J.J.; Gan, W.-B.; Yang, G. Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat. Neurosci. 2017, 20, 1122–1132. [Google Scholar] [CrossRef]
- Wei, J.-a.; Hu, X.; Zhang, B.; Liu, L.; Chen, K.; So, K.-F.; Li, M.; Zhang, L. Electroacupuncture activates inhibitory neural circuits in the somatosensory cortex to relieve neuropathic pain. iScience 2021, 24, 102066. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Z.; Ma, J.; Cui, S.; Yi, M.; Guo, H.; Wan, Y. Extracting neural oscillation signatures of laser-induced nociception in pain-related regions in rats. Front. Neural Circuits 2017, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Ploner, M.; Sorg, C.; Gross, J. Brain Rhythms of Pain. Trends Cognit. Sci. 2017, 21, 100–110. [Google Scholar] [CrossRef]
- LeBlanc, B.W.; Bowary, P.M.; Chao, Y.C.; Lii, T.R.; Saab, C.Y. Electroencephalographic signatures of pain and analgesia in rats. Pain 2016, 157, 2330–2340. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, B.W.; Lii, T.R.; Silverman, A.E.; Alleyne, R.T.; Saab, C.Y. Cortical theta is increased while thalamocortical coherence is decreased in rat models of acute and chronic pain. PAIN 2014, 155, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.L.; Oswald, M.J.; Heinl, C.; Retana Romero, O.A.; Kaushalya, S.K.; Monyer, H.; Kuner, R. Gamma oscillations in somatosensory cortex recruit prefrontal and descending serotonergic pathways in aversion and nociception. Nat. Commun. 2019, 10, 983. [Google Scholar] [CrossRef]
- Kim, S.K.; Nabekura, J. Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J. Neurosci. 2011, 31, 5477–5482. [Google Scholar] [CrossRef]
- Zikopoulos, B.; Barbas, H. Pathways for emotions and attention converge on the thalamic reticular nucleus in primates. J. Neurosci. 2012, 32, 5338–5350. [Google Scholar] [CrossRef]
- Ferreira-Gomes, J.; Neto, F.L.; Castro-Lopes, J.M. GABA(B2) receptor subunit mRNA decreases in the thalamus of monoarthritic animals. Brain Res. Bull. 2006, 71, 252–258. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, B.W.; Cross, B.; Smith, K.A.; Roach, C.; Xia, J.; Chao, Y.-C.; Levitt, J.; Koyama, S.; Moore, C.I.; Saab, C.Y. Thalamic Bursts Down-regulate Cortical Theta and Nociceptive Behavior. Sci. Rep. 2017, 7, 2482. [Google Scholar] [CrossRef]
- Eto, K.; Ishibashi, H.; Yoshimura, T.; Watanabe, M.; Miyamoto, A.; Ikenaka, K.; Moorhouse, A.J.; Nabekura, J. Enhanced GABAergic activity in the mouse primary somatosensory cortex is insufficient to alleviate chronic pain behavior with reduced expression of neuronal potassium-chloride cotransporter. J. Neurosci. 2012, 32, 16552–16559. [Google Scholar] [CrossRef]
- Bush, G.; Luu, P.; Posner, M.I. Cognitive and emotional influences in anterior cingulate cortex. Trend. Cognitive Sci. 2000, 4, 215–222. [Google Scholar] [CrossRef]
- Bush, G.; Vogt, B.A.; Holmes, J.; Dale, A.M.; Greve, D.; Jenike, M.A.; Rosen, B.R. Dorsal anterior cingulate cortex: A role in reward-based decision making. Proc. Nat. Acad. Sci. USA 2002, 99, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Decety, J.; Jackson, P.L. The functional architecture of human empathy. Behav. Cognitive Neurosci. Rev. 2004, 3, 71–100. [Google Scholar] [CrossRef]
- Rainville, P.; Duncan, G.H.; Price, D.D.; Carrier, B.; Bushnell, M.C. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 1997, 277, 968–971. [Google Scholar] [CrossRef]
- Treede, R.D.; Kenshalo, D.R.; Gracely, R.H.; Jones, A.K. The cortical representation of pain. Pain 1999, 79, 105–111. [Google Scholar] [CrossRef]
- Qu, C.; King, T.; Okun, A.; Lai, J.; Fields, H.L.; Porreca, F. Lesion of the rostral anterior cingulate cortex eliminates the aversiveness of spontaneous neuropathic pain following partial or complete axotomy. Pain 2011, 152, 1641–1648. [Google Scholar] [CrossRef]
- Johansen, J.P.; Fields, H.L.; Manning, B.H. The affective component of pain in rodents: Direct evidence for a contribution of the anterior cingulate cortex. Proc. Nat. Acad. Sci. USA 2001, 98, 8077–8082. [Google Scholar] [CrossRef]
- Rainville, P. Brain mechanisms of pain affect and pain modulation. Curr. Opin. Neurobiol. 2002, 12, 195–204. [Google Scholar] [CrossRef]
- LaGraize, S.C.; Labuda, C.J.; Rutledge, M.A.; Jackson, R.L.; Fuchs, P.N. Differential effect of anterior cingulate cortex lesion on mechanical hypersensitivity and escape/avoidance behavior in an animal model of neuropathic pain. Exp. Neurol. 2004, 188, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Martinez, E.; Kulkarni, P.M.; Zhang, Q.; Hou, Q.; Rosenberg, D.; Talay, R.; Shalot, L.; Zhou, H.; Wang, J.; et al. Cortical pain processing in the rat anterior cingulate cortex and primary somatosensory cortex. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Iwata, K.; Kamo, H.; Ogawa, A.; Tsuboi, Y.; Noma, N.; Mitsuhashi, Y.; Taira, M.; Koshikawa, N.; Kitagawa, J. Anterior cingulate cortical neuronal activity during perception of noxious thermal stimuli in monkeys. J. Neurophysiol. 2005, 94, 1980–1991. [Google Scholar] [CrossRef]
- Smith, M.L.; Asada, N.; Malenka, R.C. Anterior cingulate inputs to nucleus accumbens control the social transfer of pain and analgesia. Science 2021, 371, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bragin, E.O.; Yeliseeva, Z.V.; Vasilenko, G.F.; Meizerov, E.E.; Chuvin, B.T.; Durinyan, R.A. Cortical projections to the periaqueductal grey in the cat: A retrograde horseradish peroxidase study. Neurosci. Lett. 1984, 51, 271–275. [Google Scholar] [CrossRef]
- Peyron, R.; Faillenot, I.; Mertens, P.; Laurent, B.; Garcia-Larrea, L. Motor cortex stimulation in neuropathic pain. Correlations between analgesic effect and hemodynamic changes in the brain. A PET study. NeuroImage 2007, 34, 310–321. [Google Scholar] [CrossRef]
- Vogt, B.A. Pain and emotion interactions in subregions of the cingulate gyrus. Nat. Rev. Neurosci. 2005, 6, 533–544. [Google Scholar] [CrossRef]
- Chen, T.; Koga, K.; Descalzi, G.; Qiu, S.; Wang, J.; Zhang, L.-S.; Zhang, Z.-J.; He, X.-B.; Qin, X.; Xu, F.-Q.; et al. Postsynaptic potentiation of corticospinal projecting neurons in the anterior cingulate cortex after nerve injury. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef]
- Ko, H.-G.; Choi, J.-H.; Park, D.I.; Kang, S.J.; Lim, C.-S.; Sim, S.-E.; Shim, J.; Kim, J.-I.; Kim, S.; Choi, T.-H.; et al. Rapid Turnover of Cortical NCAM1 Regulates Synaptic Reorganization after Peripheral Nerve Injury. Cell Rep. 2018, 22, 748–759. [Google Scholar] [CrossRef]
- Li, X.Y.; Ko, H.G.; Chen, T.; Descalzi, G.; Koga, K.; Wang, H.; Kim, S.S.; Shang, Y.; Kwak, C.; Park, S.W.; et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 2010, 330, 1400–1404. [Google Scholar] [CrossRef]
- Blom, S.M.; Pfister, J.-P.; Santello, M.; Senn, W.; Nevian, T. Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. J. Neurosci. 2014, 34, 5754–5764. [Google Scholar] [CrossRef]
- Zhao, R.; Zhou, H.; Huang, L.; Xie, Z.; Wang, J.; Gan, W.-B.; Yang, G. Neuropathic pain causes pyramidal neuronal hyperactivity in the anterior cingulate cortex. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Yang, Z.; Tan, Q.; Cheng, D.; Zhang, L.; Zhang, J.; Gu, E.-w.; Fang, W.; Lu, X.; Liu, X. The changes of intrinsic excitability of pyramidal neurons in anterior cingulate cortex in neuropathic pain. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Li, X.Y.; Ko, H.G.; Chen, T.; Collingridge, G.L.; Kaang, B.K.; Zhuo, M. Erasing injury-related cortical synaptic potentiation as a new treatment for chronic pain. J. Mol. Med. 2011, 89, 847–855. [Google Scholar] [CrossRef]
- Lisman, J. Memory erasure by very high concentrations of ZIP may not be due to PKM—Zeta. Hippocampus 2012, 22, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.J.; Bachman, J.L.; Johnson, R.; Yu, Y.; Huganir, R.L. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 2013, 493, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef]
- Um, S.W.; Kim, M.J.; Leem, J.W.; Bai, S.J.; Lee, B.H. Pain-relieving effects of mTOR inhibitor in the anterior cingulate cortex of neuropathic rats. Mol. Neurobiol. 2019, 56, 2482–2494. [Google Scholar] [CrossRef]
- De Oliveira, F.R.; Fantucci, M.Z.; Adriano, L.; Valim, V.; Cunha, T.M.; Louzada-Junior, P.; Rocha, E.M. Neurological and inflammatory manifestations in Sjögren’s Syndrome: The role of the kynurenine metabolic pathway. Int. J. Mol. Sci. 2018, 19, 3953. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.-M.; Han, R.; Wang, Z.-Z.; Gao, Y.-L.; Zhu, X.-Y.; Yu, X.; Du, G.-Y.; Wang, H.-B.; Tian, J.-W.; et al. PCC0208009, an indirect IDO1 inhibitor, alleviates neuropathic pain and co-morbidities by regulating synaptic plasticity of ACC and amygdala. Biochem. Pharmacol. 2020, 177, 113926. [Google Scholar] [CrossRef]
- Kang, S.J.; Kim, S.; Lee, J.; Kwak, C.; Lee, K.; Zhuo, M.; Kaang, B.K. Inhibition of anterior cingulate cortex excitatory neuronal activity induces conditioned place preference in a mouse model of chronic inflammatory pain. Korean J. Physiol. Pharmacol. 2017, 21, 487–493. [Google Scholar] [CrossRef]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Lau, B.K.; Vaughan, C.W. Descending modulation of pain: The GABA disinhibition hypothesis of analgesia. Curr. Opin. Neurobiol. 2014, 29, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ossipov, M.H.; Dussor, G.O.; Porreca, F. Central modulation of pain. J. Clin. Investig. 2010, 120, 3779–3787. [Google Scholar] [CrossRef]
- Li, J.-N.; Sheets, P.L. The central amygdala to periaqueductal gray pathway comprises intrinsically distinct neurons differentially affected in a model of inflammatory pain. J. Physiol. 2018, 596, 6289–6305. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, J.; Sheets, P.L. Altered excitability and local connectivity of mPFC-PAG neurons in a mouse model of neuropathic pain. J. Neurosci. 2018, 38, 4829–4839. [Google Scholar] [CrossRef]
- Osborne, P.B.; Vaughan, C.W.; Wilson, H.I.; Christie, M.J. Opioid inhibition of rat periaqueductal grey neurones with identified projections to rostral ventromedial medulla in vitro. J. Physiol. 1996, 490, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.-H.; Lee, B.H.; Park, S.H.; Ryu, J.-W.; Kim, B.-O.; Park, Y.G. Microinjection of opiates into the periaqueductal gray matter attenuates neuropathic pain symptoms in rats. NeuroReport 2000, 11, 1413–1416. [Google Scholar] [CrossRef]
- Hoot, M.R.; Sim-Selley, L.J.; Selley, D.E.; Scoggins, K.L.; Dewey, W.L. Chronic neuropathic pain in mice reduces μ-opioid receptor-mediated G-protein activity in the thalamus. Brain Res. 2011, 1406, 1–7. [Google Scholar] [CrossRef]
- Maarrawi, J.; Peyron, R.; Mertens, P.; Costes, N.; Magnin, M.; Sindou, M.; Laurent, B.; Garcia-Larrea, L. Differential brain opioid receptor availability in central and peripheral neuropathic pain. Pain 2007, 127, 183–194. [Google Scholar] [CrossRef]
- Hahm, E.-T.; Kim, Y.; Lee, J.-J.; Cho, Y.-W. GABAergic synaptic response and its opioidergic modulation in periaqueductal gray neurons of rats with neuropathic pain. BMC Neurosci. 2011, 12, 41. [Google Scholar] [CrossRef]
- Li, Z.; Yin, P.; Chen, J.; Jin, S.; Liu, J.; Luo, F. CaMKIIα may modulate fentanyl-induced hyperalgesia via a CeLC-PAG-RVM-spinal cord descending facilitative pain pathway in rats. PLoS ONE 2017, 12, e0177412. [Google Scholar] [CrossRef]
- Chung, G.; Shim, H.G.; Kim, C.Y.; Ryu, H.H.; Jang, D.C.; Kim, S.H.; Lee, J.; Kim, C.E.; Kim, Y.K.; Lee, Y.S.; et al. Persistent activity of metabotropic glutamate receptor 5 in the periaqueductal gray constrains emergence of chronic neuropathic pain. Curr. Biol. CB 2020. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Guo, Y.-Y.; Zhang, X.-N.; Xu, Z.-H.; Liu, S.-B.; Guo, H.-J.; Yang, Q.; Zhang, F.-X.; Sun, X.-L.; et al. A role of periaqueductal grey NR2B-containing NMDA receptor in mediating persistent inflammatory pain. Mol. Pain 2009, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Cheng, J.K.; Chiou, L.C. Hypofunction of glutamatergic neurotransmission in the periaqueductal gray contributes to nerve-injury-induced neuropathic pain. J. Neurosci. 2013, 33, 7825–7836. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Cheng, J.K.; Chiou, L.C. Impairment of adenylyl cyclase-mediated glutamatergic synaptic plasticity in the periaqueductal grey in a rat model of neuropathic pain. J. Physiol. 2015, 593, 2955–2973. [Google Scholar] [CrossRef] [PubMed]
- Brakeman, P.R.; Lanahan, A.A.; O’Brien, R.; Roche, K.; Barnes, C.A.; Huganir, R.L.; Worley, P.F. Homer: A protein that selectively binds metabotropic glutamate receptors. Nature 1997, 386, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Borsook, D.; Upadhyay, J.; Chudler, E.H.; Becerra, L. A Key Role of the Basal Ganglia in Pain and Analgesia-Insights Gained through Human Functional Imaging. Mol. Pain 2010, 6, 27. [Google Scholar] [CrossRef]
- Haber, S.N. The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 2014, 282, 248–257. [Google Scholar] [CrossRef]
- Blanchet, P.J.; Brefel-Courbon, C. Chronic pain and pain processing in Parkinson’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 87, 200–206. [Google Scholar] [CrossRef]
- Chudler, E.H.; Dong, W.K. The role of the basal ganglia in nociception and pain. Pain 1995, 60, 3–38. [Google Scholar] [CrossRef]
- Abbadie, C.; Brown, J.L.; Mantyh, P.W.; Basbaum, A.I. Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience 1996, 70, 201–209. [Google Scholar] [CrossRef]
- Nakamura, Y.; Fukushige, R.; Watanabe, K.; Kishida, Y.; Hisaoka-Nakashima, K.; Nakata, Y.; Morioka, N. Continuous infusion of substance P into rat striatum relieves mechanical hypersensitivity caused by a partial sciatic nerve ligation via activation of striatal muscarinic receptors. Behav. Brain Res. 2020, 391, 112714. [Google Scholar] [CrossRef] [PubMed]
- Wawrzczak-Bargieła, A.; Ziółkowska, B.; Piotrowska, A.; Starnowska-Sokół, J.; Rojewska, E.; Mika, J.; Przewłocka, B.; Przewłocki, R. Neuropathic pain dysregulates gene expression of the forebrain opioid and dopamine systems. Neurotox. Res. 2020, 37, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Holmes, F.E.; Bacon, A.; Pope, R.J.P.; Vanderplank, P.A.; Kerr, N.C.H.; Sukumaran, M.; Pachnis, V.; Wynick, D. Transgenic overexpression of galanin in the dorsal root ganglia modulates pain-related behavior. Proc. Nat. Acad. Sci. USA 2003, 100, 6180–6185. [Google Scholar] [CrossRef]
- Duan, H.; Zhang, Y.; Zhang, X.-M.; Xu, H.-H.; Shu, J.; Xu, S.-L. Antinociceptive roles of galanin receptor 1 in nucleus accumbens of rats in a model of neuropathic pain. J. Neurosci. Res. 2015, 93, 1542–1551. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, Z.; Gambeta, E.; Xu, S.C.; Thomas, C.; Godfrey, N.; Chen, L.; M’Dahoma, S.; Borgland, S.L.; Zamponi, G.W. Dopamine inputs from the ventral tegmental area into the medial prefrontal cortex modulate neuropathic pain-associated behaviors in mice. Cell Rep. 2020, 31, 107812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qian, Y.-L.; Li, C.; Liu, D.; Wang, L.; Wang, X.-Y.; Liu, M.-J.; Liu, H.; Zhang, S.; Guo, X.-Y.; et al. Brain-derived neurotrophic factor in the mesolimbic reward circuitry mediates nociception in chronic neuropathic pain. Biol. Psychiatry 2017, 82, 608–618. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Zhang, G.; Qiao, X.; Zhang, M. SIRT1 mediates neuropathic pain induced by sciatic nerve chronic constrictive injury in the VTA-NAc pathway. Pain Res. Manag. 2020, 2020, 4245968. [Google Scholar] [CrossRef]
- Sagheddu, C.; Aroni, S.; de Felice, M.; Lecca, S.; Luchicchi, A.; Melis, M.; Muntoni, A.L.; Romano, R.; Palazzo, E.; Guida, F.; et al. Enhanced serotonin and mesolimbic dopamine transmissions in a rat model of neuropathic pain. Neuropharmacology 2015, 97, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Ide, S.; Minami, M. Pain relief induces dopamine release in the rat nucleus accumbens during the early but not late phase of neuropathic pain. Neurosci. Lett. 2016, 629, 73–78. [Google Scholar] [CrossRef]
- Pautrat, A.; Rolland, M.; Barthelemy, M.; Baunez, C.; Sinniger, V.; Piallat, B.; Savasta, M.; Overton, P.G.; David, O.; Coizet, V. Revealing a novel nociceptive network that links the subthalamic nucleus to pain processing. eLife 2018, 7, e36607. [Google Scholar] [CrossRef]
- Luan, Y.; Tang, D.; Wu, H.; Gu, W.; Wu, Y.; Cao, J.-L.; Xiao, C.; Zhou, C. Reversal of hyperactive subthalamic circuits differentially mitigates pain hypersensitivity phenotypes in parkinsonian mice. Proc. Nat. Acad. Sci. USA 2020, 117, 10045. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Kuner, T. Cellular circuits in the brain and their modulation in acute and chronic pain. Physiol. Rev. 2021, 101, 213–258. [Google Scholar] [CrossRef] [PubMed]
- McGuire, D.B. Comprehensive and multidimensional assessment and measurement of pain. J. Pain Symptom Manag. 1992, 7, 312–319. [Google Scholar] [CrossRef]
- Kiritoshi, T.; Neugebauer, V. Pathway-specific alterations of cortico-amygdala transmission in an arthritis pain model. ACS Chem. Neurosci. 2018, 9, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.F.; Sheets, P.L. Sex-specific disruption of distinct mpfc inhibitory neurons in spared-nerve injury model of neuropathic pain. Cell Rep. 2020, 31, 107729. [Google Scholar] [CrossRef]
- Radzicki, D.; Pollema-Mays, S.L.; Sanz-Clemente, A.; Martina, M. Loss of M1 receptor dependent cholinergic excitation contributes to mPFC deactivation in neuropathic pain. J. Neurosci. 2017, 37, 2292–2304. [Google Scholar] [CrossRef]
- Hoover, W.B.; Vertes, R.P. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 2007, 212, 149–179. [Google Scholar] [CrossRef]
- Willis, W.D., Jr. Central nervous system mechanisms for pain modulation. Appl. Neurophysiol. 1985, 48, 153–165. [Google Scholar] [CrossRef]
- Vanegas, H.; Schaible, H.G. Descending control of persistent pain: Inhibitory or facilitatory? Brain Res. Brain Res. Rev. 2004, 46, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Heinricher, M.M.; Tavares, I.; Leith, J.L.; Lumb, B.M. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Rev. 2009, 60, 214–225. [Google Scholar] [CrossRef]
- McGaraughty, S.; Reinis, S.; Tsoukatos, J. Two distinct unit activity responses to morphine in the rostral ventromedial medulla of awake rats. Brain Res. 1993, 604, 331–333. [Google Scholar] [CrossRef]
- Heinricher, M.M.; Morgan, M.M.; Tortorici, V.; Fields, H.L. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience 1994, 63, 279–288. [Google Scholar] [CrossRef]
- Cury, R.G.; Galhardoni, R.; Fonoff, E.T.; Perez Lloret, S.; Dos Santos Ghilardi, M.G.; Barbosa, E.R.; Teixeira, M.J.; Ciampi de Andrade, D. Sensory abnormalities and pain in Parkinson disease and its modulation by treatment of motor symptoms. Eur. J. Pain 2016, 20, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.A.; Walker, R.W.; Hildreth, T.J.; Prentice, W.M. A survey of pain in idiopathic Parkinson’s disease. J. Pain Symptom Manag. 2006, 32, 462–469. [Google Scholar] [CrossRef]
- Beiske, A.G.; Loge, J.H.; Rønningen, A.; Svensson, E. Pain in Parkinson’s disease: Prevalence and characteristics. Pain 2009, 141, 173–177. [Google Scholar] [CrossRef]
- Strobel, C.; Hunt, S.; Sullivan, R.; Sun, J.; Sah, P. Emotional regulation of pain: The role of noradrenaline in the amygdala. Sci. China. Life Sci. 2014, 57, 384–390. [Google Scholar] [CrossRef]
- Ikeda, R.; Takahashi, Y.; Inoue, K.; Kato, F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain 2007, 127, 161–172. [Google Scholar] [CrossRef]
- Gonçalves, L.; Silva, R.; Pinto-Ribeiro, F.; Pêgo, J.M.; Bessa, J.M.; Pertovaara, A.; Sousa, N.; Almeida, A. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp. Neurol. 2008, 213, 48–56. [Google Scholar] [CrossRef]
- Tyrtyshnaia, A.; Manzhulo, I. Neuropathic pain causes memory deficits and dendrite tree morphology changes in mouse hippocampus. J. Pain Res. 2020, 13, 345–354. [Google Scholar] [CrossRef]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef]
- Ignatowski, T.A.; Covey, W.C.; Knight, P.R.; Severin, C.M.; Nickola, T.J.; Spengler, R.N. Brain-derived TNFalpha mediates neuropathic pain. Brain Res. 1999, 841, 70–77. [Google Scholar] [CrossRef]
- Mai, C.-L.; Wei, X.; Gui, W.-S.; Xu, Y.-N.; Zhang, J.; Lin, Z.-J.; Tan, Z.; Meng, Y.-T.; Li, Y.-Y.; Zhou, L.-J.; et al. Differential regulation of GSK-3β in spinal dorsal horn and in hippocampus mediated by interleukin-1beta contributes to pain hypersensitivity and memory deficits following peripheral nerve injury. Mol. Pain 2019, 15. [Google Scholar] [CrossRef]
- Kaster, M.P.; Gadotti, V.M.; Calixto, J.B.; Santos, A.R.S.; Rodrigues, A.L.S. Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 2012, 62, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Guan, S.; Ge, H.; Xiong, W.; He, L.; Liu, L.; Yin, C.; Liu, H.; Li, G.; Xu, C.; et al. Effects of palmatine on rats with comorbidity of diabetic neuropathic pain and depression. Brain Res. Bull. 2018, 139, 56–66. [Google Scholar] [CrossRef]
- Martini, M.L.; Oermann, E.K.; Opie, N.L.; Panov, F.; Oxley, T.; Yaeger, K. Sensor modalities for brain-computer interface technology: A comprehensive literature review. Neurosurgery 2019, 86, E108–E117. [Google Scholar] [CrossRef] [PubMed]
- Pisarchik, A.N.; Maksimenko, V.A.; Hramov, A.E. From novel technology to novel applications: Comment on “An integrated brain-machine interface platform with thousands of channels” by Elon Musk and Neuralink. J. Med. Internet Res. 2019, 21, e16356. [Google Scholar] [CrossRef]
- Lefaucheur, J.-P.; Antal, A.; Ahdab, R.; Ciampi de Andrade, D.; Fregni, F.; Khedr, E.M.; Nitsche, M.; Paulus, W. The use of repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) to relieve pain. Brain Stimul. 2008, 1, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Boccard, S.G.J.; Prangnell, S.J.; Pycroft, L.; Cheeran, B.; Moir, L.; Pereira, E.A.C.; Fitzgerald, J.J.; Green, A.L.; Aziz, T.Z. Long-term results of deep brain stimulation of the anterior cingulate cortex for neuropathic pain. World Neurosurg. 2017, 106, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.C.; Lu, G.; Wang, S.; Schweder, P.M.; Hyam, J.A.; Stein, J.F.; Paterson, D.J.; Aziz, T.Z.; Green, A.L. Ventral periaqueductal grey stimulation alters heart rate variability in humans with chronic pain. Exp. Neurol. 2010, 223, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Weaver, F.M.; Follett, K.A.; Stern, M.; Luo, P.; Harris, C.L.; Hur, K.; Marks, W.J.; Rothlind, J.; Sagher, O.; Moy, C.; et al. Randomized trial of deep brain stimulation for Parkinson disease. Neurology 2012, 79, 55. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Area | Pain Model | Pathological Neural Plasticity | Modulation (M) and Its Effect (E) | Reference |
|---|---|---|---|---|
| S1 | PSL * in mice | Spine turnover rates ↑ Astrocytic calcium activity ↑ Extracellular glutamates ↑ TSP-1 ↑ | M: Infusion of MPEP, BAPTA, or siTSP-1 IP3R2 Knockout E: Spine turnover rate ↓; TSP-1 ↓ Mechanical allodynia ↓ | [35] |
| Evoked potential of the S1 ↑ Spine turnover rates ↑ | M: Immediate local nerve blockade E: Spine turnover rate ↓ Mechanical allodynia ↓ | [46] | ||
| Mushroom spines ↓ Thin spines ↑ Gain rate of axonal boutons ↑ | [37] | |||
| Transient spinal cord ischemia in mice | Spontaneous AP firing ↑ Intrinsic excitability ↑ EPSP frequency ↑ Calcium activity of pyramidal neurons ↑ | M: Optogenetic stimulation of pyramidal neurons E: Sensory-evoked potential ↓ Spontaneous firing of layer 5 pyramidal neurons ↓ Intrinsic excitability of layer 5 pyramidal neurons ↓ Bilateral mechanical allodynia ↓ | [38] | |
| SNI * in mice | Calcium activity of pyramidal neurons ↑ Calcium activity of VIP+ interneurons ↑ Calcium activity of SOM+ interneurons ↓ Calcium activity of PV+ interneurons ↓ | M: Chemogenetic activation of SOM+ interneurons E: Calcium activity of pyramidal neurons ↓ Mechanical allodynia ↓ | [39] | |
| CCI * in mice | Calcium activity of pyramidal neurons ↑ Calcium activity of VIP+ interneurons ↑ Calcium activity of SOM+ interneurons ↓ | M: EA* intervention to GB30* and GB34* E: Calcium activity of pyramidal neurons ↓ Calcium activity of SOM+ interneurons ↓ Calcium activity of VIP+ interneurons ↓ Mechanical hyperalgesia ↓, Thermal hyperalgesia ↓ | [40] | |
| ACC | CPN * ligation in mice | mEPSC amplitude of pyramidal neurons ↑ Spine turnover rate ↑ NCAM1 turnover rate ↑ | M: Anisomycin into the ACC E: Spine turnover rate ↓ Mechanical allodynia ↓ Thermal allodynia ↓ | [67] |
| CPN * ligation in mice | Evoked EPSC amplitude ↑ PKMζ and p-PKMζ ↑ | M: ZIP* into the ACC E: Evoked EPSC amplitude ↓ Mechanical allodynia ↓, CPP ↑ | [68] | |
| CCI* in mice | Connections between excitatory and inhibitory neurons ↓ Intrinsic excitability of pyramidal neurons ↑ mEPSC and mIPSC frequency in layer 5↓ | [69] | ||
| SNI * in mice | Spontaneous and evoked calcium activity of pyramidal neurons in layer 5 ↑ | [70] | ||
| SNI * in mice | sEPCS frequency of pyramidal neurons in layer 2/3 ↑ Intrinsic excitability of pyramidal neurons ↑ | [71] | ||
| SNL* in rat | Dendritic spine density in the ACC ↑ IDO1 expression in the ACC ↑ | M: Oral administration of PCC0208009* E: Mechanical allodynia ↓ Thermal hyperalgesia ↓ Conditioned place preference ↑ | [78] | |
| SNI * in rat | mTOR signaling ↑ | M: Rapamycin * into the ACC E: Mechanical allodynia ↓ PSD-95 ↓, Evoked activity of ACC ↓ | [76] | |
| PAG | CCI * in mice | G-protein activity of the μ-opioid receptor-protein in reponse to DAMGO ↓ | [87] | |
| Sacral nerve transection in rat | Frequency of GABAergic mIPSCs ↑ Frequency of IPSC response to DAMGO is not altered | [89] | ||
| SNL * in rat | mGluR5 activity ↓ Homer1a expression ↑ sEPSC frequency of PAG-RVM neurons↓ Intrinsic excitability of PAG neurons ↓ | M: DHPG into the PAG or shHomer1a E: Mechanical allodynia ↓ mGluR5 activity ↑ intrinsic excitability of PAG neurons ↑ | [91] | |
| SNL * in rat | EPSCs frequency and amplitude ↓ NR1 and NR2B subunits ↑ | [93] | ||
| SNL * in rat | Forskolin-induced EPSC potentiation ↓ | M: Infusion of forskolin into the PAG E: Mechanical allodynia ↓ | [94] | |
| CFA * | NR2B subunits ↑ mEPSCs amplitude ↑ | M: Ro 25-6981 * into the PAG Oral administration of Hyperoside* E: Thermal allodynia ↓, NR2B subunits ↓ | [92] | |
| BG | PSL * in rats | Muscarinic cholinergic neurons through NK1 receptors ↑ | M: Co-infusion of SP and CP96345* into the striatum Atropine and mecamylamine into the striatum E: Mechanical hyperalgesia ↓ | [101] |
| CCI * in mice | Prodynorphin and proenkephalin in the NAc↑ κ and δ opioid receptors in the NAc ↑ | [102] | ||
| PSL * in rat | GalR1 in the NAc ↑ | M: M617* or galanin into the NAc E: Thermal and mechanical hyperalgesia ↓ | [104] | |
| SNI * in mice | DA in the NAc ↑ | M: Optogenetic stimulation of DA terminals E: Mechanical hyperalgesia ↓, CPP ↑ Calcium activity of mPFC ↑ c-Fos in layer 5 of mPFC ↑ | [105] | |
| CCI * in mice | Firing rates of VTA DA neurons ↑ c-Fos in VTA-NAc DA neurons↑ BDNF in the NAc ↑ | M: Conditional knockdown of BDNF in the VTA-NAc pathway, Microinfusion of anisomycin into the VTA E: Thermal hyperalgesia ↓ | [106] | |
| CCI * in mice | SIRT1 in the VTA ↓ c-Fos in the VTA ↑ | M: SRT1720* into the VTA E: Thermal hyperalgesia ↓ | [107] | |
| SNI * in rat | Burst firing of VTA DA neurons ↑ D2 receptors in the NAc ↓ DA release in the NAc ↑ | [108] | ||
| SNL * in rat | DA release in the NAc in response to sucrose ↑ | [109] | ||
| 6-OHDA * lesioned rats | Firing rate of phasic response in the STN ↑ | [110] | ||
| 6-OHDA * lesioned mice | Spontaneous and evoked firing rate in the STN ↑ | M: Optogenetic inhibition of STN neurons Optogenetic stimulation of STN neurons E: Mechanical and thermal allodynia ↓ | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, M.S.; Park, H.; Kim, S.K. Neural Plasticity in the Brain during Neuropathic Pain. Biomedicines 2021, 9, 624. https://doi.org/10.3390/biomedicines9060624
Bak MS, Park H, Kim SK. Neural Plasticity in the Brain during Neuropathic Pain. Biomedicines. 2021; 9(6):624. https://doi.org/10.3390/biomedicines9060624
Chicago/Turabian StyleBak, Myeong Seong, Haney Park, and Sun Kwang Kim. 2021. "Neural Plasticity in the Brain during Neuropathic Pain" Biomedicines 9, no. 6: 624. https://doi.org/10.3390/biomedicines9060624
APA StyleBak, M. S., Park, H., & Kim, S. K. (2021). Neural Plasticity in the Brain during Neuropathic Pain. Biomedicines, 9(6), 624. https://doi.org/10.3390/biomedicines9060624