
Modeling Diet-Induced NAFLD and NASH in Rats: A Comprehensive Review

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Definition and Etiology of NAFLD and NASH
1.2. In Vivo Modeling of Human NAFLD/NASH
1.3. Rat versus Mouse in Liver Disease Modeling
2. Diet-Induced NAFLD and NASH Models in Rats
2.1. Amino Acid Modified Diets
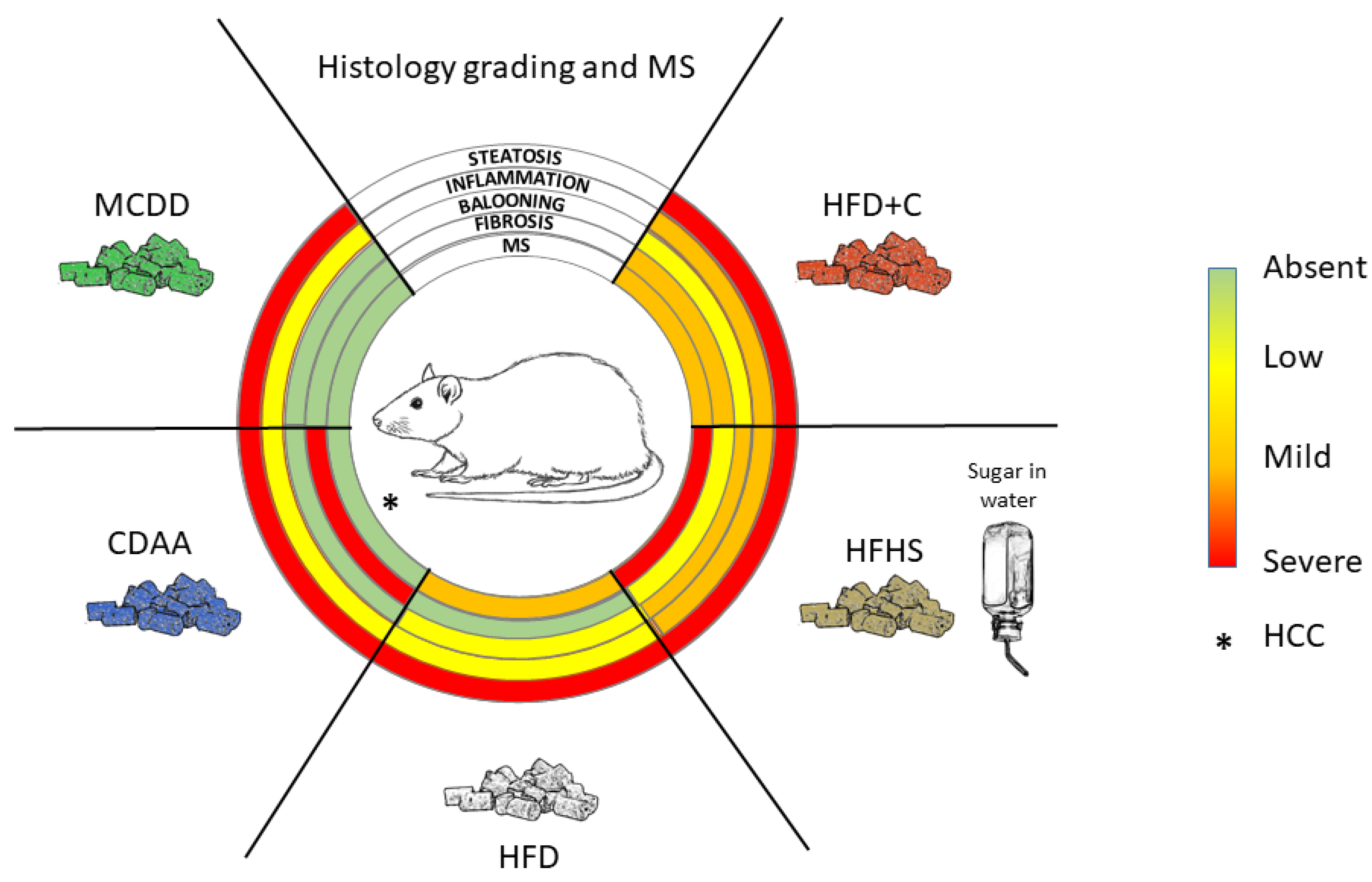
2.1.1. Methionine- and Choline-Deficient Diet
2.1.2. Choline-Deficient L-Amino Acid Defined Diet
2.2. “Western-Style” Diets
2.2.1. High-Fat Diet
2.2.2. Complemented High-Fat Diet
Atherogenic Diets (High-Fat and High-Cholesterol (HFD+C) Diet)
High-Fat and High-Sugar Diet
3. Other Factors Affecting Diet-Induced NAFLD/NASH Model Phenotype
3.1. Effects of Food Intake Methods
3.2. Effect of Housing Environmental Factors
3.3. Sex-Related Differences
3.4. The Role of Gut Microbiome
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Reccia, I.; Kumar, J.; Akladios, C.; Virdis, F.; Pai, M.; Habib, N.; Spalding, D. Non-alcoholic fatty liver disease: A sign of systemic disease. Metabolism 2017, 72, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Lequoy, M.; Gigante, E.; Couty, J.P.; Desbois-Mouthon, C. Hepatocellular carcinoma in the context of non-alcoholic steatohepatitis (NASH): Recent advances in the pathogenic mechanisms. Horm Mol. Biol Clin. Investig. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
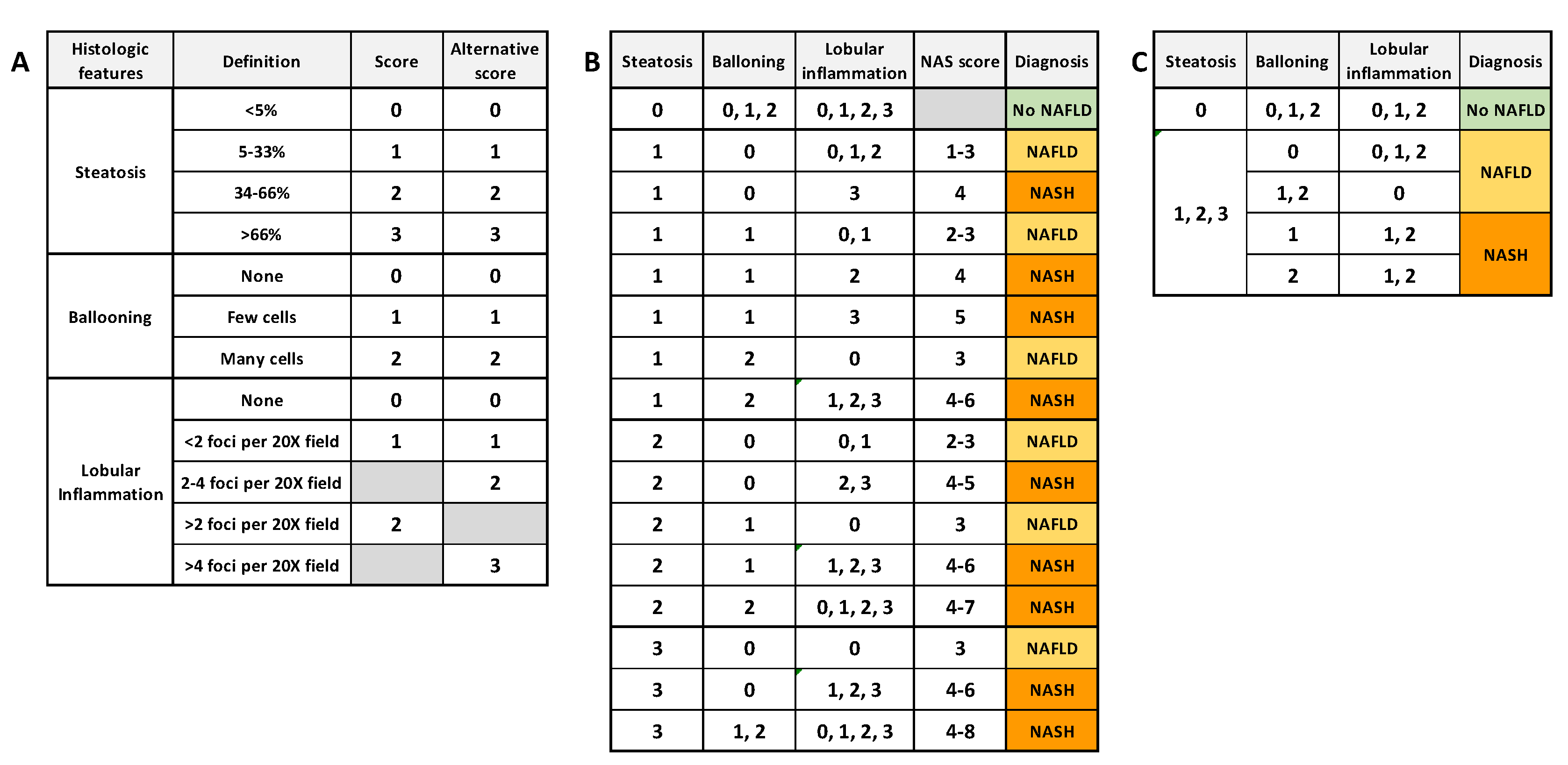
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Poitou, C.; Veyrie, N.; Bouillot, J.L.; Basdevant, A.; Paradis, V.; Tordjman, J.; Clement, K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012, 56, 1751–1759. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver organoids: From basic research to therapeutic applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992, 41, 1422–1428. [Google Scholar] [CrossRef]
- Asamoto, M.; Hokaiwado, N.; Murasaki, T.; Shirai, T. Connexin 32 dominant-negative mutant transgenic rats are resistant to hepatic damage by chemicals. Hepatology 2004, 40, 205–210. [Google Scholar] [CrossRef]
- Lu, P.; Yang, G.; Jiang, L.; He, W.; Wu, W.; Qi, L.; Shen, S.; Rao, J.; Zhang, P.; Xue, Z.; et al. Characterizing disease progression of nonalcoholic steatohepatitis in Leptin-deficient rats by integrated transcriptome analysis. Exp. Biol. Med. 2021, 246, 678–687. [Google Scholar] [CrossRef]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef]
- Naiki-Ito, A.; Kato, H.; Naiki, T.; Yeewa, R.; Aoyama, Y.; Nagayasu, Y.; Suzuki, S.; Inaguma, S.; Takahashi, S. A novel model of non-alcoholic steatohepatitis with fibrosis and carcinogenesis in connexin 32 dominant-negative transgenic rats. Arch. Toxicol. 2020, 94, 4085–4097. [Google Scholar] [CrossRef]
- Kitamori, K.; Naito, H.; Tamada, H.; Kobayashi, M.; Miyazawa, D.; Yasui, Y.; Sonoda, K.; Tsuchikura, S.; Yasui, N.; Ikeda, K.; et al. Development of novel rat model for high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis progression in SHRSP5/Dmcr. Environ. Health Prev. Med. 2012, 17, 173–182. [Google Scholar] [CrossRef]
- Shimoyama, M.; Laulederkind, S.J.; De Pons, J.; Nigam, R.; Smith, J.R.; Tutaj, M.; Petri, V.; Hayman, G.T.; Wang, S.J.; Ghiasvand, O.; et al. Exploring human disease using the Rat Genome Database. Dis. Model. Mech. 2016, 9, 1089–1095. [Google Scholar] [CrossRef]
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat. Commun. 2017, 8, 14250. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef]
- Jensen, V.S.; Tveden-Nyborg, P.; Zacho-Rasmussen, C.; Quaade, M.L.; Ipsen, D.H.; Hvid, H.; Fledelius, C.; Wulff, E.M.; Lykkesfeldt, J. Variation in diagnostic NAFLD/NASH read-outs in paired liver samples from rodent models. J. Pharmacol. Toxicol. Methods 2020, 101, 106651. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, J.; Lu, C.; Wang, J.; Ge, J.; Huang, Y.; Zhang, L.; Wang, Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006, 79, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meyer, C.; Xu, C.; Weng, H.; Hellerbrand, C.; ten Dijke, P.; Dooley, S. Animal models of chronic liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G449–G468. [Google Scholar] [CrossRef] [PubMed]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Arman, T.; Lynch, K.D.; Montonye, M.L.; Goedken, M.; Clarke, J.D. Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats. Toxins 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Canet, M.J.; Hardwick, R.N.; Lake, A.D.; Dzierlenga, A.L.; Clarke, J.D.; Cherrington, N.J. Modeling human nonalcoholic steatohepatitis-associated changes in drug transporter expression using experimental rodent models. Drug Metab. Dispos. 2014, 42, 586–595. [Google Scholar] [CrossRef]
- Xin, H.G.; Zhang, B.B.; Wu, Z.Q.; Hang, X.F.; Xu, W.S.; Ni, W.; Zhang, R.Q.; Miao, X.H. Treatment with baicalein attenuates methionine-choline deficient diet-induced non-alcoholic steatohepatitis in rats. Eur. J. Pharmacol. 2014, 738, 310–318. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- George, J.; Pera, N.; Phung, N.; Leclercq, I.; Yun Hou, J.; Farrell, G. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J. Hepatol. 2003, 39, 756–764. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Yu, H.H.; Hsieh, M.C.; Wu, S.Y.; Sy, E.D.; Shan, Y.S. Effects of duodenal-jejunal bypass surgery in ameliorating nonalcoholic steatohepatitis in diet-induced obese rats. Diabetes Metab. Syndr. Obes. 2019, 12, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, R.; Clarkson, V.; Shephard, E.G.; Marais, D.A.; Jaffer, M.A.; Woodburne, V.E.; Kirsch, R.E.; Hall Pde, L. Rodent nutritional model of non-alcoholic steatohepatitis: Species, strain and sex difference studies. J. Gastroenterol. Hepatol. 2003, 18, 1272–1282. [Google Scholar] [CrossRef]
- Liu, B.; Deng, X.; Jiang, Q.; Li, G.; Zhang, J.; Zhang, N.; Xin, S.; Xu, K. Scoparone alleviates inflammation, apoptosis and fibrosis of non-alcoholic steatohepatitis by suppressing the TLR4/NF-kappaB signaling pathway in mice. Int. Immunopharmacol. 2019, 75, 105797. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Tsuchida, T.; Yasuda, S.I.; Oka, K. Dietary iron restriction leads to a reduction in hepatic fibrosis in a rat model of non-alcoholic steatohepatitis. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Nakade, Y.; Yamauchi, T.; Sakamoto, K.; Inoue, T.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Ito, K.; et al. Influence of nicotine on choline-deficient, L-amino acid-defined diet-induced non-alcoholic steatohepatitis in rats. PLoS ONE 2017, 12, e0180475. [Google Scholar] [CrossRef] [PubMed]
- Namisaki, T.; Moriya, K.; Kitade, M.; Takeda, K.; Kaji, K.; Okura, Y.; Shimozato, N.; Sato, S.; Nishimura, N.; Seki, K.; et al. Effect of combined farnesoid X receptor agonist and angiotensin II type 1 receptor blocker on hepatic fibrosis. Hepatol. Commun. 2017, 1, 928–945. [Google Scholar] [CrossRef]
- Shimozato, N.; Namisaki, T.; Kaji, K.; Kitade, M.; Okura, Y.; Sato, S.; Moriya, K.; Seki, K.; Kawaratani, H.; Takaya, H.; et al. Combined effect of a farnesoid X receptor agonist and dipeptidyl peptidase-4 inhibitor on hepatic fibrosis. Hepatol. Res. 2019, 49, 1147–1161. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Rolo, A.P.; Duarte, F.V.; Simoes, A.M.; Palmeira, C.M. Differential alterations in mitochondrial function induced by a choline-deficient diet: Understanding fatty liver disease progression. Mitochondrion 2008, 8, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Kotake, Y.; Sang, H.; Pye, Q.N.; Wallis, G.L.; Kolker, L.M.; Tabatabaie, T.; Stewart, C.A.; Konishi, Y.; Nakae, D.; et al. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis 2000, 21, 983–989. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Tamaki, Y.; Nakade, Y.; Yamauchi, T.; Makino, Y.; Yokohama, S.; Okada, M.; Aso, K.; Kanamori, H.; Ohashi, T.; Sato, K.; et al. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J. Gastroenterol. 2013, 48, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, U.; Redgrave, T.G.; Oates, P.S. Effect of dietary fat to produce non-alcoholic fatty liver in the rat. J. Gastroenterol. Hepatol. 2009, 24, 1463–1471. [Google Scholar] [CrossRef]
- Kucera, O.; Garnol, T.; Lotkova, H.; Stankova, P.; Mazurova, Y.; Hroch, M.; Bolehovska, R.; Rousar, T.; Cervinkova, Z. The effect of rat strain, diet composition and feeding period on the development of a nutritional model of non-alcoholic fatty liver disease in rats. Physiol. Res. 2011, 60, 317–328. [Google Scholar] [CrossRef]
- Wang, Y.; Ausman, L.M.; Russell, R.M.; Greenberg, A.S.; Wang, X.D. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J. Nutr. 2008, 138, 1866–1871. [Google Scholar] [CrossRef]
- Vial, G.; Dubouchaud, H.; Couturier, K.; Cottet-Rousselle, C.; Taleux, N.; Athias, A.; Galinier, A.; Casteilla, L.; Leverve, X.M. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J. Hepatol. 2011, 54, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Emamat, H.; Foroughi, F.; Eini-Zinab, H.; Hekmatdoost, A. The Effects of Onion Consumption on Prevention of Nonalcoholic Fatty Liver Disease. Indian J. Clin. Biochem. 2018, 33, 75–80. [Google Scholar] [CrossRef]
- Su, Y.B.; Li, T.H.; Huang, C.C.; Tsai, H.C.; Huang, S.F.; Hsieh, Y.C.; Yang, Y.Y.; Huang, Y.H.; Hou, M.C.; Lin, H.C. Chronic calcitriol supplementation improves the inflammatory profiles of circulating monocytes and the associated intestinal/adipose tissue alteration in a diet-induced steatohepatitis rat model. PLoS ONE 2018, 13, e0194867. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Candelaresi, C.; Saccomanno, S.; Ferretti, G.; Bachetti, T.; Marzioni, M.; De Minicis, S.; Nobili, L.; Salzano, R.; Omenetti, A.; et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: Role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am. J. Pathol. 2006, 169, 846–860. [Google Scholar] [CrossRef]
- Fujita, M.; Kuraji, R.; Ito, H.; Hashimoto, S.; Toen, T.; Fukada, T.; Numabe, Y. Histological effects and pharmacokinetics of lipopolysaccharide derived from Porphyromonas gingivalis on rat maxilla and liver concerning with progression into non-alcoholic steatohepatitis. J. Periodontol. 2018, 89, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yuan, L.; Yu, H.; Xi, Y.; Xiao, R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not in diet-resistant rats. Life Sci. 2014, 110, 53–60. [Google Scholar] [CrossRef]
- Gauthier, M.S.; Favier, R.; Lavoie, J.M. Time course of the development of non-alcoholic hepatic steatosis in response to high-fat diet-induced obesity in rats. Br. J. Nutr. 2006, 95, 273–281. [Google Scholar] [CrossRef]
- Ichimura, M.; Kawase, M.; Masuzumi, M.; Sakaki, M.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tamaru, S.; Kato, S.; Tsuneyama, K.; et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol. Res. 2015, 45, 458–469. [Google Scholar] [CrossRef]
- Haldrup, D.; Heeboll, S.; Thomsen, K.L.; Andersen, K.J.; Meier, M.; Mortensen, F.V.; Nyengaard, J.R.; Hamilton-Dutoit, S.; Gronbaek, H. Preserved liver regeneration capacity after partial hepatectomy in rats with non-alcoholic steatohepatitis. World J. Hepatol. 2018, 10, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, M.; Masuzumi, M.; Kawase, M.; Sakaki, M.; Tamaru, S.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tsuneyama, K.; Matsuda, S.; et al. A diet-induced Sprague-Dawley rat model of nonalcoholic steatohepatitis-related cirrhosis. J. Nutr. Biochem. 2017, 40, 62–69. [Google Scholar] [CrossRef]
- Cai, C.X.; Buddha, H.; Castelino-Prabhu, S.; Zhang, Z.; Britton, R.S.; Bacon, B.R.; Neuschwander-Tetri, B.A. Activation of Insulin-PI3K/Akt-p70S6K Pathway in Hepatic Stellate Cells Contributes to Fibrosis in Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2017, 62, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.J.; Fan, J.G.; Ding, X.D.; Qiao, L.; Wang, G.L. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig. Dis. Sci. 2010, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, D.; Lukomska, A.; Dec, K.; Skonieczna-Zydecka, K.; Gutowska, I.; Skorka-Majewicz, M.; Styburski, D.; Misiakiewicz-Has, K.; Pilutin, A.; Palma, J.; et al. Diet-Induced Rat Model of Gradual Development of Non-Alcoholic Fatty Liver Disease (NAFLD) with Lipopolysaccharides (LPS) Secretion. Diagnostics 2019, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Sasao, M.; Asakawa, E.; Narita, S.; Hisano, M.; Suruga, K.; Ichimura, M.; Tsuneyama, K.; Tanaka, K.; Omagari, K. Dietary fat, cholesterol, and cholic acid affect the histopathologic severity of nonalcoholic steatohepatitis in Sprague-Dawley rats. Pathol. Res. Pract. 2019, 215, 152599. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Z.; Gregg, E.W.; Flanders, W.D.; Merritt, R.; Hu, F.B. Added sugar intake and cardiovascular diseases mortality among US adults. JAMA Intern. Med. 2014, 174, 516–524. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Garcia-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; Del Rosario-Tamayo, B.A.; Guajardo-Lopez, C.; Saavedra-Molina, A.; Rodriguez-Orozco, A.R.; Cortes-Rojo, C. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Buchheim-Dieckow, K.; Castro, J.P.; Laeger, T.; Wardelmann, K.; Kleinridders, A.; Johrens, K.; Puschel, G.P. Reduced Oxidative Stress and Enhanced FGF21 Formation in Livers of Endurance-Exercised Rats with Diet-Induced NASH. Nutrients 2019, 11, 2709. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Liu, H.; Zheng, Z.; Lu, R.; Jiang, Z. Genistein can ameliorate hepatic inflammatory reaction in nonalcoholic steatohepatitis rats. Biomed. Pharmacother. 2019, 111, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lezana, T.; Raurell, I.; Bravo, M.; Torres-Arauz, M.; Salcedo, M.T.; Santiago, A.; Schoenenberger, A.; Manichanh, C.; Genesca, J.; Martell, M.; et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018, 67, 1485–1498. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Naitoh, T.; Nagao, M.; Tanaka, N.; Watanabe, K.; Imoto, H.; Miyachi, T.; Motoi, F.; Unno, M. Increased Bile Acid Signals After Duodenal-Jejunal Bypass Improve Non-alcoholic Steatohepatitis (NASH) in a Rodent Model of Diet-Induced NASH. Obes. Surg. 2018, 28, 1643–1652. [Google Scholar] [CrossRef]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, C.; Lin, H.; Zhang, X.; Wen, H.; Shen, Y.; Wang, T.; Chen, S.; Liu, Y.; Chen, X. Evaluation of Non-alcoholic Fatty Liver Disease Using Acoustic Radiation Force Impulse Imaging Elastography in Rat Models. Ultrasound Med. Biol. 2017, 43, 2619–2628. [Google Scholar] [CrossRef]
- Karimi-Sales, E.; Ebrahimi-Kalan, A.; Alipour, M.R. Preventive effect of trans-chalcone on non-alcoholic steatohepatitis: Improvement of hepatic lipid metabolism. Biomed. Pharmacother. 2019, 109, 1306–1312. [Google Scholar] [CrossRef]
- Sampey, B.P.; Vanhoose, A.M.; Winfield, H.M.; Freemerman, A.J.; Muehlbauer, M.J.; Fueger, P.T.; Newgard, C.B.; Makowski, L. Cafeteria diet is a robust model of human metabolic syndrome with liver and adipose inflammation: Comparison to high-fat diet. Obesity 2011, 19, 1109–1117. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Graspeuntner, S.; Cappelletti, M.; Wu, D.; Mukherjee, R.; Chan, C.C.; Lawson, M.J.; Klarquist, J.; et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat. Med. 2017, 23, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Albustanji, L.; Perez, G.S.; AlHarethi, E.; Aldiss, P.; Bloor, I.; Barreto-Medeiros, J.M.; Budge, H.; Symonds, M.E.; Dellschaft, N. Housing Temperature Modulates the Impact of Diet-Induced Rise in Fat Mass on Adipose Tissue Before and During Pregnancy in Rats. Front. Physiol. 2019, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Esquirol, Y.; Bongard, V.; Mabile, L.; Jonnier, B.; Soulat, J.M.; Perret, B. Shift work and metabolic syndrome: Respective impacts of job strain, physical activity, and dietary rhythms. Chronobiol. Int. 2009, 26, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, B.; Knutsson, A.; Lindahl, B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup. Environ. Med. 2001, 58, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Rousso-Noori, L.; Zwighaft, Z.; Neufeld-Cohen, A.; Golik, M.; Kraut-Cohen, J.; Wang, M.; Han, X.; Asher, G. Circadian clocks and feeding time regulate the oscillations and levels of hepatic triglycerides. Cell Metab. 2014, 19, 319–330. [Google Scholar] [CrossRef]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef]
- Chen, K.; Ma, J.; Jia, X.; Ai, W.; Ma, Z.; Pan, Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 117–125. [Google Scholar] [CrossRef]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef]
- Yamajuku, D.; Okubo, S.; Haruma, T.; Inagaki, T.; Okuda, Y.; Kojima, T.; Noutomi, K.; Hashimoto, S.; Oda, H. Regular feeding plays an important role in cholesterol homeostasis through the liver circadian clock. Circ. Res. 2009, 105, 545–548. [Google Scholar] [CrossRef]
- Shimizu, H.; Hanzawa, F.; Kim, D.; Sun, S.; Laurent, T.; Umeki, M.; Ikeda, S.; Mochizuki, S.; Oda, H. Delayed first active-phase meal, a breakfast-skipping model, led to increased body weight and shifted the circadian oscillation of the hepatic clock and lipid metabolism-related genes in rats fed a high-fat diet. PLoS ONE 2018, 13, e0206669. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Csak, T.; Szabo, G. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World, J. Gastroenterol. 2014, 20, 8525–8534. [Google Scholar] [CrossRef] [PubMed]
- Stoppeler, S.; Palmes, D.; Fehr, M.; Holzen, J.P.; Zibert, A.; Siaj, R.; Schmidt, H.H.; Spiegel, H.U.; Bahde, R. Gender and strain-specific differences in the development of steatosis in rats. Lab. Anim. 2013, 47, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Medrikova, D.; Jilkova, Z.M.; Bardova, K.; Janovska, P.; Rossmeisl, M.; Kopecky, J. Sex differences during the course of diet-induced obesity in mice: Adipose tissue expandability and glycemic control. Int. J. Obes. 2012, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, V.; Kaakoush, N.O.; Maloney, C.A.; Raipuria, M.; Huinao, K.D.; Mitchell, H.M.; Morris, M.J. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS ONE 2015, 10, e0126931. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreres, L.; Jílková, Z.M.; Vial, G.; Marche, P.N.; Decaens, T.; Lerat, H. Modeling Diet-Induced NAFLD and NASH in Rats: A Comprehensive Review. Biomedicines 2021, 9, 378. https://doi.org/10.3390/biomedicines9040378
Carreres L, Jílková ZM, Vial G, Marche PN, Decaens T, Lerat H. Modeling Diet-Induced NAFLD and NASH in Rats: A Comprehensive Review. Biomedicines. 2021; 9(4):378. https://doi.org/10.3390/biomedicines9040378
Chicago/Turabian StyleCarreres, Lydie, Zuzana Macek Jílková, Guillaume Vial, Patrice N. Marche, Thomas Decaens, and Hervé Lerat. 2021. "Modeling Diet-Induced NAFLD and NASH in Rats: A Comprehensive Review" Biomedicines 9, no. 4: 378. https://doi.org/10.3390/biomedicines9040378
APA StyleCarreres, L., Jílková, Z. M., Vial, G., Marche, P. N., Decaens, T., & Lerat, H. (2021). Modeling Diet-Induced NAFLD and NASH in Rats: A Comprehensive Review. Biomedicines, 9(4), 378. https://doi.org/10.3390/biomedicines9040378