
Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults

 ,
,
Abstract
1. Introduction
1.1. Background
1.2. Historical Context
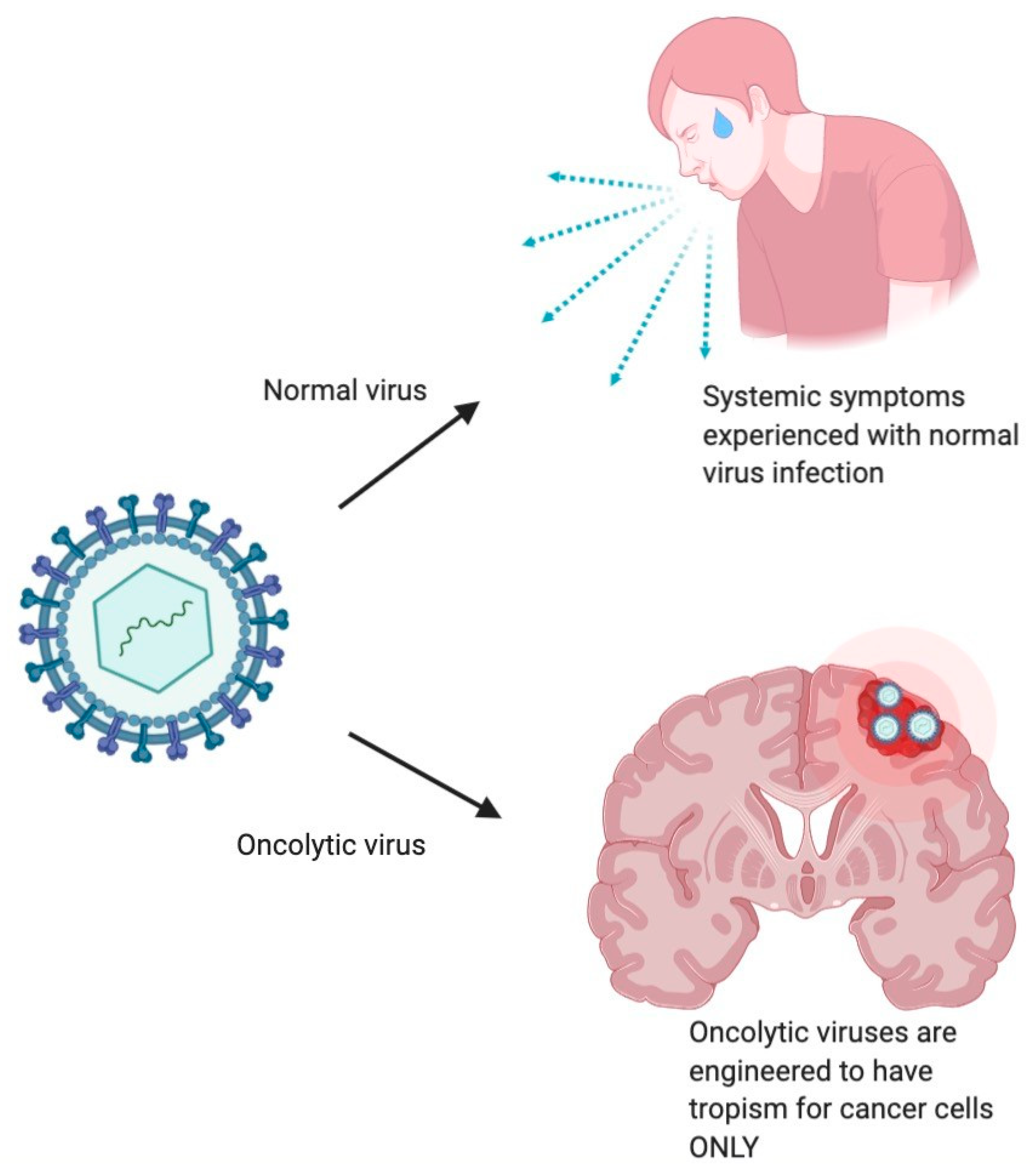
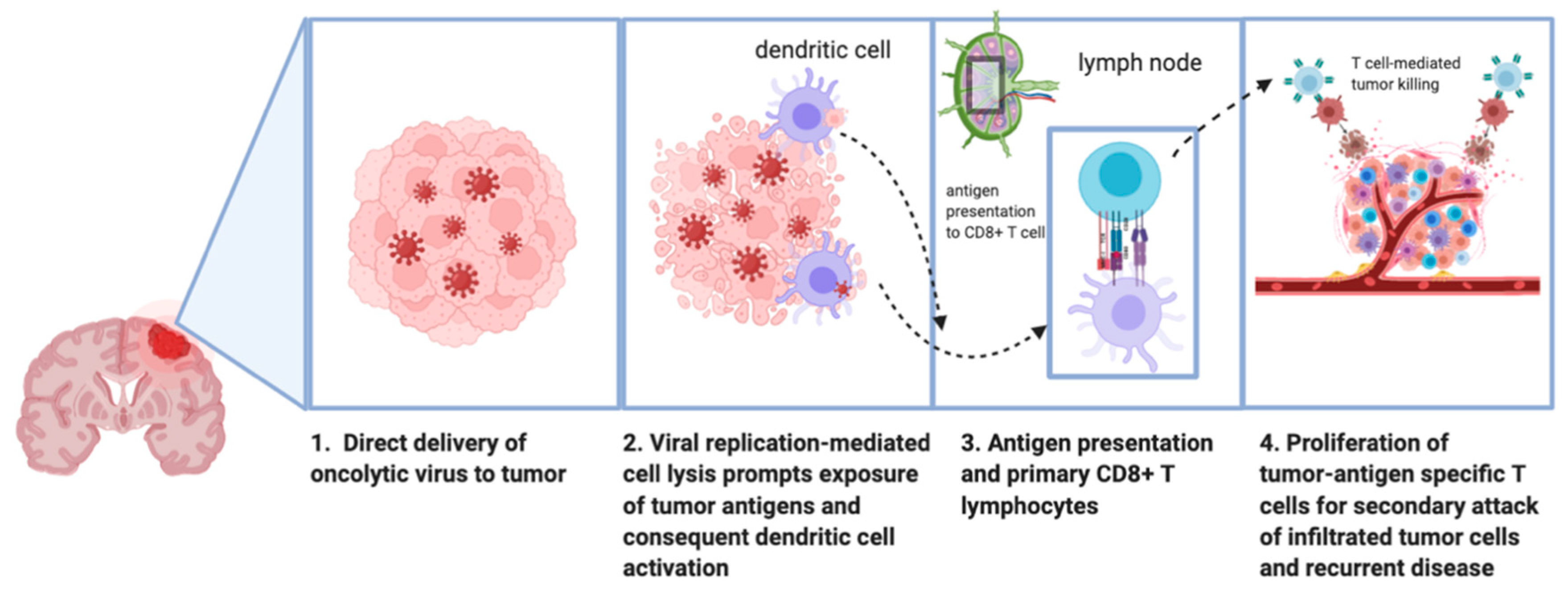
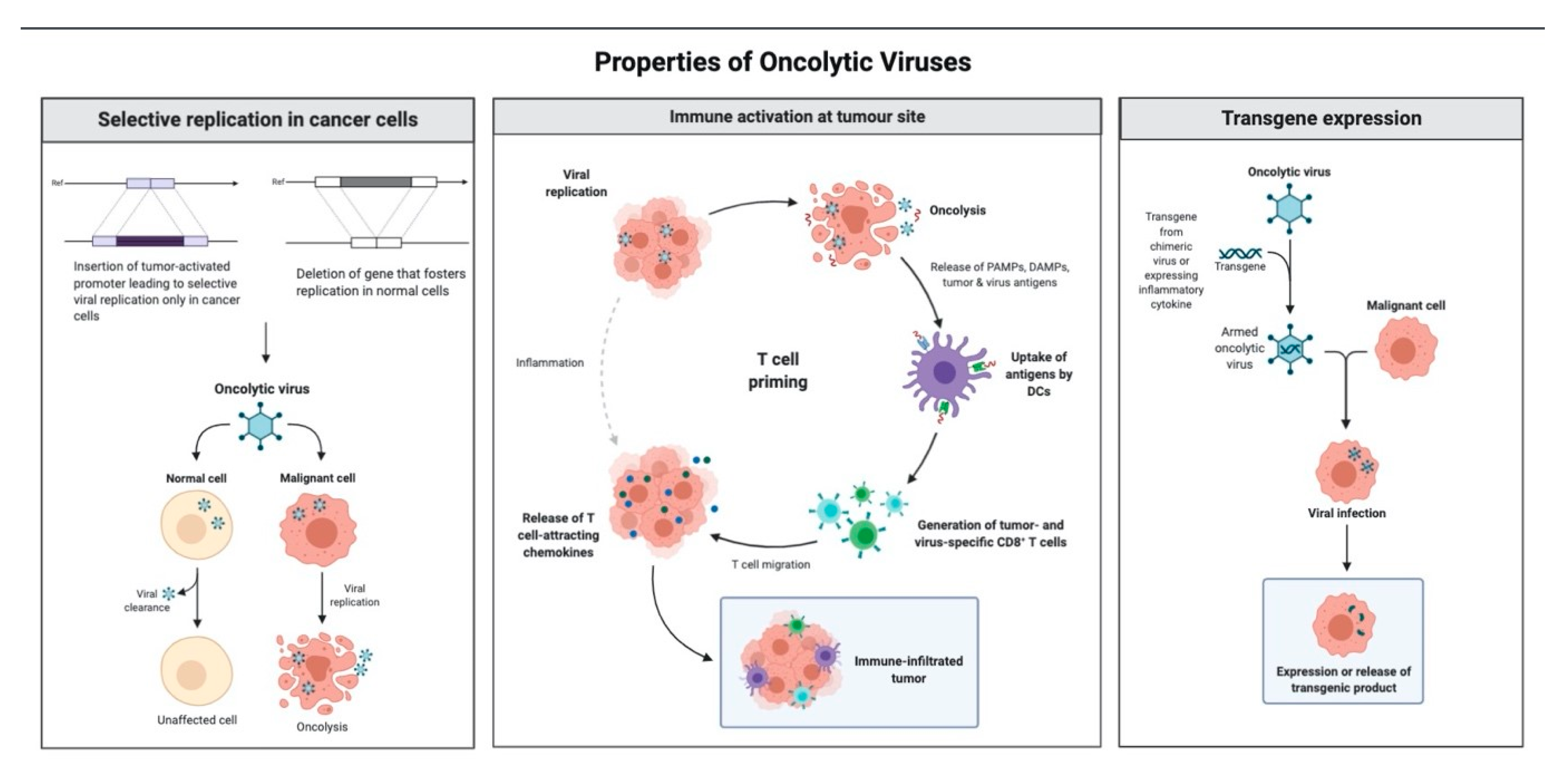
1.3. Mechanism of Antitumor Effect of Oncolytic Viruses
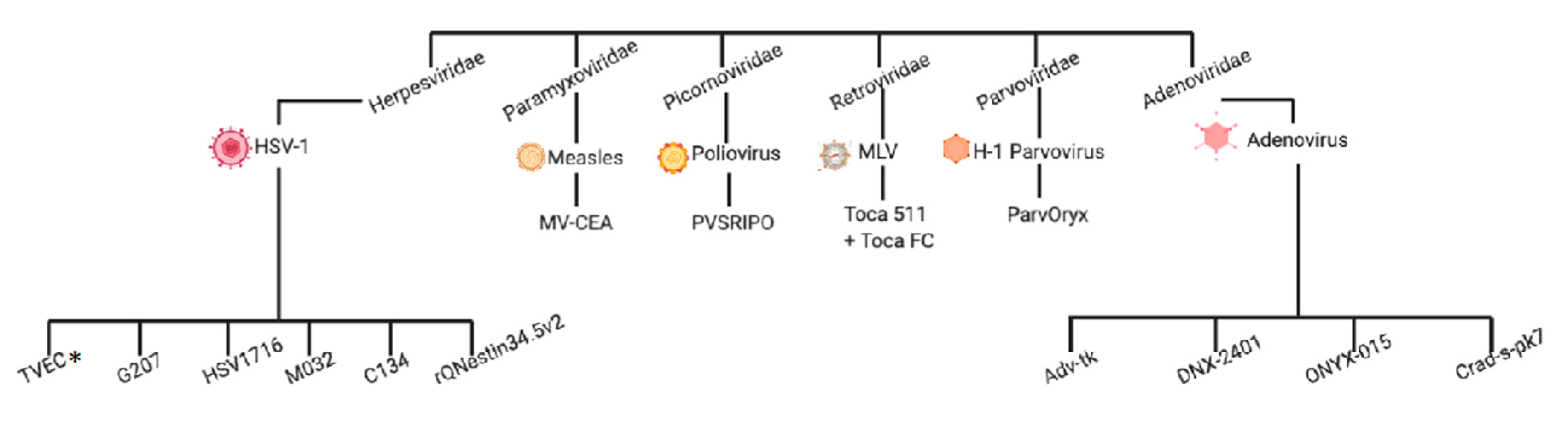
2. Clinical Experiences with Virotherapy in High-Grade Glioma
2.1. Herpesviridae
2.1.1. Talimogene Laherparepvec (TVEC, OncoVexGM-CSF, or IMLYGIC)
2.1.2. HSV G207
2.1.3. HSV1716
2.1.4. rQNestin34.5v.2
2.1.5. M032
2.1.6. C134
2.2. Adenoviridae
2.2.1. Aglatimagene Besadenovec (AdV-tk)
2.2.2. DNX-2401 (Tasadenoturev, Formerly Delta-24-RGD)
2.2.3. ONYX-015
2.2.4. CRAd-S-pk7
2.3. Retroviridae
Vocimagene Amiretrorepvec + (5-fluorocytosine(6-amino-5fluoro-1H-pyrimidin-2-one)) − (Toca 511 + Toca FC)
2.4. Picornoviridae
PVSRIPO
2.5. Reoviridae
Pelareorep (REOLYSIN)
2.6. Paramyxoviridae
MV-CEA
2.7. Parvoviridae
Parvovirus H-1 (H-1PV, Parv-Oryx)
2.8. Summary of Clinical Experiences Using OVs in High-Grade Glioma
3. Summary of Preclinical Experiences with Combinatory Virotherapy in Glioblastoma
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| HGG | high-grade glioma |
| CNS | central nervous system |
| BBB | blood–brain barrier |
| OV | oncolytic virus |
| Treg | regulatory T cell |
| MDSC | myeloid-derived suppressor cell |
| NK | natural killer |
| PAMP | pathogen-associated molecular pattern |
| PRR | pattern recognition receptor |
| DAMP | damage-associated molecular patterns |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| CTLA-4 | cytotoxic T-lymphocyte associated protein 4 |
| PD-1 | programmed cell death protein-1 |
| HSV | herpes simplex virus |
| FDA | Food and Drug Administration (of the United States) |
| CPA | cyclophosphamide |
| TMZ | temozolomide |
| CD | cytosine deaminase |
| 5-FU | 5-fluorouracil |
| CEA | carcinoembryogenic antigen |
| ECM | extracellular matrix |
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neuro Oncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.S.; Khatri, D.; Reichman, N.; Patel, N.V.; Wong, T.; Fralin, S.R.; Li, M.; Ellis, J.A.; Ortiz, R.; Langer, D.J.; et al. Super selective intra-arterial cerebral infusion of modern chemotherapeutics after blood–brain barrier disruption: Where are we now, and where we are going. J. Neuro Oncol. 2020, 147, 261–278. [Google Scholar] [CrossRef]
- Etame, A.B.; Diaz, R.J.; Smith, C.A.; Mainprize, T.G.; Hynynen, K.; Rutka, J.T. Focused ultrasound disruption of the blood-brain barrier: A new frontier for therapeutic delivery in molecular neurooncology. Neurosurg. Focus 2012, 32, E3. [Google Scholar] [CrossRef]
- De Pace, N. Sulla Scomparsa di un Enorme Cancro Vegetante del Collo Dell’utero Senza Cura Chirurgica. Ginecologia 1912, 9, 82–89. [Google Scholar]
- Higgins, G.K.; Pack, G.T. Virus therapy in the treatment of tumors. Bull. Hosp. Jt. Dis. 1951, 12, 379–382. [Google Scholar]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Pearl, T.M.; Markert, J.M.; Cassady, K.A.; Ghonime, M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics 2019, 13, 14–21. [Google Scholar] [CrossRef]
- Papanastassiou, V.; Rampling, R.; Fraser, M.; Petty, R.; Hadley, D.; Nicoll, J.; Harland, J.; Mabbs, R.; Brown, M. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: A proof of principle study. Gene Ther. 2002, 9, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEX GM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of Human Glioblastoma with a Live Attenuated Zika Virus Vaccine Candidate. MBio 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Groeneveldt, C.; van Hall, T.; van der Burg, S.H.; Ten Dijke, P.; van Montfoort, N. Immunotherapeutic Potential of TGF-beta Inhibition and Oncolytic Viruses. Trends Immunol. 2020, 41, 404–418. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virotherapy 2018, 7, 65–77. [Google Scholar] [CrossRef]
- Soldozy, S.; Skaff, A.; Soldozy, K.; Sokolowski, J.D.; Norat, P.; Yagmurlu, K.; Sharifi, K.A.; Tvrdik, P.; Park, M.S.; Kalani, M.Y.S.; et al. From Bench to Bedside, the Current State of Oncolytic Virotherapy in Pediatric Glioma. Neurosurgery 2020, 87, 1091–1097. [Google Scholar] [CrossRef]
- Kemp, V.; Lamfers, M.L.M.; van der Pluijm, G.; van den Hoogen, B.G.; Hoeben, R.C. Developing oncolytic viruses for clinical use: A consortium approach. Cytokine Growth Factor Rev. 2020, 56, 133–140. [Google Scholar] [CrossRef]
- Rius-Rocabert, S.; García-Romero, N.; García, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic virotherapy in glioma tumors. Int. J. Mol. Sci. 2020, 21, 7604. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, g207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Federoff, H.J.; Schoeniger, L.O. G207, Modified Herpes Simplex Virus Type 1, Kills Human Pancreatic Cancer Cells In Vitro. J. Gastrointest. Surg. 1999, 3, 127–133. [Google Scholar] [CrossRef]
- Brown, S.M.; MacLean, A.R.; Aitken, J.D.; Harland, J. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J. Gen. Virol. 1994, 75, 3679–3686. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef]
- Lamfers, M.L.M.; Fulci, G.; Gianni, D.; Tang, Y.; Kurozumi, K.; Kaur, B.; Moeniralm, S.; Saeki, Y.; Carette, J.E.; Weissleder, R.; et al. Cyclophosphamide Increases Transgene Expression Mediated by an Oncolytic Adenovirus in Glioma-Bearing Mice Monitored by Bioluminescence Imaging. Mol. Ther. 2006, 14, 779–788. [Google Scholar] [CrossRef]
- Qing, L.X.; Jang, J.-H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of Systemically Administered Oncolytic Vaccinia Virotherapy for Malignant Gliomas Is Enhanced by Combination Therapy with Rapamycin or Cyclophosphamide. Clin. Cancer Res. 2009, 15, 2777–2788. [Google Scholar] [CrossRef]
- Wakimoto, H.; Fulci, G.; Tyminski, E.; Chiocca, E.A. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004, 11, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the Safety and Biodistribution of M032, an Attenuated Herpes Simplex Virus Type 1 Expressing hIL-12, After Intracerebral Administration to Aotus Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Manetti, R.; Parronchi, P.; Giudizi, M.G.; Piccinni, M.; Maggi, E.; Trinchieri, G.; Romagnani, S. Natural Killer Cell Stimulatory Factory (Interleucin 12 [IL-12]) Induces Responses and Inhibits the Dedevelopment of IL-4-producing Th Cells. J. Exp. Med. 1993, 177, 1199–1204. [Google Scholar] [CrossRef]
- Voest, E.E.; Kenyon, B.M.; O’Reilly, M.S.; Truitt, G.; D’Amato, R.J.; Folkman, J. Inhibition of angiogenesis in vivo by interleukin 12 [see comments]. J. Natl. Cancer Inst. 1995, 87, 581–586. [Google Scholar] [CrossRef]
- Friedman, G.K.; Nan, L.; Haas, M.C.; Kelly, V.M.; Moore, B.P.; Langford, C.P.; Xu, H.; Han, X.; Beierle, E.A.; Markert, J.M.; et al. γ 1 34.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 2015, 22, 348–355. [Google Scholar] [CrossRef]
- Cassady, K.A.; Bauer, D.F.; Roth, J.; Chambers, M.R.; Shoeb, T.; Coleman, J.; Prichard, M.; Gillespie, G.Y.; Markert, J.M. Pre-clinical Assessment of C134, a Chimeric Oncolytic Herpes Simplex Virus, in Mice and Non-human Primates. Mol. Ther. Oncolytics 2017, 5, 1–10. [Google Scholar] [CrossRef]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef]
- Ghonime, M.G.; Jackson, J.; Shah, A.; Roth, J.; Li, M.; Saunders, U.; Coleman, J.; Gillespie, G.Y.; Markert, J.M.; Cassady, K.A. Chimeric HCMV/HSV-1 and Δγ134.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl. Oncol. 2018, 11, 86–93. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016, 18, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.Y.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Alemany, R.; Gomez-Manzano, C.; Fuller, G.N.; Khan, A.; Conrad, C.A.; Liu, T.-J.; Jiang, H.; Lemoine, M.G.; Suzuki, K.; et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J. Natl. Cancer Inst. 2003, 95, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Lang, F.F.; Tran, N.D.; Puduvalli, V.K.; Elder, J.B.; Fink, K.L.; Conrad, C.A.; Yung, W.K.A.; Penas-Prado, M.; Gomez-Manzano, C.; Peterkin, J.; et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J. Clin. Oncol. 2017, 35 (Suppl. S15), 2002. [Google Scholar] [CrossRef]
- Alonso, M.M.; García-Moure, M.; Gonzalez-Huarriz, M.; Marigil, M.; Hernandez-Alcoceba, R.; Buñales, M.; Hervás, S.; Gallego, J.; Gomez-Manzano, C.; Fueyo, J.; et al. Abstract CT027: Oncolytic virus DNX-2401 with a short course of temozolomide for glioblastoma at first recurrence: Clinical data and prognostic biomarkers. Cancer Res. 2017, 77, CT027. [Google Scholar] [CrossRef]
- Zadeh, G.; Daras, M.; Cloughesy, T.F.; Colman, H.; Kumthekar, P.U.; Chen, C.C.; Aiken, R.; Groves, M.D.; Ong, S.; Ramakrishna, R.; et al. Ltbk-04. Phase 2 multicenter study of the oncolytic adenovirus dnx-2401 (tasadenoturev) in combination with pembrolizumab for recurrent glioblastoma; captive study (keynote-192). Neuro Oncol. 2020, 22 (Suppl. S2), ii237. [Google Scholar] [CrossRef]
- Dobner, T.; Horikoshi, N.; Rubenwolf, S.; Shenk, T. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science 1996, 272, 1470–1473. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-johannes, A.; Fattaey, A.; et al. Adenovirus Mutant Replicates Selectively Deficient Human Tumor That in p53- Cells. Science 2014, 274, 373–376. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.J.; Haviv, Y.S.; Lu, B.; Wang, M.; Rivera, A.A.; Ulasov, I.V.; Lamfers, M.L.M.; Rein, D.; Lesniak, M.S.; Siegal, G.P.; et al. The human survivin promoter: A novel transcriptional targeting strategy for treatment of glioma. J. Neurosurg. 2008, 104, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Borovjagin, A.V.; Schroeder, B.A.; Baryshnikov, A.Y. Oncolytic adenoviruses: A thorny path to glioma cure. Genes Dis. 2014, 1, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Kawaji, H.; Yamasaki, T. Use of genetically engineered stem cells for Glioma therapy (Review). Oncol. Lett. 2016, 11, 9–15. [Google Scholar] [CrossRef]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and selection of toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Mitchell, L.A.; Lopez Espinoza, F.; Mendoza, D.; Kato, Y.; Inagaki, A.; Hiraoka, K.; Kasahara, N.; Gruber, H.E.; Jolly, D.J.; Robbins, J.M. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro Oncol. 2017, 19, 930–939. [Google Scholar] [CrossRef]
- Takahashi, M.; Valdes, G.; Hiraoka, K.; Inagaki, A.; Kamijima, S.; Micewicz, E.; Gruber, H.E.; Robbins, J.M.; Jolly, D.J.; McBride, W.H.; et al. Radiosensitization of gliomas by intracellular generation of 5-fluorouracil potentiates prodrug activator gene therapy with a retroviral replicating vector. Cancer Gene Ther. 2014, 21, 405–410. [Google Scholar] [CrossRef]
- Carter, B.; Jolly, D.J.; Nghiemphu, P.L.; Chen, C.C.; Piccioni, D.; Cloughesy, T.F.; Lai, A.; Kalkanis, S.N.; Hanna, M.; Liau, L.M.; et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Alexandert, L.; Wimmer, E.; Chanock, R.M. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants (neuropathogenicity/attenuation). Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.W.; Brown, M.C.; Sacco, M.T.; Gromeier, M. Engineered Oncolytic Poliovirus PVSRIPO Subverts MDA5-Dependent Innate Immune Responses in Cancer Cells. J. Virol. 2018, 92, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Nair, S.K. Recombinant Poliovirus for Cancer Immunotherapy. Annu. Rev. Med. 2018, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W.K. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W.K. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef]
- Maitra, R.; Ghalib, M.H.; Goel, S. Reovirus: A Targeted Therapeutic--Progress And Potential. Mol. Cancer Res. 2012, 10, 1514–1525. [Google Scholar] [CrossRef]
- Forsyth, P.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Kemp, V.; van den Wollenberg, D.J.M.; Camps, M.G.M.; van Hall, T.; Kinderman, P.; Pronk-van Montfoort, N.; Hoeben, R.C. Arming oncolytic reovirus with GM-CSF gene to enhance immunity. Cancer Gene Ther. 2019, 26, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Phuong, L.K.; Allen, C.; Peng, K.-W.; Giannini, C.; Greiner, S.; TenEyck, C.J.; Mishra, P.K.; Macura, S.I.; Russell, S.J.; Galanis, E.C. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003, 63, 2462–2469. [Google Scholar] [PubMed]
- Myers, R.; Harvey, M.; Kaufmann, T.J.; Greiner, S.M.; Krempski, J.W.; Raffel, C.; Shelton, S.E.; Soeffker, D.; Zollman, P.; Federspiel, M.J.; et al. Toxicology Study of Repeat Intracerebral Administration of a Measles Virus Derivative Producing Carcinoembryonic Antigen in Rhesus Macaques in Support of a Phase I/II Clinical Trial for Patients with Recurrent Gliomas. Hum. Gene Ther. 2008, 19, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Herrero YCalle, M.; Rommelaere, J.; Schlehofer, J.R. Oncolytic potential of rodent parvoviruses for cancer therapy in humans: A brief review. J. Vet. Med. Ser. B Infect. Dis. Vet. Public Health 2005, 52, 327–330. [Google Scholar] [CrossRef]
- Rommelaere, J.; Cornelis, J.J. Antineoplasic activity of parvoviruses. J. Virol. Methods 1991, 33, 233–251. [Google Scholar] [CrossRef]
- Geletneky, K.; Leoni, A.L.; Pohlmeyer-Esch, G.; Loebhard, S.; Baetz, A.; Leuchs, B.; Roscher, M.; Hoefer, C.; Jochims, K.; Dahm, M.; et al. Pathology, organ distribution, and immune response after single and repeated intravenous injection of rats with clinical-grade parvovirus H1. Comp. Med. 2015, 65, 23–35. [Google Scholar]
- Geletneky, K.; Leoni, A.L.; Pohlmeyer-Esch, G.; Loebhard, S.; Leuchs, B.; Hoefer, C.; Jochims, K.; Dahm, M.; Huber, B.; Rommelaere, J.; et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp. Med. 2015, 65, 36–45. [Google Scholar]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic Activation of Cathepsins Mediates Parvovirus H-1-Induced Killing of Cisplatin and TRAIL-Resistant Glioma Cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef]
- Geletneky, K.; Huesing, J.; Rommelaere, J.; Schlehofer, J.R.; Leuchs, B.; Dahm, M.; Krebs, O.; von Knebel Doeberitz, M.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; de López-Diaz Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017, 19, 493–502. [Google Scholar] [CrossRef]
- Errington-Mais, F.; Cockle, J.V.; Furness, A.J.; Collinson, F.J.; Pandha, H.; Griffin, S.D.; Ilett, E.J.; Wurdak, H.; Kottke, T.J.; Rose, A.S.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial effects of vegfr kinase inhibitor axitinib and oncolytic virotherapy in mouse and human glioblastoma stem-like cell models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Cockle, J.V.; Brüning-Richardson, A.; Scott, K.J.; Thompson, J.; Kottke, T.; Morrison, E.; Ismail, A.; Carcaboso, A.M.; Rose, A.; Selby, P.; et al. Oncolytic Herpes Simplex Virus Inhibits Pediatric Brain Tumor Migration and Invasion. Mol. Ther. Oncolytics 2017, 5, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Clise-Dwyer, K.; Ruisaard, K.E.; Fan, X.; Tian, W.; Gumin, J.; Lamfers, M.L.; Kleijn, A.; Lang, F.F.; Yung, W.K.A.; et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS ONE 2014, 9, e97407. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Raffel, C. Using Cystine Knot Proteins as a Novel Approach to Retarget Oncolytic Measles Virus. Mol. Ther. Oncolytics 2017, 7, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yao, T.; Zheng, Y.; Li, Z.; Zhang, Q.; Zhang, L.; Zhou, D. Development of next generation adeno-associated viral vectors capable of selective tropism and efficient gene delivery. Biomaterials 2016, 80, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Nair, M.; Banasavadi-Siddegowda, Y.K.; Liu, J.; Nallanagulagari, T.; Jaime-Ramirez, A.C.; Guo, J.Y.; Quadri, H.; Zhang, J.; Bockhorst, K.H.; et al. Enhancing Therapeutic Efficacy of Oncolytic Herpes Simplex Virus-1 with Integrin β1 Blocking Antibody OS2966. Mol. Cancer Ther. 2019, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- GuhaSarkar, D.; Neiswender, J.; Su, Q.; Gao, G.; Sena-Esteves, M. Intracranial AAV-IFN-β gene therapy eliminates invasive xenograft glioblastoma and improves survival in orthotopic syngeneic murine model. Mol. Oncol. 2017, 11, 180–193. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | NCT | Study Phase | Published Results | n | Study Population | Outcomes |
|---|---|---|---|---|---|---|
| G207 | NCT00157703 | Phase I | Markert et al. 2014 | 9 | Recurrent malignant glioma | Safety demonstrated (AEs) Median survival from inoculation = 7.5 months mPFS = 2.5 months |
| NCT00028158 | Phase Ib/II | Markert et al. 2000 | 21 | Recurrent malignant glioma | Safety demonstrated (AEs) Mean TTP = 3.5 months Mean OS = 15.9 (glioblastoma) and 40.5 (anaplastic astrocytoma) | |
| HSV1716 | (UK) | Phase I | Rampling et al. 2000 | 9 | Recurrent malignant glioma | Safety demonstrated (AEs) |
| (UK) | Phase I | Papanastassiou et al. 2002 | 12 | Malignant glioma | Safety demonstrated (AEs) | |
| (UK) | Phase I | Harrow et al. 2004 | 12 | Recurrent or newly diagnosed high grade glioma | Safety demonstrated (AEs) | |
| AdV-tk | NCT00589875 | Phase II | Wheeler et al. 2016 | 48 | Newly diagnosed glioblastoma | Safety demonstrated (AEs, DLTs) mOS = 17.1 months mPFS = 8.1 months Surival at 1, 2, 3 years = 67%, 35%, 19% |
| DNX-2401 | NCT00805376 | Phase I | Lang et al. 2018 | 37 | Recurrent malignant glioma | Safety demonstrated (AEs, DLTs) Study arm A (single injection)- Tumor reduction in 72% of patients mOS = 9.5 months Study arm B (infusion and resection) mOS = 13 months |
| NCT02197169 (TARGET-1) | Phase Ib | Lang et al. 2017 | 27 | Recurrent glioblastoma | Tolerability of DNX-2401 as monotherapy (compared to combination with IFN-gamma) demonstrated (AEs) OS-12 (33%) OS-18 (22%) | |
| NCT01956734 (Spain) | Phase I | Alonso et al. 2017 | 31 | Glioblastoma at first recurrence | Safety demonstrated when combined with TMZ (AEs), efficacy endpoints not yet reported | |
| NCT02798406 (CAPTIVE/KEYNOTE-192) | Phase II | Zadeh et al. 2020 | 49 | Recurrent glioblastoma | Safety demonstrated when combined with pembrolizumab (AEs) mOS = 12.5 months OS12 = 54.5%, OS18 = 20.8% | |
| ONYX-015 | - | Phase I | Chiocca et al. 2004 | 24 | Recurrent malignant glioma | Safety demonstrated (AEs, DLTs) Median survival = 6.2 months (4.9 months for glioblastoma patients, 11.4 in AA/AO) |
| Toca511 + TocaFC | NCT01470794 | Phase I | Cloughesy et al. 2016 | 43 | Recurrent high grade glioma | Safety (AEs, DLTs) OS (HGG) = 13.6 months OS (glioblastoma) = 11.6 months For all evaluable patients: OS6 (87.9%), OS9 (72.4%), OS12 (52.5%), OS24 (29.1%) PFS = 3.2 months, PFS6 = 16.3% |
| NCT01156584 | Phase I | - | 54 | Recurrent high grade glioma | - | |
| NCT01985256 | Phase I | - | 17 | Recurrent or progressive high grade glioma | - | |
| NCT02414165 (Toca 5) | Phase II/III | Cloughesy et al. 2020 | 201 | Recurrent glioblastoma/anaplastic astrocytoma | Safety (AEs) mOS = 11.1 months Efficacy was not demonstrated over control arm | |
| PVSRIPO | NCT01491893 | Phase I | Desjardins et al. 2018 | 61 | Recurrent glioblastoma | mOS = 12.5 months OS 24 M and 36 M = 21% |
| REOLYSIN | NCT00528684 | Phase I/II | Forsyth et al. 2008 | 12 | Recurrent malignant glioma | mOS = 21 weeks (range 6–234) mTTP 4.3 weeks (range 2.6–39) MTD not reached |
| NCT00528684 | Phase I/II | Kickielinski et al. 2014 | 15 | Recurrent malignant glioma | mOS = 140 days mTTP = 61 days |
| Agent | NCT | Study Phase | n | Trial Design/Population | Outcomes (Safety, Efficacy) |
|---|---|---|---|---|---|
| G207 | NCT02457845 | Phase I | 12 | Pediatric progressive or recurrent supratentorial tumors | Safety, tolerability (AEs) PFS, OS |
| NCT03911388 | Phase I | 15 | Pediatric recurrent or refractory cerebellar tumors | Safety, tolerability (AEs) PFS, OS | |
| HSV1716 | NCT02031965 | Phase I Terminated by sponsor | 2 | Pediatric refractory/recurrent high grade glioma | MTD, PFS, and OS up to 15 years |
| rQNestin | NCT03152318 | Phase I | 108 | Malignant glioma | MTD |
| M032 | NCT02062827 | Phase I | 36 | Recurrent malignant glioma | MTD TTP and survival up to 12 months |
| C134 | NCT03657576 | Phase I | 24 | Recurrent glioblastoma | Safety, tolerability (AEs) PFS- 3 d, 28 d, 3 M, 6 M, 12 M, OS up to 12 M |
| DNX-2401 | NCT03896568 | Phase I | 36 | Recurrent high-grade glioma | MTD, AEs Tumor response, TTP for 1 year |
| CRAd-S-pk7 | NCT03072134 | Phase I | 12 | Newly diagnosed malignant glioma | Neurological side effects, MRIs for progression |
| Toca 511 + Toca FC | NCT02598011 (Toca 7) | Phase Ib Terminated by sponsor | 18 | Newly diagnosed high grade glioma | DLTs |
| PVSRIPO | NCT03043391 | Phase Ib | 12 | Pediatric recurrent malignant glioma | Toxicity, 24 month OS |
| PVSRIPO + atezolizumab | NCT03973879 | Phase Ib/2 Withdrawn, resubmission expected | _ | Recurrent malignant glioma | Safety (AEs), survival at 24 M |
| PVSRIPO + lomustine | NCT02986178 | Phase II | 122 | Recurrent malignant glioma | Objective response (iRANO) at 24 and 36 M, duration of ORR, OS at 24 and 36 M, safety (AEs) |
| REOLYSIN + GM-CSF | NCT02444546 | Phase I | 6 | Pediatric relapsed/refractory brain tumors | MTD (DLT), AE, mOS, OR, TTP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Immidisetti, A.V.; Nwagwu, C.D.; Adamson, D.C.; Patel, N.V.; Carbonell, A.-M. Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults. Biomedicines 2021, 9, 138. https://doi.org/10.3390/biomedicines9020138
Immidisetti AV, Nwagwu CD, Adamson DC, Patel NV, Carbonell A-M. Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults. Biomedicines. 2021; 9(2):138. https://doi.org/10.3390/biomedicines9020138
Chicago/Turabian StyleImmidisetti, Amanda V., Chibueze D. Nwagwu, David C. Adamson, Nitesh V. Patel, and Anne-Marie Carbonell. 2021. "Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults" Biomedicines 9, no. 2: 138. https://doi.org/10.3390/biomedicines9020138
APA StyleImmidisetti, A. V., Nwagwu, C. D., Adamson, D. C., Patel, N. V., & Carbonell, A.-M. (2021). Clinically Explored Virus-Based Therapies for the Treatment of Recurrent High-Grade Glioma in Adults. Biomedicines, 9(2), 138. https://doi.org/10.3390/biomedicines9020138