
Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions

Abstract
1. Introduction
1.1. Meningioma Cell of Origin
1.2. WHO Grading
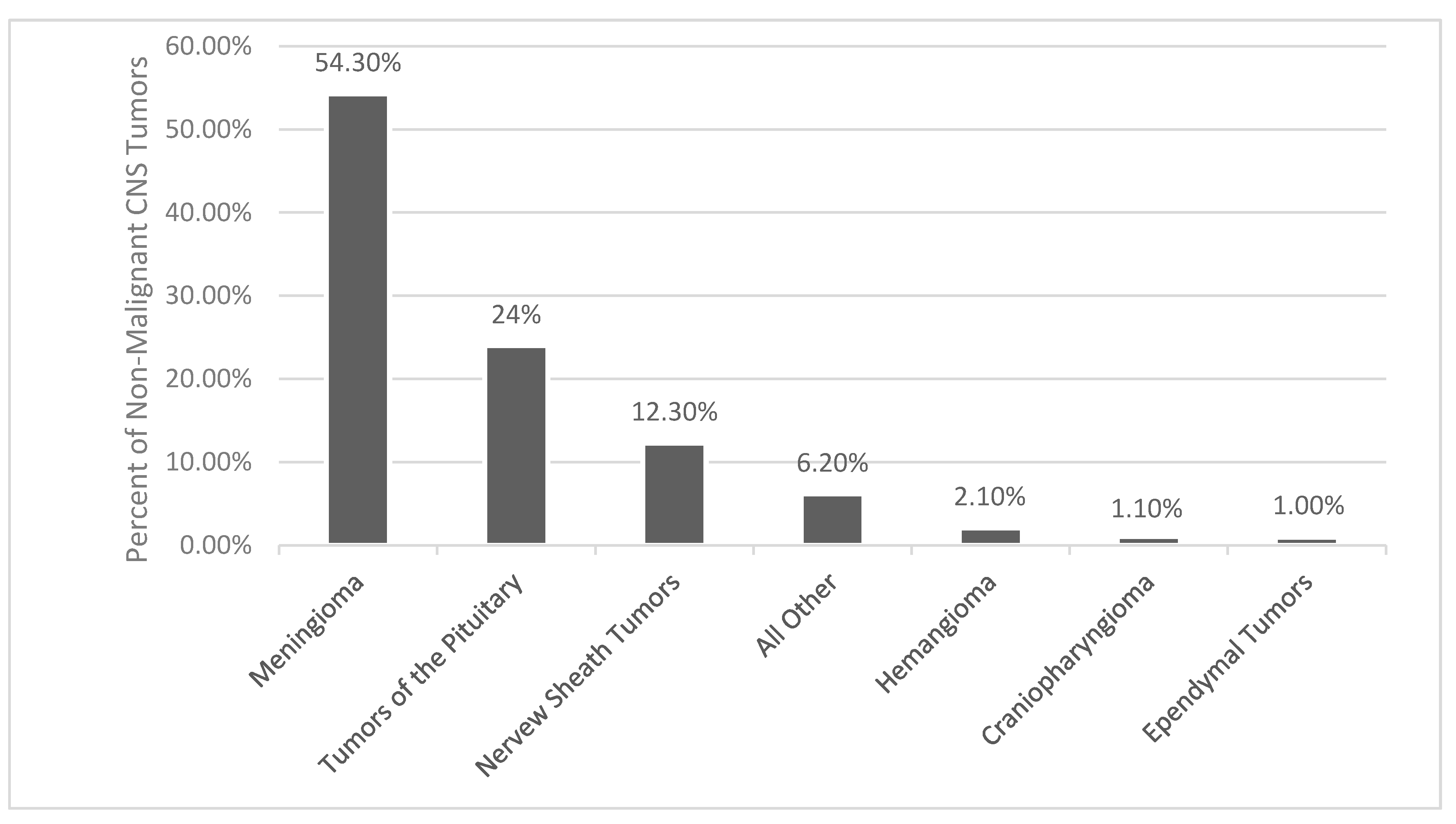
1.3. Epidemiology
1.4. Etiology/Risk Factors
1.4.1. Ionizing Radiation
1.4.2. Obesity
1.4.3. Occupational (Pesticide/Herbicide)/Diet/Allergies
1.4.4. Hormones
1.5. Molecular Characteristics
1.5.1. Cytogenetics
1.5.2. Familial Syndromes
Neurofibromatosis Type 2 (NF2)
Gorlin Syndrome
Cowden Syndrome
Werner Syndrome
BAP1 Tumor Predisposition Syndrome
Familial Syndromes Associated with SMARCB1 and SMARCE1
Other Familial Syndrome
1.5.3. Somatic Mutations
NF2
Non-NF2 Mutations
1.5.4. Epigenetic Modifications
2. Clinical Features
2.1. Presenting Locations
2.2. Signs and Symptoms
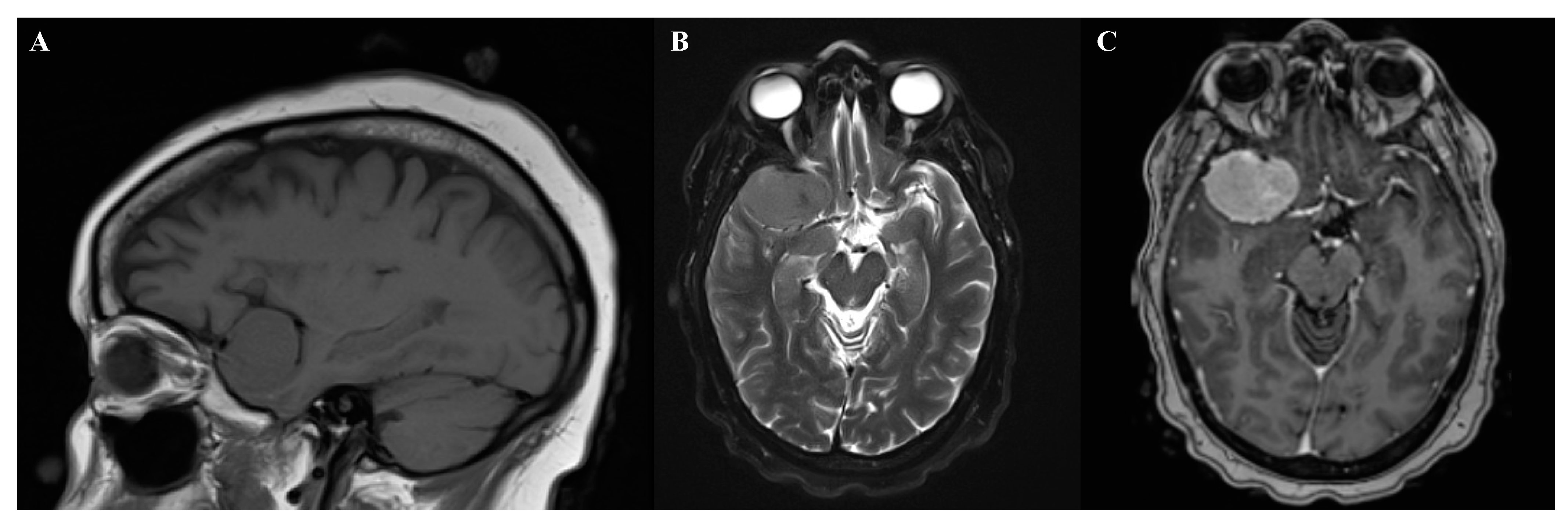
2.3. Diagnostics
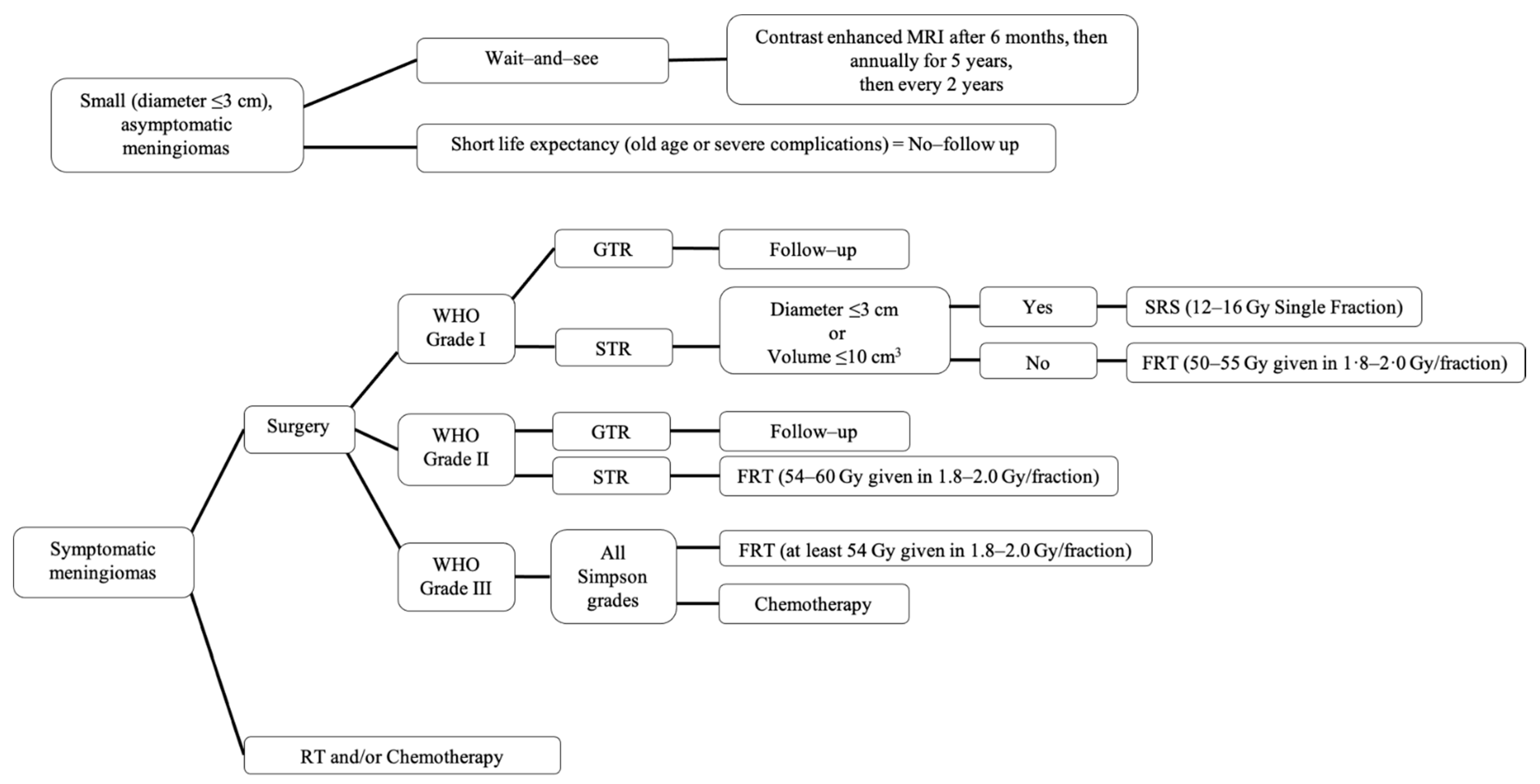
3. Treatment
4. Outcomes and Natural History of Meningioma
5. Current Areas of Research
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Huntoon, K.; Toland, A.M.S.; Dahiya, S. Meningioma: A Review of Clinicopathological and Molecular Aspects. Front. Oncol. 2020, 10, 579599. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System; World Health Organization Classification of Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016; ISBN 978-92-832-4492-9. [Google Scholar]
- Weller, R.O. Microscopic Morphology and Histology of the Human Meninges. Morphologie 2005, 89, 22–34. [Google Scholar] [CrossRef]
- Nabeshima, S.; Reese, T.S.; Landis, D.M.; Brightman, M.W. Junctions in the Meninges and Marginal Glia. J. Comp. Neurol. 1975, 164, 127–169. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fang, L.; Meyer, P.; Killer, H.E.; Flammer, J.; Neutzner, A. Anti-Inflammatory Response Following Uptake of Apoptotic Bodies by Meningothelial Cells. J. Neuroinflam. 2014, 11, 35. [Google Scholar] [CrossRef]
- Li, J.; Fang, L.; Killer, H.E.; Flammer, J.; Meyer, P.; Neutzner, A. Meningothelial Cells as Part of the Central Nervous System Host Defence. Biol. Cell 2013, 105, 304–315. [Google Scholar] [CrossRef]
- Hemion, C.; Li, J.; Kohler, C.; Scholl, H.P.N.; Meyer, P.; Killer, H.E.; Neutzner, A. Clearance of Neurotoxic Peptides and Proteins by Meningothelial Cells. Exp. Cell Res. 2020, 396, 112322. [Google Scholar] [CrossRef]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Niwa-Kawakita, M.; Chareyre, F.; Taranchon, E.; Han, Z.-Y.; Martinelli, C.; Lusis, E.A.; Hegedus, B.; Gutmann, D.H.; et al. Identification of a Progenitor Cell of Origin Capable of Generating Diverse Meningioma Histological Subtypes. Oncogene 2011, 30, 2333–2344. [Google Scholar] [CrossRef]
- Perry, A.; Gutmann, D.H.; Reifenberger, G. Molecular Pathogenesis of Meningiomas. J. Neurooncol. 2004, 70, 183–202. [Google Scholar] [CrossRef]
- Preusser, M.; Brastianos, P.K.; Mawrin, C. Advances in Meningioma Genetics: Novel Therapeutic Opportunities. Nat. Rev. Neurol. 2018, 14, 106–115. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, Y.S. Molecular Characteristics of Meningiomas. J. Pathol. Transl. Med. 2020, 54, 45–63. [Google Scholar] [CrossRef]
- Thevandiran, D.; Nga, V.; Chang, K.T.E.; Ng, L.P.; Seow, W.T.; Low, D.C.Y.; Yeo, T.T.; Low, S.Y.Y. Paediatric Meningiomas in Singapore—Case Series of a Rare Entity. J. Clin. Neurosci. 2020, 73, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.; Auguste, K.I.; Gupta, N. Meningiomas in children. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 169, pp. 253–259. ISBN 978-0-12-804280-9. [Google Scholar]
- Nakasu, S.; Hirano, A.; Shimura, T.; Llena, J.F. Incidental Meningiomas in Autopsy Study. Surg. Neurol. 1987, 27, 319–322. [Google Scholar] [CrossRef]
- Johnson, M.D.; Abu-Farsakh, S. Clinicopathologic Features of Incidental Meningiomas: A Review of the Literature and the University of Rochester Autopsy Experience. Clin. Neuropathol. 2019, 38, 118–121. [Google Scholar] [CrossRef]
- Wiemels, J.; Wrensch, M.; Claus, E.B. Epidemiology and Etiology of Meningioma. J. Neurooncol. 2010, 99, 307–314. [Google Scholar] [CrossRef]
- Al-Mefty, O.; Kersh, J.E.; Routh, A.; Smith, R.R. The Long-Term Side Effects of Radiation Therapy for Benign Brain Tumors in Adults. J. Neurosurg. 1990, 73, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Umansky, F.; Shoshan, Y.; Rosenthal, G.; Fraifeld, S.; Spektor, S. Radiation-Induced Meningioma. Neurosurg. Focus 2008, 24, E7. [Google Scholar] [CrossRef] [PubMed]
- Ron, E.; Modan, B.; Boice, J.D.; Alfandary, E.; Stovall, M.; Chetrit, A.; Katz, L. Tumors of the Brain and Nervous System after Radiotherapy in Childhood. N. Engl. J. Med. 1988, 319, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Sadetzki, S.; Chetrit, A.; Freedman, L.; Stovall, M.; Modan, B.; Novikov, I. Long-Term Follow-up for Brain Tumor Development after Childhood Exposure to Ionizing Radiation for Tinea Capitis. Radiat. Res. 2005, 163, 424–432. [Google Scholar] [CrossRef]
- Sadamori, N.; Shibata, S.; Mine, M.; Miyazaki, H.; Miyake, H.; Kurihara, M.; Tomonaga, M.; Sekine, I.; Okumura, Y. Incidence of Intracranial Meningiomas in Nagasaki Atomic-Bomb Survivors. Int. J. Cancer 1996, 67, 318–322. [Google Scholar] [CrossRef]
- Shintani, T.; Hayakawa, N.; Hoshi, M.; Sumida, M.; Kurisu, K.; Oki, S.; Kodama, Y.; Kajikawa, H.; Inai, K.; Kamada, N. High Incidence of Meningioma among Hiroshima Atomic Bomb Survivors. J. Radiat. Res. 1999, 40, 49–57. [Google Scholar] [CrossRef]
- Modan, B.; Baidatz, D.; Mart, H.; Steinitz, R.; Levin, S.G. Radiation-Induced Head and Neck Tumours. Lancet 1974, 1, 277–279. [Google Scholar] [CrossRef]
- Takahashi, H.; Cornish, A.J.; Sud, A.; Law, P.J.; Disney-Hogg, L.; Calvocoressi, L.; Lu, L.; Hansen, H.M.; Smirnov, I.; Walsh, K.M.; et al. Mendelian Randomization Provides Support for Obesity as a Risk Factor for Meningioma. Sci. Rep. 2019, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chen, J.; Wang, J.; Gong, S.; Jin, H.; Sheng, P.; Qi, X.; Lv, L.; Dong, Y.; Hou, L. Body Mass Index and Risk of Brain Tumors: A Systematic Review and Dose-Response Meta-Analysis. Eur. J. Clin. Nutr. 2016, 70, 757–765. [Google Scholar] [CrossRef]
- Samkange-Zeeb, F.; Schlehofer, B.; Schüz, J.; Schlaefer, K.; Berg-Beckhoff, G.; Wahrendorf, J.; Blettner, M. Occupation and Risk of Glioma, Meningioma and Acoustic Neuroma: Results from a German Case-Control Study (Interphone Study Group, Germany). Cancer Epidemiol. 2010, 34, 55–61. [Google Scholar] [CrossRef]
- Khuder, S.A.; Mutgi, A.B.; Schaub, E.A. Meta-Analyses of Brain Cancer and Farming. Am. J. Ind. Med. 1998, 34, 252–260. [Google Scholar] [CrossRef]
- Provost, D.; Cantagrel, A.; Lebailly, P.; Jaffré, A.; Loyant, V.; Loiseau, H.; Vital, A.; Brochard, P.; Baldi, I. Brain Tumours and Exposure to Pesticides: A Case-Control Study in Southwestern France. Occup. Environ. Med. 2007, 64, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Samanic, C.M.; De Roos, A.J.; Stewart, P.A.; Rajaraman, P.; Waters, M.A.; Inskip, P.D. Occupational Exposure to Pesticides and Risk of Adult Brain Tumors. Am. J. Epidemiol. 2008, 167, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.B.; Howe, G.; Pogoda, J.M.; Zhang, F.F.; Ahlbom, A.; Choi, W.; Giles, G.G.; Little, J.; Lubin, F.; Menegoz, F.; et al. An International Case-Control Study of Adult Diet and Brain Tumor Risk: A Histology-Specific Analysis by Food Group. Ann. Epidemiol. 2009, 19, 161–171. [Google Scholar] [CrossRef]
- Wang, P.-F.; Ji, W.-J.; Zhang, X.-H.; Li, S.-W.; Yan, C.-X. Allergy Reduces the Risk of Meningioma: A Meta-Analysis. Sci. Rep. 2017, 7, 40333. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Wrensch, M.; Sison, J.D.; Zhou, M.; Bondy, M.; Calvocoressi, L.; Black, P.M.; Yu, H.; Schildkraut, J.M.; Claus, E.B. Reduced Allergy and Immunoglobulin E among Adults with Intracranial Meningioma Compared to Controls. Int. J. Cancer 2011, 129, 1932–1939. [Google Scholar] [CrossRef]
- Korhonen, K.; Salminen, T.; Raitanen, J.; Auvinen, A.; Isola, J.; Haapasalo, H. Female Predominance in Meningiomas Can Not Be Explained by Differences in Progesterone, Estrogen, or Androgen Receptor Expression. J. Neurooncol. 2006, 80, 1–7. [Google Scholar] [CrossRef]
- Baldi, I.; Engelhardt, J.; Bonnet, C.; Bauchet, L.; Berteaud, E.; Grüber, A.; Loiseau, H. Epidemiology of Meningiomas. Neurochirurgie 2018, 64, 5–14. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, Y.; Wang, Y.; Liu, L.; Lou, J.; Deng, Y.; Zhao, P.; Shao, A. Clinical Significance of Somatostatin Receptor (SSTR) 2 in Meningioma. Front. Oncol. 2020, 10, 1633. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Cubo-Delgado, E.; González-Bernal, J.J.; Martín-Alonso, J. A Practical Overview on the Molecular Biology of Meningioma. Curr. Neurol. Neurosci. Rep. 2020, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-Y.; Shao, C.; Huang, Y.-L.; Hui, G.-Z.; Zhou, Y.-X.; Wang, Z. Reproductive and Exogenous Hormone Factors in Relation to Risk of Meningioma in Women: A Meta-Analysis. PLoS ONE 2013, 8, e83261. [Google Scholar] [CrossRef] [PubMed]
- Benson, V.S.; Pirie, K.; Green, J.; Casabonne, D.; Beral, V. Million Women Study Collaborators Lifestyle Factors and Primary Glioma and Meningioma Tumours in the Million Women Study Cohort. Br. J. Cancer 2008, 99, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Benson, V.S.; Kirichek, O.; Beral, V.; Green, J. Menopausal Hormone Therapy and Central Nervous System Tumor Risk: Large UK Prospective Study and Meta-Analysis. Int. J. Cancer 2015, 136, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Jiang, Y.; Wen, T.; Lu, S.; Yao, L.; Meng, F. Association of Hormone Replacement Therapy with Increased Risk of Meningioma in Women: A Hospital-Based Multicenter Study with Propensity Score Matching. Asia Pac. J. Clin. Oncol. 2019, 15, e147–e153. [Google Scholar] [CrossRef]
- Muskens, I.S.; Wu, A.H.; Porcel, J.; Cheng, I.; Le Marchand, L.; Wiemels, J.L.; Setiawan, V.W. Body Mass Index, Comorbidities, and Hormonal Factors in Relation to Meningioma in an Ethnically Diverse Population: The Multiethnic Cohort. Neuro Oncol. 2019, 21, 498–507. [Google Scholar] [CrossRef]
- Michaud, D.S.; Gallo, V.; Schlehofer, B.; Tjønneland, A.; Olsen, A.; Overvad, K.; Dahm, C.C.; Kaaks, R.; Lukanova, A.; Boeing, H.; et al. Reproductive Factors and Exogenous Hormone Use in Relation to Risk of Glioma and Meningioma in a Large European Cohort Study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Rankin, C.; Grunberg, S.; Sherrod, A.E.; Ahmadi, J.; Townsend, J.J.; Feun, L.G.; Fredericks, R.K.; Russell, C.A.; Kabbinavar, F.F.; et al. Double-Blind Phase III Randomized Trial of the Antiprogestin Agent Mifepristone in the Treatment of Unresectable Meningioma: SWOG S9005. J. Clin. Oncol. 2015, 33, 4093–4098. [Google Scholar] [CrossRef]
- Wen, P.Y.; Quant, E.; Drappatz, J.; Beroukhim, R.; Norden, A.D. Medical Therapies for Meningiomas. J. Neurooncol. 2010, 99, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.T.; Ramachandran, S.; Lama, S.; Sutherland, G.R. Towards Molecular Classification of Meningioma: Evolving Treatment and Diagnostic Paradigms. World Neurosurg. 2018, 119, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Aizer, A.A.; Abedalthagafi, M.; Bi, W.L.; Horvath, M.C.; Arvold, N.D.; Al-Mefty, O.; Lee, E.Q.; Nayak, L.; Rinne, M.L.; Norden, A.D.; et al. A Prognostic Cytogenetic Scoring System to Guide the Adjuvant Management of Patients with Atypical Meningioma. Neuro Oncol. 2016, 18, 269–274. [Google Scholar] [CrossRef]
- Nassiri, F.; Mamatjan, Y.; Suppiah, S.; Badhiwala, J.H.; Mansouri, S.; Karimi, S.; Saarela, O.; Poisson, L.; Gepfner-Tuma, I.; Schittenhelm, J.; et al. DNA Methylation Profiling to Predict Recurrence Risk in Meningioma: Development and Validation of a Nomogram to Optimize Clinical Management. Neuro Oncol. 2019, 21, 901–910. [Google Scholar] [CrossRef]
- Lamszus, K. Meningioma Pathology, Genetics, and Biology. J. Neuropathol. Exp. Neurol. 2004, 63, 275–286. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic Analysis of Non-NF2 Meningiomas Reveals Mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Mawrin, C.; Perry, A. Pathological Classification and Molecular Genetics of Meningiomas. J. Neurooncol. 2010, 99, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.G.; Boström, J.; Wolter, M.; Baudis, M.; Collins, V.P.; Reifenberger, G.; Lichter, P. Analysis of Genomic Alterations in Benign, Atypical, and Anaplastic Meningiomas: Toward a Genetic Model of Meningioma Progression. Proc. Natl. Acad. Sci. USA 1997, 94, 14719–14724. [Google Scholar] [CrossRef]
- Al-Rashed, M.; Foshay, K.; Abedalthagafi, M. Recent Advances in Meningioma Immunogenetics. Front. Oncol. 2019, 9, 1472. [Google Scholar] [CrossRef]
- Mei, Y.; Bi, W.L.; Greenwald, N.F.; Agar, N.Y.; Beroukhim, R.; Dunn, G.P.; Dunn, I.F. Genomic Profile of Human Meningioma Cell Lines. PLoS ONE 2017, 12, e0178322. [Google Scholar] [CrossRef] [PubMed]
- Al-Mefty, O.; Kadri, P.A.S.; Pravdenkova, S.; Sawyer, J.R.; Stangeby, C.; Husain, M. Malignant Progression in Meningioma: Documentation of a Series and Analysis of Cytogenetic Findings. J. Neurosurg. 2004, 101, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic Landscape of High-Grade Meningiomas. NPJ Genom. Med. 2017, 2. [Google Scholar] [CrossRef]
- Domingues, P.H.; Sousa, P.; Otero, Á.; Gonçalves, J.M.; Ruiz, L.; de Oliveira, C.; Lopes, M.C.; Orfao, A.; Tabernero, M.D. Proposal for a New Risk Stratification Classification for Meningioma Based on Patient Age, WHO Tumor Grade, Size, Localization, and Karyotype. Neuro Oncol. 2014, 16, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Azab, M.A.; Abou-Al-Shaar, H.; Guan, J.; Eli, I.; Jensen, R.L.; Ormond, D.R. Clinical Potential of Meningioma Genomic Insights: A Practical Review for Neurosurgeons. Neurosurg. Focus 2018, 44, E10. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.S.; Merrill, P.H.; Bi, W.L.; Jones, R.T.; Listewnik, M.L.; Ramkissoon, S.H.; Thorner, A.R.; Dunn, I.F.; Beroukhim, R.; Alexander, B.M.; et al. Angiomatous Meningiomas Have a Distinct Genetic Profile with Multiple Chromosomal Polysomies Including Polysomy of Chromosome 5. Oncotarget 2014, 5, 10596–10606. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Hsu, N.; Wolf, Z.; Bryke, C.; Varma, H. Multiple Whole Chromosomal Gains Define Angiomatous Meningiomas and Are Absent From the Tumor Vasculature. J. Neuropathol. Exp. Neurol. 2020, 79, 618–625. [Google Scholar] [CrossRef]
- Lusis, E.A.; Watson, M.A.; Chicoine, M.R.; Lyman, M.; Roerig, P.; Reifenberger, G.; Gutmann, D.H.; Perry, A. Integrative Genomic Analysis Identifies NDRG2 as a Candidate Tumor Suppressor Gene Frequently Inactivated in Clinically Aggressive Meningioma. Cancer Res. 2005, 65, 7121–7126. [Google Scholar] [CrossRef]
- Zhang, X.; Gejman, R.; Mahta, A.; Zhong, Y.; Rice, K.A.; Zhou, Y.; Cheunsuchon, P.; Louis, D.N.; Klibanski, A. Maternally Expressed Gene 3, an Imprinted Noncoding RNA Gene, Is Associated with Meningioma Pathogenesis and Progression. Cancer Res. 2010, 70, 2350–2358. [Google Scholar] [CrossRef]
- Och, W.; Szmuda, T.; Sikorska, B.; Springer, J.; Jaskólski, D.; Zakrzewska, M.; Liberski, P.P. Recurrence-Associated Chromosomal Anomalies in Meningiomas: Single-Institution Study and a Systematic Review with Meta-Analysis. Neurol. Neurochir. Pol. 2016, 50, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Wani, K.M.; Wilson, C.D.; Zadeh, G.; DeMonte, F.; Jones, D.T.W.; Pfister, S.M.; Sulman, E.P.; Aldape, K.D. Global Epigenetic Profiling Identifies Methylation Subgroups Associated with Recurrence-Free Survival in Meningioma. Acta Neuropathol. 2017, 133, 431–444. [Google Scholar] [CrossRef]
- Kerr, K.; Qualmann, K.; Esquenazi, Y.; Hagan, J.; Kim, D.H. Familial Syndromes Involving Meningiomas Provide Mechanistic Insight Into Sporadic Disease. Neurosurgery 2018, 83, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Osswald, M. Meningiomas: Overview and New Directions in Therapy. Semin. Neurol. 2018, 38, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J. Germline and Somatic Mutations in Meningiomas. Cancer Genet. 2015, 208, 107–114. [Google Scholar] [CrossRef]
- Smith, M.J.; Higgs, J.E.; Bowers, N.L.; Halliday, D.; Paterson, J.; Gillespie, J.; Huson, S.M.; Freeman, S.R.; Lloyd, S.; Rutherford, S.A.; et al. Cranial Meningiomas in 411 Neurofibromatosis Type 2 (NF2) Patients with Proven Gene Mutations: Clear Positional Effect of Mutations, but Absence of Female Severity Effect on Age at Onset. J. Med. Genet. 2011, 48, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo Model: Hedgehog-Induced Medulloblastoma with 90% Incidence and Leptomeningeal Spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef]
- Aavikko, M.; Li, S.-P.; Saarinen, S.; Alhopuro, P.; Kaasinen, E.; Morgunova, E.; Li, Y.; Vesanen, K.; Smith, M.J.; Evans, D.G.R.; et al. Loss of SUFU Function in Familial Multiple Meningioma. Am. J. Hum. Genet. 2012, 91, 520–526. [Google Scholar] [CrossRef]
- Evans, D.G.; Oudit, D.; Smith, M.J.; Rutkowski, D.; Allan, E.; Newman, W.G.; Lear, J.T. First Evidence of Genotype-Phenotype Correlations in Gorlin Syndrome. J. Med. Genet. 2017, 54, 530–536. [Google Scholar] [CrossRef]
- Yakubov, E.; Ghoochani, A.; Buslei, R.; Buchfelder, M.; Eyüpoglu, I.Y.; Savaskan, N. Hidden Association of Cowden Syndrome, PTEN Mutation and Meningioma Frequency. Oncoscience 2016, 3, 149–155. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The Functions and Regulation of the PTEN Tumour Suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J. Spectrum and Risk of Neoplasia in Werner Syndrome: A Systematic Review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef]
- Shankar, G.M.; Santagata, S. BAP1 Mutations in High-Grade Meningioma: Implications for Patient Care. Neuro Oncol. 2017, 19, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and Somatic BAP1 Mutations in High-Grade Rhabdoid Meningiomas. Neuro Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.J.A.; Oba-Shinjo, S.M.; de Almeida, A.N.; Marie, S.K.N. Molecular Alterations in Meningiomas: Literature Review. Clin. Neurol. Neurosurg. 2019, 176, 89–96. [Google Scholar] [CrossRef]
- van den Munckhof, P.; Christiaans, I.; Kenter, S.B.; Baas, F.; Hulsebos, T.J.M. Germline SMARCB1 Mutation Predisposes to Multiple Meningiomas and Schwannomas with Preferential Location of Cranial Meningiomas at the Falx Cerebri. Neurogenetics 2012, 13, 1–7. [Google Scholar] [CrossRef]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An Overview of Meningiomas. Future Oncol. 2018, 14, 2161–2177. [Google Scholar] [CrossRef]
- Lu, J.-Q.; Reddy, K. Letter: Familial Syndromes Involving Meningiomas Provide Mechanistic Insight Into Sporadic Disease. Neurosurgery 2019, 85, E396. [Google Scholar] [CrossRef]
- Ruttledge, M.H.; Sarrazin, J.; Rangaratnam, S.; Phelan, C.M.; Twist, E.; Merel, P.; Delattre, O.; Thomas, G.; Nordenskjöld, M.; Collins, V.P. Evidence for the Complete Inactivation of the NF2 Gene in the Majority of Sporadic Meningiomas. Nat. Genet. 1994, 6, 180–184. [Google Scholar] [CrossRef]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 Inactivation in Tumor Biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Perry, A.; Reifenberger, G. Histological Classification and Molecular Genetics of Meningiomas. Lancet Neurol. 2006, 5, 1045–1054. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmancı, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent Somatic Mutations in POLR2A Define a Distinct Subset of Meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K Mutations Are as Common as AKT1 and SMO Mutations in Meningioma. Neuro Oncol. 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zotti, T.; Scudiero, I.; Vito, P.; Stilo, R. The Emerging Role of TRAF7 in Tumor Development. J. Cell Physiol. 2017, 232, 1233–1238. [Google Scholar] [CrossRef]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.W.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory Meningiomas Are Defined by Combined KLF4 K409Q and TRAF7 Mutations. Acta Neuropathol. 2013, 125, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Olar, A.; Koelsche, C.; Reuss, D.; Bissel, J.; Kratz, A.; Capper, D.; Schefzyk, S.; Hielscher, T.; et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Juratli, T.A.; Castro, B.A.; Lazaro, T.T.; Gill, C.M.; Nayyar, N.; Strickland, M.R.; Babinski, M.; Johnstone, S.E.; Frosch, M.P.; et al. Genomic Analysis of Posterior Fossa Meningioma Demonstrates Frequent AKT1 E17K Mutations in Foramen Magnum Meningiomas. J. Neurol. Surg. B Skull Base 2019, 80, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA Methylation-Based Classification and Grading System for Meningioma: A Multicentre, Retrospective Analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Castro, M.R.H.; Lee, J.C.; Villanueva-Meyer, J.E.; Bush, N.A.O.; McDermott, M.W.; Solomon, D.A.; Perry, A.; Magill, S.T.; Raleigh, D.R. DNA Methylation Profiling Demonstrates Superior Diagnostic Classification to RNA-Sequencing in a Case of Metastatic Meningioma. Acta Neuropathol. Commun. 2020, 8, 82. [Google Scholar] [CrossRef]
- He, S.; Pham, M.H.; Pease, M.; Zada, G.; Giannotta, S.L.; Wang, K.; Mack, W.J. A Review of Epigenetic and Gene Expression Alterations Associated with Intracranial Meningiomas. Neurosurg. Focus 2013, 35, E5. [Google Scholar] [CrossRef] [PubMed]
- Galani, V.; Lampri, E.; Varouktsi, A.; Alexiou, G.; Mitselou, A.; Kyritsis, A.P. Genetic and Epigenetic Alterations in Meningiomas. Clin. Neurol. Neurosurg. 2017, 158, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Kishida, Y.; Natsume, A.; Kondo, Y.; Takeuchi, I.; An, B.; Okamoto, Y.; Shinjo, K.; Saito, K.; Ando, H.; Ohka, F.; et al. Epigenetic Subclassification of Meningiomas Based on Genome-Wide DNA Methylation Analyses. Carcinogenesis 2012, 33, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Magill, S.T.; Young, J.S.; Chae, R.; Aghi, M.K.; Theodosopoulos, P.V.; McDermott, M.W. Relationship between Tumor Location, Size, and WHO Grade in Meningioma. Neurosurg. Focus 2018, 44, E4. [Google Scholar] [CrossRef] [PubMed]
- Voß, K.M.; Spille, D.C.; Sauerland, C.; Suero Molina, E.; Brokinkel, C.; Paulus, W.; Stummer, W.; Holling, M.; Jeibmann, A.; Brokinkel, B. The Simpson Grading in Meningioma Surgery: Does the Tumor Location Influence the Prognostic Value? J. Neurooncol. 2017, 133, 641–651. [Google Scholar] [CrossRef]
- Zouaoui, S.; Darlix, A.; Rigau, V.; Mathieu-Daudé, H.; Bauchet, F.; Bessaoud, F.; Fabbro-Peray, P.; Trétarre, B.; Figarella-Branger, D.; Taillandier, L.; et al. Descriptive Epidemiology of 13,038 Newly Diagnosed and Histologically Confirmed Meningiomas in France: 2006–2010. Neurochirurgie 2018, 64, 15–21. [Google Scholar] [CrossRef]
- Meling, T.R.; Da Broi, M.; Scheie, D.; Helseth, E. Meningiomas: Skull Base versus Non-Skull Base. Neurosurg. Rev. 2019, 42, 163–173. [Google Scholar] [CrossRef]
- Englot, D.J.; Magill, S.T.; Han, S.J.; Chang, E.F.; Berger, M.S.; McDermott, M.W. Seizures in Supratentorial Meningioma: A Systematic Review and Meta-Analysis. J. Neurosurg. 2016, 124, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Whittle, I.R.; Smith, C.; Navoo, P.; Collie, D. Meningiomas. Lancet 2004, 363, 1535–1543. [Google Scholar] [CrossRef]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef]
- Pintea, B.; Kandenwein, J.A.; Lorenzen, H.; Blume, C.; Daher, F.; Kristof, R.A. Differences in Clinical Presentation, Intraoperative Findings and Outcome between Petroclival and Lateral Posterior Pyramid Meningioma. Clin. Neurol. Neurosurg. 2016, 141, 122–128. [Google Scholar] [CrossRef]
- Bosnjak, R.; Derham, C.; Popović, M.; Ravnik, J. Spontaneous Intracranial Meningioma Bleeding: Clinicopathological Features and Outcome. J. Neurosurg. 2005, 103, 473–484. [Google Scholar] [CrossRef]
- Nowosielski, M.; Galldiks, N.; Iglseder, S.; Kickingereder, P.; von Deimling, A.; Bendszus, M.; Wick, W.; Sahm, F. Diagnostic Challenges in Meningioma. Neuro Oncol. 2017, 19, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Goldbrunner, R.; Minniti, G.; Preusser, M.; Jenkinson, M.D.; Sallabanda, K.; Houdart, E.; von Deimling, A.; Stavrinou, P.; Lefranc, F.; Lund-Johansen, M.; et al. EANO Guidelines for the Diagnosis and Treatment of Meningiomas. Lancet Oncol. 2016, 17, e383–e391. [Google Scholar] [CrossRef]
- Huang, R.Y.; Bi, W.L.; Griffith, B.; Kaufmann, T.J.; la Fougère, C.; Schmidt, N.O.; Tonn, J.C.; Vogelbaum, M.A.; Wen, P.Y.; Aldape, K.; et al. Imaging and Diagnostic Advances for Intracranial Meningiomas. Neuro Oncol. 2019, 21, i44–i61. [Google Scholar] [CrossRef]
- Lyndon, D.; Lansley, J.A.; Evanson, J.; Krishnan, A.S. Dural Masses: Meningiomas and Their Mimics. Insights Imaging 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Nagai Yamaki, V.; de Souza Godoy, L.F.; Alencar Bandeira, G.; Tavares Lucato, L.; Correa Lordelo, G.; Fontoura Solla, D.J.; Santana Neville, I.; Jacobsen Teixeira, M.; Silva Paiva, W. Dural-Based Lesions: Is It a Meningioma? Neuroradiology 2021. [Google Scholar] [CrossRef] [PubMed]
- Meling, T.R.; Da Broi, M.; Scheie, D.; Helseth, E.; Smoll, N.R. Meningioma Surgery–Are We Making Progress? World Neurosurg. 2019, 125, e205–e213. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Park, J.H.; Park, E.S.; Kim, J.H. “Wait-and-See” Strategies for Newly Diagnosed Intracranial Meningiomas Based on the Risk of Future Observation Failure. World Neurosurg. 2017, 107, 604–611. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, W.; Hou, Y.; Wen, C.; Wang, J.; Wu, P.; Guo, Z. An Overview of Managements in Meningiomas. Front. Oncol. 2020, 10, 1523. [Google Scholar] [CrossRef]
- Karsy, M.; Guan, J.; Cohen, A.; Colman, H.; Jensen, R.L. Medical Management of Meningiomas: Current Status, Failed Treatments, and Promising Horizons. Neurosurg. Clin. N. Am. 2016, 27, 249–260. [Google Scholar] [CrossRef]
- Kessler, R.A.; Garzon-Muvdi, T.; Yang, W.; Weingart, J.; Olivi, A.; Huang, J.; Brem, H.; Lim, M. Metastatic Atypical and Anaplastic Meningioma: A Case Series and Review of the Literature. World Neurosurg. 2017, 101, 47–56. [Google Scholar] [CrossRef] [PubMed]
- van der Vossen, S.; Schepers, V.P.M.; Berkelbach van der Sprenkel, J.W.; Visser-Meily, J.M.A.; Post, M.W.M. Cognitive and Emotional Problems in Patients after Cerebral Meningioma Surgery. J. Rehabil. Med. 2014, 46, 430–437. [Google Scholar] [CrossRef]
- Wilson, T.A.; Huang, L.; Ramanathan, D.; Lopez-Gonzalez, M.; Pillai, P.; De Los Reyes, K.; Kumal, M.; Boling, W. Review of Atypical and Anaplastic Meningiomas: Classification, Molecular Biology, and Management. Front. Oncol. 2020, 10, 565582. [Google Scholar] [CrossRef]
- Rogers, L.; Zhang, P.; Vogelbaum, M.A.; Perry, A.; Ashby, L.S.; Modi, J.M.; Alleman, A.M.; Galvin, J.; Brachman, D.; Jenrette, J.M.; et al. Intermediate-Risk Meningioma: Initial Outcomes from NRG Oncology RTOG 0539. J. Neurosurg. 2018, 129, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.C.; Ares, C.; Villa, S.; Peerdeman, S.M.; Renard, L.; Baumert, B.G.; Lucas, A.; Veninga, T.; Pica, A.; Jefferies, S.; et al. Adjuvant Postoperative High-Dose Radiotherapy for Atypical and Malignant Meningioma: A Phase-II Parallel Non-Randomized and Observation Study (EORTC 22042-26042). Radiother. Oncol. 2018, 128, 260–265. [Google Scholar] [CrossRef]
- Jenkinson, M.D.; Javadpour, M.; Haylock, B.J.; Young, B.; Gillard, H.; Vinten, J.; Bulbeck, H.; Das, K.; Farrell, M.; Looby, S.; et al. The ROAM/EORTC-1308 Trial: Radiation versus Observation Following Surgical Resection of Atypical Meningioma: Study Protocol for a Randomised Controlled Trial. Trials 2015, 16, 519. [Google Scholar] [CrossRef] [PubMed]
- Graillon, T.; Sanson, M.; Campello, C.; Idbaih, A.; Peyre, M.; Peyrière, H.; Basset, N.; Autran, D.; Roche, C.; Kalamarides, M.; et al. Everolimus and Octreotide for Patients with Recurrent Meningioma: Results from the Phase II CEVOREM Trial. Clin. Cancer Res. 2020, 26, 552–557. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grade I | Grade II | Grade III | |
|---|---|---|---|
| Histologic Subtypes | Meningothelial Fibrous Transitional Psammomatous Angiomatous Microcystic Secretory Lymphoplasmacyte-rich Metaplastic | Atypical Clear cell Choroid | Anaplastic Rhabdoid Papillary |
| Diagnostic Criteria | -Presence of <4 mitoses per 10 HPF | -Presence of 4–19 mitoses per 10 HPF or -Brain invasion or -At least 3/5 of the following: -Patternless sheeting architecture -Small cell formation with high N/C ratio -Prominent nucleoli -Hypercellularity -Spontaneous intratumoral micronecrosis | -Presence of ≥20 mitoses per 10 HPF or -Overtly malignant morphology (carcinomatous, sarcomatous, and melanomatous cytology) |
| Grade I | Grade II | Grade III | |
|---|---|---|---|
| Subtype (WHO grade) | Meningothelial (TRAF7, AKT1, SMO, PIK3CA, POLR2A) Fibrous (NF2) Transitional (NF2, PIK3CA, AKT1) Psammomatous (NF2) Angiomatous Microcystic Secretory (KLF4, TRAF7) Lymphoplasmacyte-rich Metaplastic | Atypical (NF2, TRAF7, AKT1, TERT) Clear cell (SMARCE1) Choroid | Anaplastic (NF2, TERT) Rhadoid (BAP1) Papillary |
| Gene Mutation |  | ||
Chromosomal Alterations | Loss: 22q  | Loss: 1p, 6q, 10, 14q, 18q Gain: 1q, 9q, 12q, 15q, 17q, 20q | Loss: 9p Amplification: 17q |
| Affected Genes | Possible Role in Meningioma |
|---|---|
| Methylation/up/down regulation | |
| ALPL | Progression |
| CCND1 | Growth and aggressiveness |
| CDK5R1 | Growth and aggressiveness |
| CTGF | Recurrence |
| CTNNB1 | Growth and aggressiveness |
| ENC1 | Growth and aggressiveness |
| GSTP1 | Higher grade |
| HIF-3α4 | Progression |
| HOXA5 | Tumorigenesis, higher grade |
| HOXA6 | Tumorigenesis, progression, higher grade |
| HOXA9 | Tumorigenesis, progression, higher grade |
| HOXA11 | Tumorigenesis, higher grade |
| IGFBP2 | Growth and aggressiveness, progression |
| IGFBP3 | Growth and aggressiveness |
| IGF2 | Growth and aggressiveness |
| IGF2BP1 | Tumorigenesis |
| LMO4 | Tumorigenesis |
| MAL2 | Higher grade, malignant transformation |
| MEG3 | Tumorigenesis, higher grade |
| NDRG2 | Growth and aggressiveness |
| PENK | Tumorigenesis, progression |
| RASSF1A | Malignant transformation |
| THBS1 | Angiogenesis |
| TIMP3 | Progression, higher grade, recurrence |
| TP53 | Progression |
| TP73 | Tumorigenesis, malignant transformation |
| UPK3A | Tumorigenesis, progression |
| WNK2 | Tumorigenesis, progression, higher grade |
| Histone Modifications | |
| HIST1HIc | Tumorigenesis, progression |
| H3K27me3 | Recurrence |
| KDM5C | Grade 1, III |
| KDM6A | Grade II |
| Chromatin Remodeler | |
| SMARCB1 | Higher grade |
| SMARCE1 | Higher grade |
| microRNA | |
| miR-21 | Tumorigenesis, progression |
| miR-29c-3p | Recurrence |
| miR-145 | Higher grade |
| miR-190a | Progression, recurrence |
| miR-200a | Tumorigenesis |
| miR-219-5p | Recurrence |
| miR-224 | Progression |
| miR-335 | Progression |
| Location | Frequency (%) |
|---|---|
| Convexity | 20–37% |
| Parasagittal (NF2) | 13–22% |
| 5% |
| Spine (AKT1) | 7–12% |
| Skull Base | 43–51% |
| 10–20% |
| 9–36% |
| 6–15% |
| 2–4% |
| 5% |
| 2–11% |
| 3% |
| <1–9% |
| Intraventricular (NF2) | 1–5% |
| Orbital | <1–2% |
| Ectopic locations | <1% |
| Extent of Resection | Simpson Grade | Description |
|---|---|---|
| Gross Total Resection (GTR) | Grade 1 | Gross total resection of tumor, dural attachment, and involved bone (extradural extension) |
| Grade 2 | Gross total resection of tumor, coagulation of dural attachment | |
| Grade 3 | Gross total resection of tumor without resection of coagulation of dural and extradural components | |
| Subtotal Resection (STR) | Grade 4 | Partial (subtotal) resection of tumor |
| -- | Grade 5 | Biopsy only |
| Type of Therapy | Study/Agent | Dose/Target | Results | Trial ID |
|---|---|---|---|---|
| Radiotherapy | WHO grade II GTR meningiomas w/adjuvant RT | 54 Gy in 1.8 Gy per fraction | -- | NCT04127760 |
| 59.4 Gy in 33 fractions of 1.8 Gy each | -- | NCT03180268 | ||
| 60 Gy in 30 fractions | -- | ISRCTN71502099 | ||
| 54 Gy in 30 fractions of 1.8 Gy each | 3-year PFS = 98.3% 3-year OS = 96% | NCT00895622 (RTOG 0539) | ||
| 60 Gy in 30 fractions | 3-year PFS = 88.7% 3-year OS = 98.2% | NCT00626730 (EORTC 22042-26042) | ||
| Chemotherapy | Vismodegib GSK2256098 | SMO FAK | -- | NCT02523014 |
| Selumetinib | MEK pathway | -- | NCT03095248 | |
| Ribociclib | CDK-p16-Rb pathway | -- | NCT02933736 | |
| Everolimus | mTOR-pathway | -- | NCT01880749 NCT01419639 | |
| Everolimus + Octreotide | mTOR + SSTR2A | 6-month PFS = 55% 6-month OS = 90% 12-month OS = 75% | NCT02333565 (CEVOREM) | |
| Vistusertib (AZD2014) | mTOR-pathway | -- | NCT03071874 | |
| 6-month PFS = 88.9% Decrease in tumor volume of at least 20% = 5.6% | NCT02831257 | |||
| Alpelisib Trametinib | Pi3Kα inhibitor MEK inhibitor | -- | NCT03631953 | |
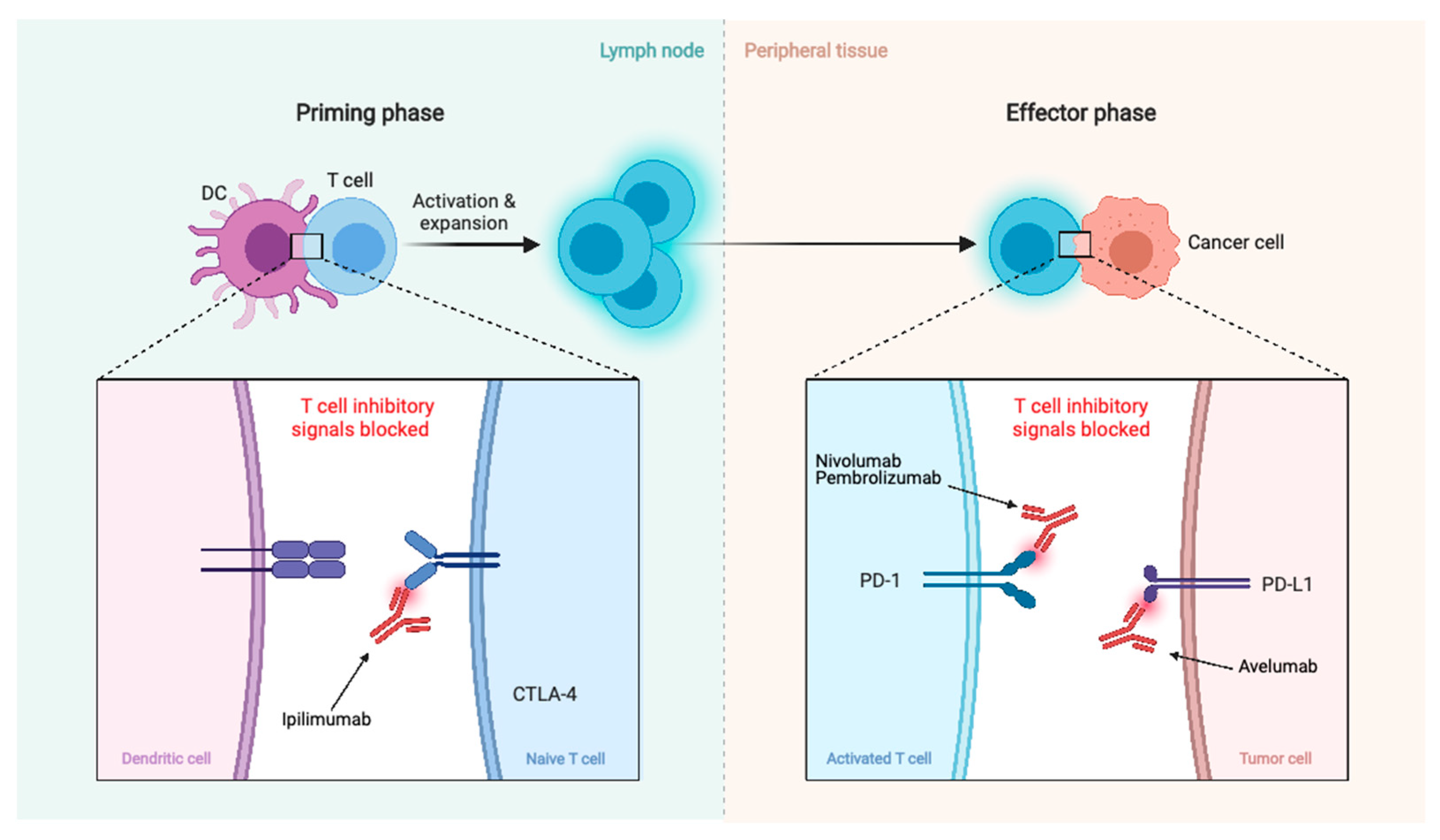
| Immunotherapy | Nivolumab | PD-1 | -- | NCT02648997 NCT03173950 |
| Nivolumab w/Multi-Fraction SRS ± Ipilimumab | PD-1 ± CTLA-4 | -- | NCT03604978 | |
| Pembrolizumab | PD-1 | -- | NCT03279692 NCT03016091 | |
| Pembrolizumab w/SRS | PD-1 | -- | NCT04659811 | |
| Avelumab w/Proton radiotherapy | PD-L1 | -- | NCT03267836 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogasawara, C.; Philbrick, B.D.; Adamson, D.C. Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions. Biomedicines 2021, 9, 319. https://doi.org/10.3390/biomedicines9030319
Ogasawara C, Philbrick BD, Adamson DC. Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions. Biomedicines. 2021; 9(3):319. https://doi.org/10.3390/biomedicines9030319
Chicago/Turabian StyleOgasawara, Christian, Brandon D. Philbrick, and D. Cory Adamson. 2021. "Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions" Biomedicines 9, no. 3: 319. https://doi.org/10.3390/biomedicines9030319
APA StyleOgasawara, C., Philbrick, B. D., & Adamson, D. C. (2021). Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions. Biomedicines, 9(3), 319. https://doi.org/10.3390/biomedicines9030319