
Anti-NLRP3 Inflammasome Natural Compounds: An Update

Abstract
1. Introduction
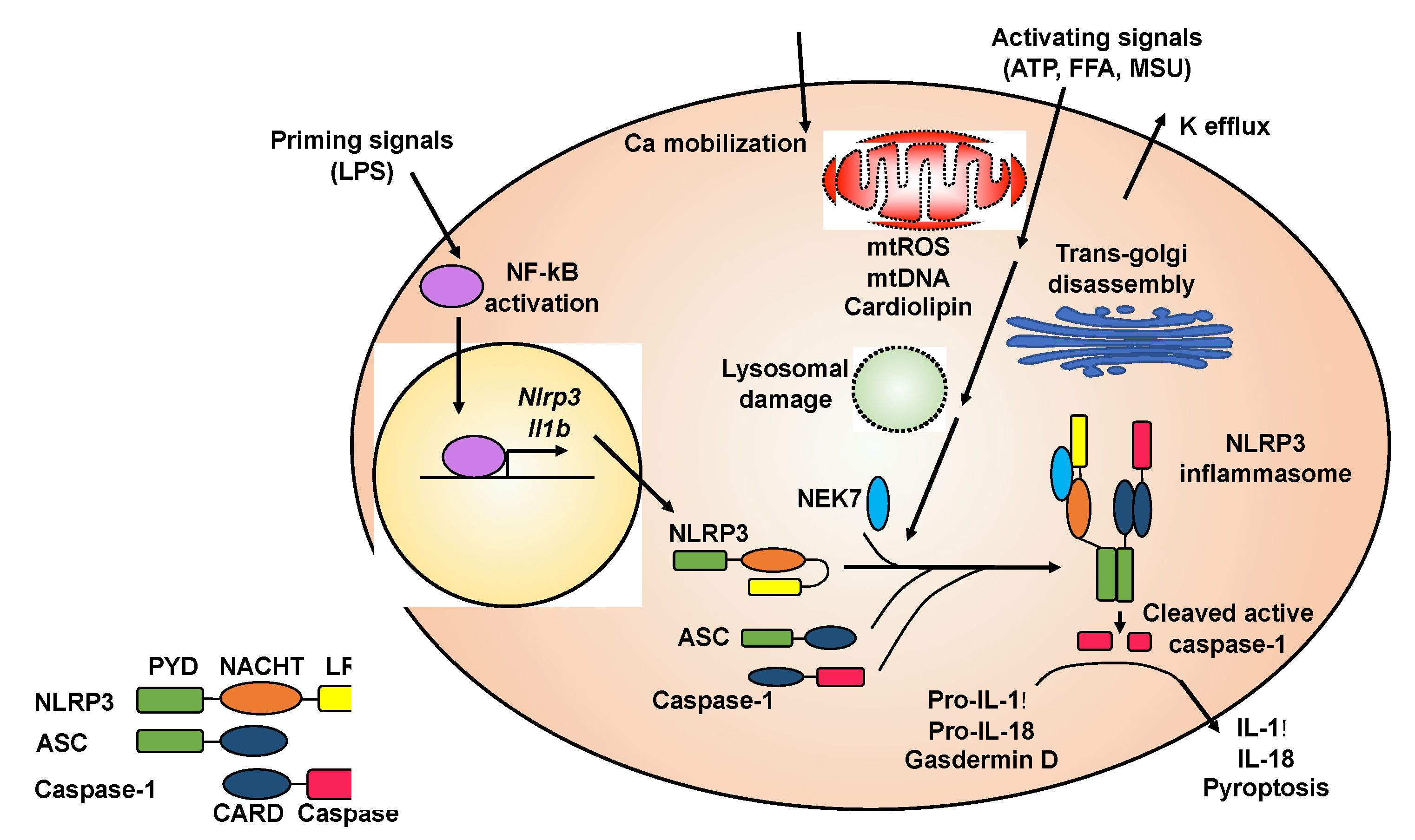
2. NLRP3 Inflammasome and Its Involvement in Diseases
2.1. Activation of the NLRP3 Inflammasome
2.2. Involvement of the NLRP3 Inflammasome in Diseases
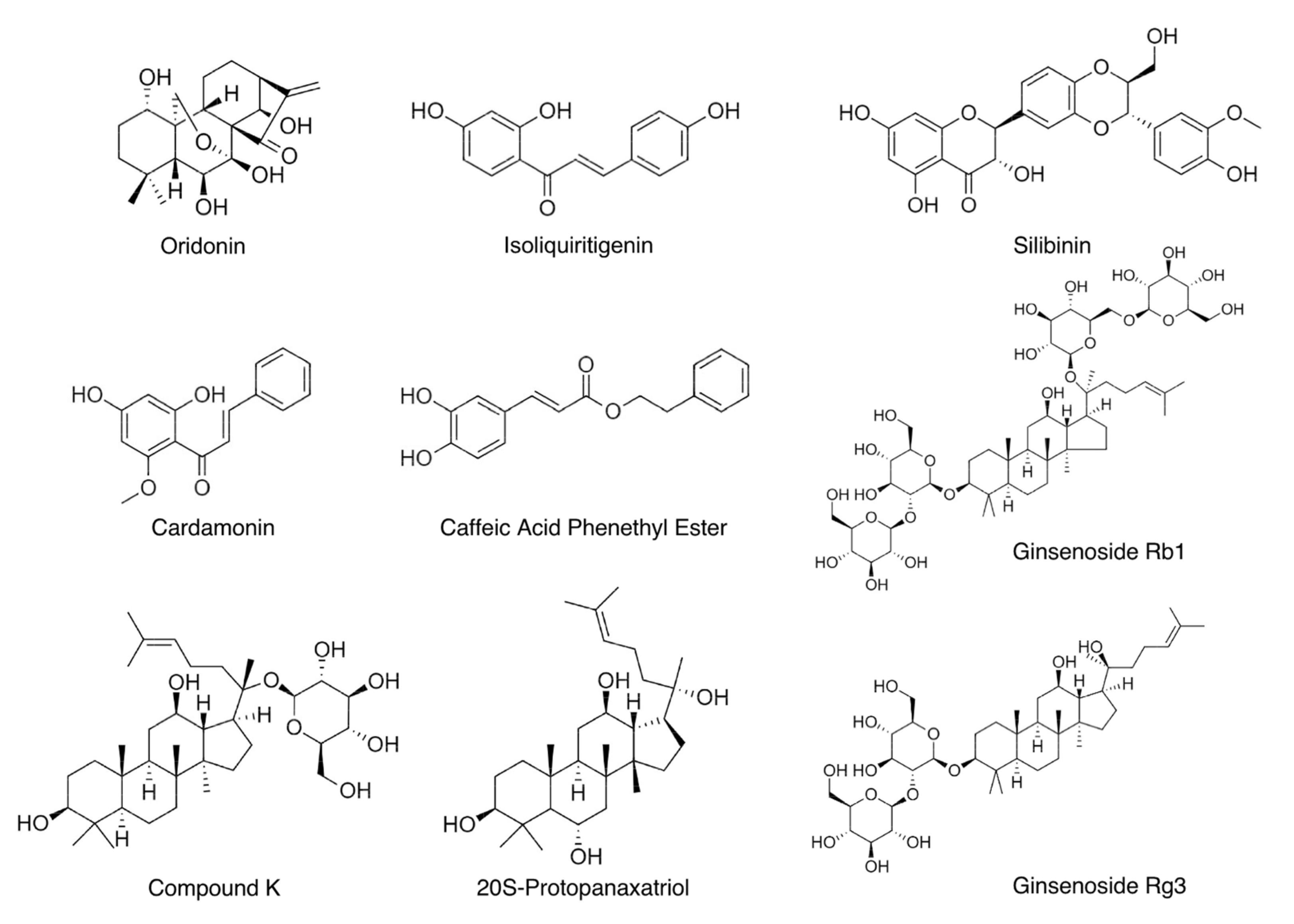
3. Recently Identified Natural Compounds with Anti-NLRP3 Inflammasome Functions
3.1. Oridonin
3.2. Isoliquiritigenin
3.3. Silibinin
3.4. Cardamonin
3.5. Caffeic Acid Phenethyl Ester
3.6. Ginsenosides
4. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Ehses, J.A.; Donath, M.Y.; Mandrup-Poulsen, T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 2009, 32, 1663–1668. [Google Scholar] [CrossRef]
- So, A.; Dumusc, A.; Nasi, S. The role of IL-1 in gout: From bench to bedside. Rheumatology 2018, 57, i12–i19. [Google Scholar] [CrossRef]
- Jahan, S.; Kumar, D.; Chaturvedi, S.; Rashid, M.; Wahajuddin, M.; Khan, Y.A.; Goyal, S.N.; Patil, C.R.; Mohanraj, R.; Subramanya, S.; et al. Therapeutic targeting of NLRP3 inflammasomes by natural products and pharmaceuticals: A novel mechanistic approach for inflammatory diseases. Curr. Med. Chem. 2017, 24, 1645–1670. [Google Scholar] [CrossRef]
- Tozser, J.; Benko, S. Natural compounds as regulators of NLRP3 inflammasome-mediated IL-1beta production. Mediat. Inflamm. 2016, 2016, 5460302. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Yu, J.R.; Leslie, K.S. Cryopyrin-associated periodic syndrome: An update on diagnosis and treatment response. Curr. Allergy Asthma Rep. 2011, 11, 12–20. [Google Scholar] [CrossRef]
- Tartey, S.; Kanneganti, T.D. Inflammasomes in the pathophysiology of autoinflammatory syndromes. J. Leukoc. Biol. 2020, 107, 379–391. [Google Scholar] [CrossRef]
- Brydges, S.D.; Broderick, L.; McGeough, M.D.; Pena, C.A.; Mueller, J.L.; Hoffman, H.M. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J. Clin. Invest. 2013, 123, 4695–4705. [Google Scholar] [CrossRef]
- Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.A.; Misaghi, A.; Gandhi, C.; Putnam, C.D.; Boyle, D.L.; Firestein, G.S.; Horner, A.A.; et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 2009, 30, 875–887. [Google Scholar] [CrossRef]
- Busso, N.; So, A. Mechanisms of inflammation in gout. Arthritis Res. Ther. 2010, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.X.; Huang, P.; Hillert, J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult. Scler. 2004, 10, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 10480–10485. [Google Scholar] [CrossRef]
- Matsuki, T.; Nakae, S.; Sudo, K.; Horai, R.; Iwakura, Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int. Immunol. 2006, 18, 399–407. [Google Scholar] [CrossRef]
- Furlan, R.; Martino, G.; Galbiati, F.; Poliani, P.L.; Smiroldo, S.; Bergami, A.; Desina, G.; Comi, G.; Flavell, R.; Su, M.S.; et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J. Immunol. 1999, 163, 2403–2409. [Google Scholar]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef]
- Weiner, H.L.; Frenkel, D. Immunology and immunotherapy of Alzheimer’s disease. Nat. Rev. Immunol. 2006, 6, 404–416. [Google Scholar] [CrossRef]
- Del Rey, N.L.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernandez-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s disease: 200 years later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Haslam, D.W.; James, W.P. Obesity. Lancet 2005, 366, 1197–1209. [Google Scholar] [CrossRef]
- Saliba, L.J.; Maffett, S. Hypertensive heart disease and obesity: A review. Heart Fail. Clin. 2019, 15, 509–517. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Boulange, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Goossens, G.H.; Blaak, E.E.; Theunissen, R.; Duijvestijn, A.M.; Clement, K.; Tervaert, J.W.; Thewissen, M.M. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol. Immunol. 2012, 50, 142–149. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; Joosten, L.A.; van Velzen, J.F.; Hijmans, A.; Pol, J.A.; van der Vliet, J.A.; Netea, M.G.; Tack, C.J.; et al. The inflammasome and caspase-1 activation: A new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 2011, 152, 3769–3778. [Google Scholar] [CrossRef]
- Esser, N.; L’Homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Xing, Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013, 22, 746–750. [Google Scholar] [CrossRef]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse models for atherosclerosis research—Which is my line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Menu, P.; Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef]
- Olokoba, A.B.; Obateru, O.A.; Olokoba, L.B. Type 2 diabetes mellitus: A review of current trends. Oman Med. J. 2012, 27, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Commane, M.; Jiang, Z.; Stark, G.R. IL-1-induced NFkappa B and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK). Proc. Natl. Acad. Sci. USA 2001, 98, 4461–4465. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Verma, G.; Datta, M. IL-1beta induces ER stress in a JNK dependent manner that determines cell death in human pancreatic epithelial MIA PaCa-2 cells. Apoptosis 2010, 15, 864–876. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Donath, M.Y.; Mandrup-Poulsen, T. Role of IL-1beta in type 2 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 314–321. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug. Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Raskin, I.; Ribnicky, D.M.; Komarnytsky, S.; Ilic, N.; Poulev, A.; Borisjuk, N.; Brinker, A.; Moreno, D.A.; Ripoll, C.; Yakoby, N.; et al. Plants and human health in the twenty-first century. Trends Biotechnol. 2002, 20, 522–531. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, C.; Ye, N.; Liu, Z.; Wold, E.A.; Chen, H.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, H.; Li, H.; Ci, X.; Peng, L. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-kappaB pathways. Cell Commun. Signal. 2019, 17, 62. [Google Scholar] [CrossRef]
- Yan, C.; Yan, H.; Mao, J.; Liu, Y.; Xu, L.; Zhao, H.; Shen, J.; Cao, Y.; Gao, Y.; Li, K.; et al. Neuroprotective effect of oridonin on traumatic brain injury via inhibiting NLRP3 inflammasome in experimental mice. Front. Neurosci. 2020, 14, 557170. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Zhou, J.; Shen, Q. Oridonin and its derivatives for cancer treatment and overcoming therapeutic resistance. Genes Dis. 2020. [Google Scholar] [CrossRef]
- Tian, T.; Jin, Y.; Ma, Y.; Xie, W.; Xu, H.; Zhang, K.; Zhang, L.; Du, Y. Identification of metabolites of oridonin in rats with a single run on UPLC-Triple-TOF-MS/MS system based on multiple mass defect filter data acquisition and multiple data processing techniques. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 1006, 80–92. [Google Scholar] [CrossRef]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef]
- Lee, Y.; Kwon, E.Y.; Choi, M.S. Dietary isoliquiritigenin at a low dose ameliorates insulin resistance and NAFLD in diet-induced obesity in C57BL/6J mice. Int. J. Mol. Sci. 2018, 19, 3281. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-kappaB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lv, H.; Wen, Z.; Ci, X.; Peng, L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-kappaB pathway in macrophages and in acute lung injury. Front. Immunol. 2017, 8, 1518. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lv, X.; Yang, H.; Peng, L.; Ci, X. Isoliquiritigenin exerts antioxidative and anti-inflammatory effects via activating the KEAP-1/Nrf2 pathway and inhibiting the NF-kappaB and NLRP3 pathways in carrageenan-induced pleurisy. Food Funct. 2020, 11, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, B.; Cao, S.; Wang, Y.; Wu, D. Silybin attenuates LPS-induced lung injury in mice by inhibiting NF-kappaB signaling and NLRP3 activation. Int. J. Mol. Med. 2017, 39, 1111–1118. [Google Scholar] [CrossRef]
- Matias, M.L.; Gomes, V.J.; Romao-Veiga, M.; Ribeiro, V.R.; Nunes, P.R.; Romagnoli, G.G.; Peracoli, J.C.; Peracoli, M.T.S. Silibinin downregulates the NF-kappaB pathway and NLRP1/NLRP3 inflammasomes in monocytes from pregnant women with preeclampsia. Molecules 2019, 24, 1548. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, D.; She, L.; Wang, Z.; Yang, N.; Sun, R.; Zhang, Y.; Yan, C.; Wei, Q.; Aa, J.; et al. Silybin inhibits NLRP3 inflammasome assembly through the NAD(+)/SIRT2 pathway in mice with nonalcoholic fatty liver disease. FASEB J. 2018, 32, 757–767. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, G.; Gao, Y.; Zhan, X.; Qin, N.; Fu, S.; Li, R.; Niu, M.; Wang, J.; Liu, Y.; et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm. Sin. B 2019, 9, 734–744. [Google Scholar] [CrossRef]
- Wang, K.; Lv, Q.; Miao, Y.M.; Qiao, S.M.; Dai, Y.; Wei, Z.F. Cardamonin, a natural flavone, alleviates inflammatory bowel disease by the inhibition of NLRP3 inflammasome activation via an AhR/Nrf2/NQO1 pathway. Biochem. Pharmacol. 2018, 155, 494–509. [Google Scholar] [CrossRef]
- Lee, H.E.; Yang, G.; Kim, N.D.; Jeong, S.; Jung, Y.; Choi, J.Y.; Park, H.H.; Lee, J.Y. Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: A novel strategy to treat acute gout. Sci. Rep. 2016, 6, 38622. [Google Scholar] [CrossRef]
- Dai, G.; Jiang, Z.; Sun, B.; Liu, C.; Meng, Q.; Ding, K.; Jing, W.; Ju, W. Caffeic acid phenethyl ester prevents colitis-associated cancer by inhibiting NLRP3 inflammasome. Front. Oncol. 2020, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, F.; Huang, Y.; Zhou, T.; Chen, S.; Li, G.; Shi, J.; Dong, N.; Xu, K. Caffeic acid phenethyl ester ameliorates calcification by inhibiting activation of the AKT/NF-kappaB/NLRP3 inflammasome pathway in human aortic valve interstitial cells. Front. Pharmacol. 2020, 11, 826. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S. A drug over the millennia: Pharmacognosy, chemistry, and pharmacology of licorice. Yakugaku Zasshi 2000, 120, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Du, Q.; Peng, C.; Wang, N.; Tang, H.; Xie, X.; Shen, J.; Chen, J. A review: The pharmacology of isoliquiritigenin. Phytother. Res. 2015, 29, 969–977. [Google Scholar] [CrossRef]
- Jhang, J.J.; Yen, G.C. The role of Nrf2 in NLRP3 inflammasome activation. Cell. Mol. Immunol. 2017, 14, 1011–1012. [Google Scholar] [CrossRef]
- Gaur, R.; Yadav, K.S.; Verma, R.K.; Yadav, N.P.; Bhakuni, R.S. In vivo anti-diabetic activity of derivatives of isoliquiritigenin and liquiritigenin. Phytomedicine 2014, 21, 415–422. [Google Scholar] [CrossRef]
- Peng, F.; Xiong, L.; Xie, X.; Tang, H.; Huang, R.; Peng, C. Isoliquiritigenin derivative regulates miR-374a/BAX axis to suppress triple-negative breast cancer tumorigenesis and development. Front. Pharmacol. 2020, 11, 378. [Google Scholar] [CrossRef]
- Selvaraj, B.; Kim, D.W.; Huh, G.; Lee, H.; Kang, K.; Lee, J.W. Synthesis and biological evaluation of isoliquiritigenin derivatives as a neuroprotective agent against glutamate mediated neurotoxicity in HT22 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127058. [Google Scholar] [CrossRef]
- Guo, J.; Liu, D.; Nikolic, D.; Zhu, D.; Pezzuto, J.M.; van Breemen, R.B. In vitro metabolism of isoliquiritigenin by human liver microsomes. Drug Metab. Dispos. 2008, 36, 461–468. [Google Scholar] [CrossRef]
- Chen, C.; Shenoy, A.K.; Padia, R.; Fang, D.; Jing, Q.; Yang, P.; Su, S.B.; Huang, S. Suppression of lung cancer progression by isoliquiritigenin through its metabolite 2, 4, 2’, 4’-Tetrahydroxychalcone. J. Exp. Clin. Cancer Res. 2018, 37, 243. [Google Scholar] [CrossRef]
- Abenavoli, L.; Izzo, A.A.; Milic, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Vue, B.; Zhang, S.; Zhang, X.; Parisis, K.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.H. Silibinin derivatives as anti-prostate cancer agents: Synthesis and cell-based evaluations. Eur. J. Med. Chem. 2016, 109, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Romanucci, V.; Agarwal, C.; Agarwal, R.; Pannecouque, C.; Iuliano, M.; De Tommaso, G.; Caruso, T.; Di Fabio, G.; Zarrelli, A. Silibinin phosphodiester glyco-conjugates: Synthesis, redox behaviour and biological investigations. Bioorg. Chem. 2018, 77, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M. Silybin, a major bioactive component of milk thistle (Silybum marianum L. Gaernt.)—Chemistry, bioavailability, and metabolism. Molecules 2017, 22, 1942. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiu, R.; Yuan, L.; Meng, F.; Tang, Q. Analysis on the Alpinia katsumadai components of Zingiberaceae plants and their functions on myeloma resistance. Pak. J. Pharm. Sci. 2015, 28, 1065–1068. [Google Scholar]
- Break, M.K.B.; Hossan, M.S.; Khoo, Y.; Qazzaz, M.E.; Al-Hayali, M.Z.K.; Chow, S.C.; Wiart, C.; Bradshaw, T.D.; Collins, H.; Khoo, T.J. Discovery of a highly active anticancer analogue of cardamonin that acts as an inducer of caspase-dependent apoptosis and modulator of the mTOR pathway. Fitoterapia 2018, 125, 161–173. [Google Scholar] [CrossRef]
- Dong, F.; Wang, S.; Yang, A.; Li, Q.; Wang, Y.; Dai, L.; Tao, Y.; Wei, X.; Zhang, J. Systematic screening and characterization of cardamonin metabolites using UHPLC-Q-Exactive Orbitrap MS after oral administration to rats. Arab. J. Chem. 2020, 13, 8768–8782. [Google Scholar] [CrossRef]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asad, M.H. Caffeic acid phenethyl ester and therapeutic potentials. Biomed. Res. Int. 2014, 2014, 145342. [Google Scholar] [CrossRef]
- Zhang, P.; Tang, Y.; Li, N.G.; Zhu, Y.; Duan, J.A. Bioactivity and chemical synthesis of caffeic acid phenethyl ester and its derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef]
- Chen, L.; Jin, Y.; Chen, H.; Sun, C.; Fu, W.; Zheng, L.; Lu, M.; Chen, P.; Chen, G.; Zhang, Y.; et al. Discovery of caffeic acid phenethyl ester derivatives as novel myeloid differentiation protein 2 inhibitors for treatment of acute lung injury. Eur. J. Med. Chem. 2018, 143, 361–375. [Google Scholar] [CrossRef]
- Celli, N.; Dragani, L.K.; Murzilli, S.; Pagliani, T.; Poggi, A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J. Agric. Food. Chem. 2007, 55, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Wargovich, M.J. Inflammation, cancer, and targets of ginseng. J. Nutr. 2007, 137, 183S–185S. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Roles of ginsenosides in inflammasome activation. J. Ginseng. Res. 2019, 43, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, J.; Luo, Y.; Wang, T.; Li, X.; Li, A.; Li, J.; Liu, K.; Liu, B. Ginsenoside Rb1 and compound K improve insulin signaling and inhibit ER stress-associated NLRP3 inflammasome activation in adipose tissue. J. Ginseng Res. 2016, 40, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Deng, M.Z.; Gao, Z.J.; Dang, Y.Y.; Zheng, G.D.; Yang, X.J.; Chao, Y.X.; Cai, Y.F.; Wu, X.L. Effects of compound K, a metabolite of ginsenosides, on memory and cognitive dysfunction in db/db mice involve the inhibition of ER stress and the NLRP3 inflammasome pathway. Food Funct. 2020, 11, 4416–4427. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, H.; Zheng, M.; Xu, W.; Yang, Y.; Shi, F. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 2020, 34, 208–221. [Google Scholar] [CrossRef]
- Jiang, J.; Sun, X.; Akther, M.; Lian, M.L.; Quan, L.H.; Koppula, S.; Han, J.H.; Kopalli, S.R.; Kang, T.B.; Lee, K.H. Ginsenoside metabolite 20(S)-protopanaxatriol from Panax ginseng attenuates inflammation-mediated NLRP3 inflammasome activation. J. Ethnopharmacol. 2020, 251, 112564. [Google Scholar] [CrossRef]
- Song, J.; Cui, Z.Y.; Lian, L.H.; Han, X.; Hou, L.S.; Wang, G.; Gao, L.; Zhu, Y.; Jiang, Y.C.; Dou, J.Y.; et al. 20S-Protopanaxatriol Ameliorates Hepatic Fibrosis, Potentially Involving FXR-Mediated Inflammatory Signaling Cascades. J. Agric. Food Chem. 2020, 68, 8195–8204. [Google Scholar] [CrossRef]
- Akao, T.; Kida, H.; Kanaoka, M.; Hattori, M.; Kobashi, K. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J. Pharm. Pharmacol. 1998, 50, 1155–1160. [Google Scholar] [CrossRef]
- Qian, T.; Cai, Z.; Wong, R.N.; Mak, N.K.; Jiang, Z.H. In vivo rat metabolism and pharmacokinetic studies of ginsenoside Rg3. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 816, 223–232. [Google Scholar] [CrossRef]
- Attele, A.S.; Wu, J.A.; Yuan, C.S. Ginseng pharmacology: Multiple constituents and multiple actions. Biochem. Pharmacol. 1999, 58, 1685–1693. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, H.; Bai, L.P.; Law, B.Y.K.; Xiong, H.; Zhou, X.; Xiao, R.; Qu, Y.Q.; Mok, S.W.F.; Liu, L.; et al. Novel ginsenoside derivative 20(S)-Rh2E2 suppresses tumor growth and metastasis in vivo and in vitro via intervention of cancer cell energy metabolism. Cell Death Dis. 2020, 11, 621. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liang, D.; Li, C.; Xu, G.; Jiang, M.; Li, H.; Yin, J.; Song, Y. 2-Deoxy-Rh2: A novel ginsenoside derivative, as dual-targeting anti-cancer agent via regulating apoptosis and glycolysis. Biomed. Pharmacother. 2020, 124, 109891. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.P.; Brantner, V.V. Estimating the cost of new drug development: Is it really 802 million dollars? Health Aff. (Millwood) 2006, 25, 420–428. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Aggarwal, B.B. Serendipity in cancer drug discovery: Rational or coincidence? Trends Pharmacol. Sci. 2016, 37, 435–450. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Tartey, S.; Kanneganti, T.D. Differential role of the NLRP3 inflammasome in infection and tumorigenesis. Immunology 2019, 156, 329–338. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name (Natural Source) | In Vitro Model and Dose | Animal Model and Dose | Disease Model | Inhibitory Mechanism | References |
|---|---|---|---|---|---|
| Oridonin (Rabdosia rubescens) | BMDMs, human PBMCs: 0.5–2 μM | WT and Nlrp3−/− C57BL/6J mice: 3–20 mg/kg | Peritonitis, Gouty arthritis, HFD-induced obesity | Bound to cysteine 279 of the NLRP3 protein, impeded interaction between NEK7 and NLRP3 and thus blocked NLRP3 inflammasome assembly | He [75] |
| RAW 264.7: 2.5–10 μM | C57BL/6 mice: 20–40 mg/kg | Acute lung injury | Reduced protein levels of NLRP3, ASC, caspase-1, and IL-1β in lung | Yang [76] | |
| C57BL/6 mice: 10 mg/kg | Traumatic brain injury | Reduced both mRNA and protein levels of the Nlrp3, Pycard, Casp1 genes in cerebral cortex | Yan [77] | ||
| Isoliquiritigenin (Glycyrrhiza uralensis) | BMDMs, THP-1: 1–10 μM | C57BL/6 mice: 0.5% w/w in the diet | HFD-induced obesity | Blocked assembly of the NLRP3 inflammasome; reduced expression of the Nlrp3, Pycard, Casp1, and Il1b genes in eWAT | Honda [80] |
| C57BL/6J mice: 0.02% w/w in the diet | HFD-induced obesity | Lee [81] | |||
| Sprague-Dawley rats: 10–40 mg/kg | Intracerebral hemorrhage | Reduced both mRNA and protein levels of the Nlrp3, Pycard, Casp1 in ipsilateral hemisphere; induced transcription of the Nrf2 gene; suppressed NF-κB activation | Zeng [82] | ||
| RAW 264.7: 20 μM | C57BL/6J mice: 30 mg/kg | Acute lung injury | Reduced protein levels of NLRP3, ASC, cleaved caspase-1, and pro-IL-1β in lung | Liu [83] | |
| BALB/c mice: 30 mg/kg | Pleurisy and lung injury | Reduced protein levels of NLRP3, cleaved caspase-1, and mature IL-1β in lung | Gao [84] | ||
| Silibinin (Silybum marianum) | THP-1, RAW 264.7: 25–100 μM | C57BL/6 mice: 50–100 mg/kg | Acute lung injury | Reduced ROS production; blocked assembly of NLRP3 inflammasome; suppressed NF-κB activation | Zhang [85] |
| THP-1, human PBMCs: 50 μM | Reduced transcription of the NLPR3, CASP1, and IL1B genes | Matias [86] | |||
| HepG2, primary hepatocytes: 0.2–50 μM | C57BL/6 mice: 50–100 mg/kg | HFD-induced liver steatosis | Reduced protein levels of NLRP3 and cleaved caspase-1 in liver | Zhang [87] | |
| Cardamonin (Alpinia Katsumadai) | BMDMs, iBMDMs, THP-1, human PBMCs: 1–20 μM | C57BL/6 mice: 25–50 mg/kg | Endotoxic shock | Blocked assembly of the NLRP3 inflammasome | Wang [88] |
| BMDMs, THP-1: 1–30 μM | C57BL/6 mice, BALB/c mice: 15–60 mg/kg | Colitis | Reduced NLRP3 protein level and transcript of the Il1b gene; activated AhR/Nrf2/NQO1 pathway | Wang [89] | |
| Caffeic acid phenethyl ester (Honeybee propolis) | BMDMs: 0.5–10 μM | WT and Nlrp3−/− C57BL/6 mice: 30 mg/kg | Gouty arthritis | Bound to ASC, abrogated interaction between ASC and NLRP3, and therefore blocked NLRP3 inflammasome assembly | Lee [90] |
| BMDMs, THP-1: 5–20 μM | C57BL/6 mice: 5–45 mg/kg | Colitis-associated cancer | Reduced NLRP3 protein level, but had no impact on transcription of the Nlrp3 gene; suppressed deubiquitination of the NLRP3 protein through enhancing the interaction between NLRP3 and Cullin1 and decreasing the interaction between NLRP3 and CSN5 | Dai [91] | |
| Human aortic valve interstitial cells: 10 μM | Reduced protein levels of NLRP3 and ASC; inhibited NF-κB pathway | Liu [92] |
| Name of Ginsenosides | In Vitro Model and Dose | Animal Model and Dose | Disease Model | Inhibitory Mechanism | Refe-rences |
|---|---|---|---|---|---|
| Ginsenoside Rb1 | eWAT: 10 μM | Reduced protein levels of NLRP3 and cleaved caspase-1 in eWAT cultured ex vivo | Chen [114] | ||
| Compound K | db/db mice: 10 mg/kg | Diabetes | Reduced protein levels of NLRP3, ASC, cleaved caspase-1, and IL-1β in hippocampus | Li [115] | |
| eWAT: 10 μM | Reduced protein levels of NLRP3 and cleaved caspase1 in eWAT cultured ex vivo | Chen [114] | |||
| Ginsenoside Rg3 | THP-1, RAW 264.7, BMDMs: 1–10 μg/mL | C57BL/6J mice: 10 mg/kg | Endotoxic shock | Impaired the interaction of NEK7 and NLRP3 and therefore blocked assembly of the NLRP3 inflammasome | Shi [116] |
| 20S-protopanax-atriol | Mouse peritoneal macrophages: 10–40 μM | C57BL/6 mice: 5–10 mg/kg | Peritonitis, Endotoxic shock | Blocked assembly of the NLRP3 inflammasome | Jiang [117] |
| HSC-T6: 10–20 μM | C57BL/6J mice: 10–20 mg/kg | Hepatic fibrosis | Reduced protein levels of NLRP3, cleaved caspase-1, and mature IL-1β in fibrotic liver | Song [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Yu, J. Anti-NLRP3 Inflammasome Natural Compounds: An Update. Biomedicines 2021, 9, 136. https://doi.org/10.3390/biomedicines9020136
Liu B, Yu J. Anti-NLRP3 Inflammasome Natural Compounds: An Update. Biomedicines. 2021; 9(2):136. https://doi.org/10.3390/biomedicines9020136
Chicago/Turabian StyleLiu, Baolong, and Jiujiu Yu. 2021. "Anti-NLRP3 Inflammasome Natural Compounds: An Update" Biomedicines 9, no. 2: 136. https://doi.org/10.3390/biomedicines9020136
APA StyleLiu, B., & Yu, J. (2021). Anti-NLRP3 Inflammasome Natural Compounds: An Update. Biomedicines, 9(2), 136. https://doi.org/10.3390/biomedicines9020136