
Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides

 , and
, and 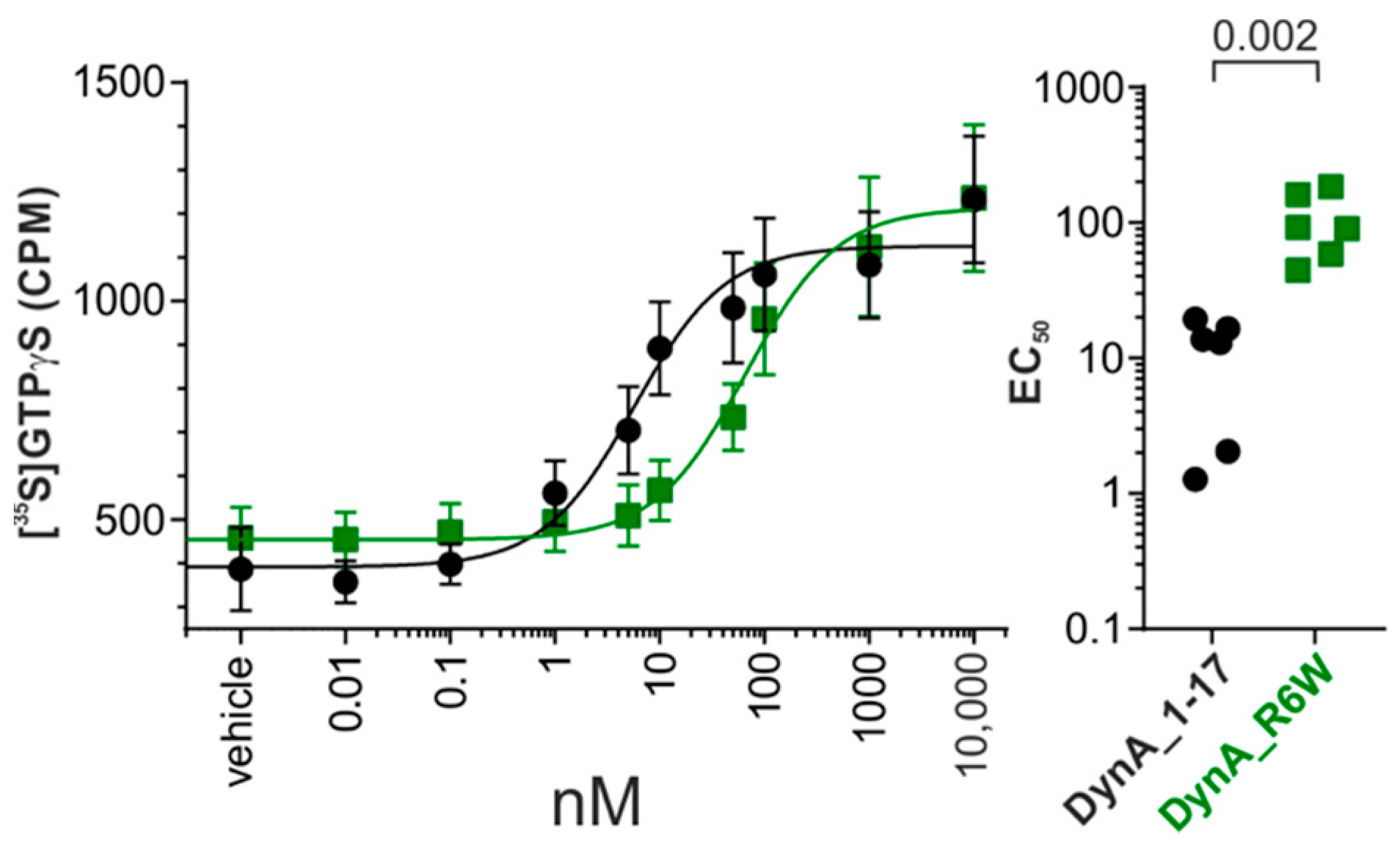{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptides
2.2. Molecular Biology
2.3. Cell Culture
2.4. [35S] Guanosine 5′-O-[γ-thio]triphosphate Functional Assays for κ Opioid Receptors
2.5. β-Arrestin Recruitment Assay
2.6. G-Protein Transducerome Assay
2.7. Modelling
3. Results
3.1. SCA23 DynA Mutant Exhibit Reduced hKOR Activation
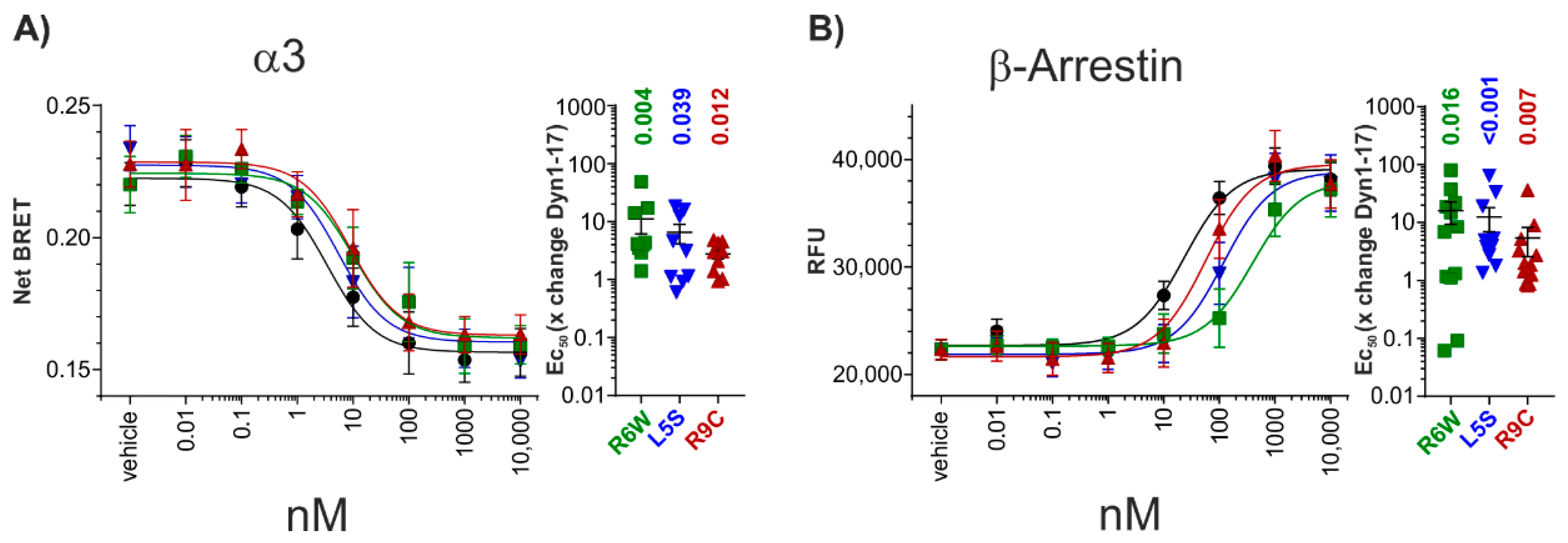
3.2. G-Protein Transducerome Activation by DynA_R6W
3.3. Investigating Signal Bias at hKOR
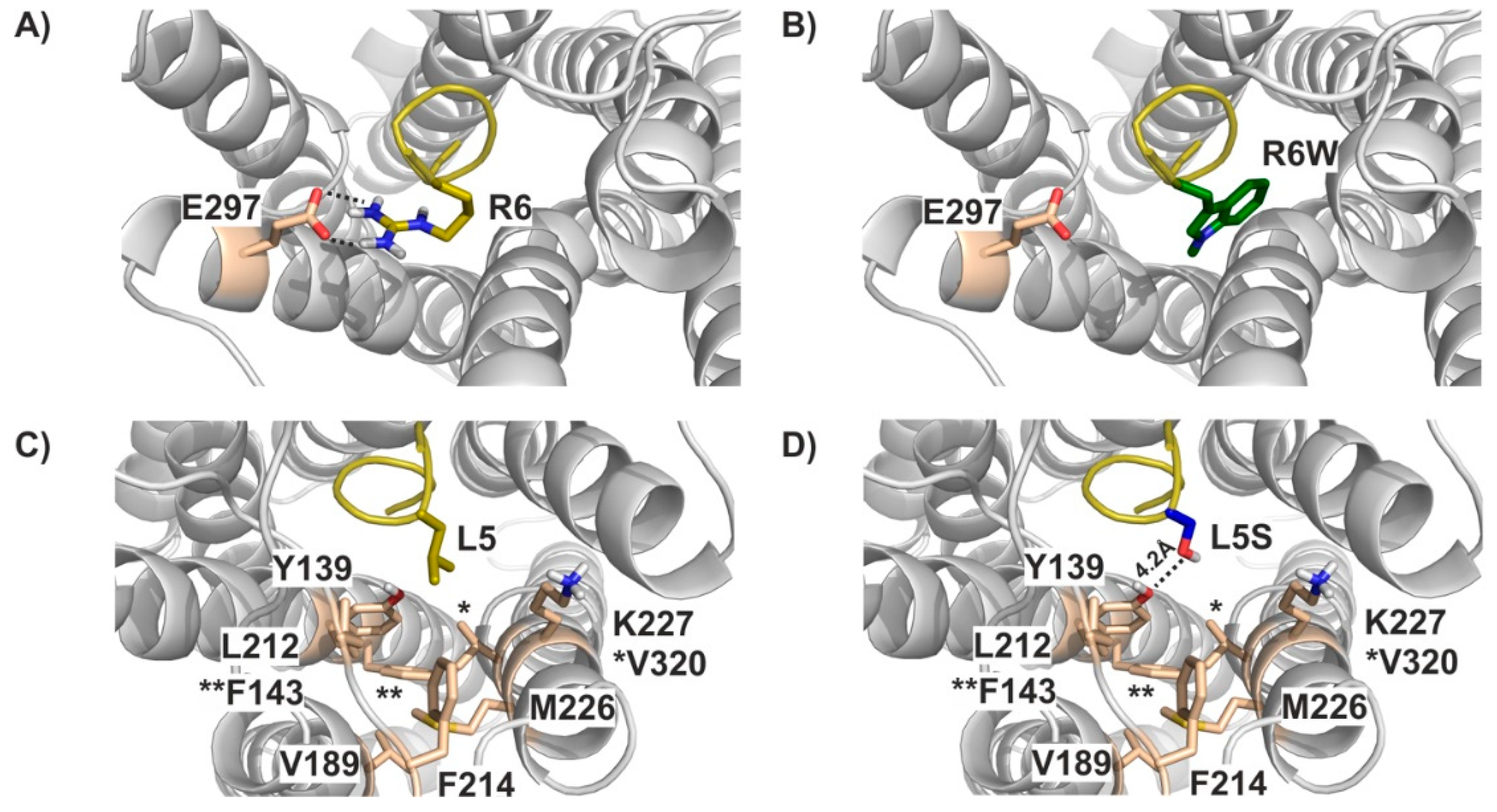
3.4. Molecular Modelling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Verbeek, D.S.; van de Warrenburg, B.P.; Wesseling, P.; Pearson, P.L.; Kremer, H.P.; Sinke, R.J. Mapping of the SCA23 locus involved in autosomal dominant cerebellar ataxia to chromosome region 20p13-12.3. Brain J. Neurol. 2004, 127, 2551–2557. [Google Scholar] [CrossRef]
- Bakalkin, G.; Watanabe, H.; Jezierska, J.; Depoorter, C.; Verschuuren-Bemelmans, C.; Bazov, I.; Artemenko, K.A.; Yakovleva, T.; Dooijes, D.; Van de Warrenburg, B.P.C.; et al. Prodynorphin Mutations Cause the Neurodegenerative Disorder Spinocerebellar Ataxia Type 23. Am. J. Hum. Genet. 2010, 87, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Jezierska, J.; Stevanin, G.; Watanabe, H.; Fokkens, M.R.; Zagnoli, F.; Kok, J.; Goas, J.Y.; Bertrand, P.; Robin, C.; Brice, A.; et al. Identification and characterization of novel PDYN mutations in dominant cerebellar ataxia cases. J. Neurol. 2013, 260, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Mizoguchi, H.; Verbeek, D.S.; Kuzmin, A.; Nyberg, F.; Krishtal, O.; Sakurada, S.; Bakalkin, G. Non-opioid nociceptive activity of human dynorphin mutants that cause neurodegenerative disorder spinocerebellar ataxia type 23. Peptides 2012, 35, 306–310. [Google Scholar] [CrossRef]
- Smeets, C.J.L.M.; Jezierska, J.; Watanabe, H.; Duarri, A.; Fokkens, M.R.; Meijer, M.; Zhou, Q.; Yakovleva, T.; Boddeke, E.; Den Dunnen, W.; et al. Elevated mutant dynorphin A causes Purkinje cell loss and motor dysfunction in spinocerebellar ataxia type 23. Brain J. Neurol. 2015, 138, 2537–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, C.J.L.M.; Zmorzyńska, J.; Melo, M.N.; Stargardt, A.; Dooley, C.; Bakalkin, G.; McLaughlin, J.; Sinke, R.J.; Marrink, S.J.; Reits, E.; et al. Altered secondary structure of Dynorphin A associates with loss of opioid signalling and NMDA-mediated excitotoxicity in SCA23. Hum. Mol. Genet. 2016, 25, 2728–2737. [Google Scholar] [CrossRef] [Green Version]
- Björnerås, J.; Gräslund, A.; Mäler, L. Membrane interaction of disease-related dynorphin A variants. Biochemistry 2013, 52, 4157–4167. [Google Scholar] [CrossRef]
- Madani, F.; Taqi, M.M.; Wärmländer, S.K.T.S.; Verbeek, D.S.; Bakalkin, G.; Gräslund, A. Perturbations of model membranes induced by pathogenic dynorphin A mutants causing neurodegeneration in human brain. Biochem. Biophys. Res. Commun. 2011, 411, 111–114. [Google Scholar] [CrossRef]
- Maximyuk, O.; Khmyz, V.; Lindskog, C.J.; Vukojević, V.; Ivanova, T.; Bazov, I.; Hauser, K.F.; Bakalkin, G.; Krishtal, O. Plasma membrane poration by opioid neuropeptides: A possible mechanism of pathological signal transduction. Cell Death Dis. 2015, 6, e1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merg, F.; Filliol, D.; Usynin, I.; Bazov, I.; Bark, N.; Hurd, Y.L.; Yakovleva, T.; Kieffer, B.L.; Bakalkin, G. Big dynorphin as a putative endogenous ligand for the kappa-opioid receptor. J. Neurochem. 2006, 97, 292–301. [Google Scholar] [CrossRef]
- Chavkin, C. Dynorphin–Still an Extraordinarily Potent Opioid Peptide. Mol. Pharmacol. 2013, 83, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Tejeda, H.A.; Shippenberg, T.S.; Henriksson, R. The dynorphin/κ-opioid receptor system and its role in psychiatric disorders. Cell. Mol. Life Sci. 2012, 69, 857–896. [Google Scholar] [CrossRef]
- Van’t Veer, A.; Carlezon, W.A. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Transduction of Receptor Signals by -Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Defea, K. Beta-arrestins and heterotrimeric G-proteins: Collaborators and competitors in signal transduction. Br. J. Pharmacol. 2008, 153, S298–S309. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Lefkowitz, R.J. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef]
- Kenakin, T. Functional Selectivity and Biased Receptor Signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleyard, S.M.; Celver, J.; Pineda, V.; Kovoor, A.; Wayman, G.A.; Chavkin, C. Agonist-dependent Desensitization of the κ Opioid Receptor by G Protein Receptor Kinase and β-Arrestin. J. Biol. Chem. 1999, 274, 23802–23807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.G.; Luo, L.Y.; Krupnick, J.G.; Benovic, J.L.; Liu-Chen, L.Y. U50,488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J. Biol. Chem. 1999, 274, 12087–12094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Sierra, S.; Lueptow, L.; Gupta, A.; Gouty, S.; Margolis, E.B.; Cox, B.M.; Devi, L.A. Biased signaling by endogenous opioid peptides. Proc. Natl. Acad. Sci. USA 2020, 117, 11820–11828. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Macey, T.A.; Lowe, J.D.; Chavkin, C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Schindler, A.G.; Shankar, H.; Messinger, D.I.; Miyatake, M.; Land, B.B.; Lemos, J.C.; Hagan, C.E.; Neumaier, J.F.; Quintana, A.; et al. Selective p38α MAPK Deletion in Serotonergic Neurons Produces Stress Resilience in Models of Depression and Addiction. Neuron 2011, 71, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrich, J.M.; Messinger, D.I.; Knakal, C.R.; Kuhar, J.R.; Schattauer, S.S.; Bruchas, M.R.; Zweifel, L.S.; Kieffer, B.L.; Phillips, P.E.M.; Chavkin, C. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J. Neurosci. 2015, 35, 12917–12931. [Google Scholar] [CrossRef]
- Land, B.B.; Bruchas, M.R.; Schattauer, S.; Giardino, W.J.; Aita, M.; Messinger, D.; Hnasko, T.S.; Palmiter, R.D.; Chavkin, C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc. Natl. Acad. Sci. USA 2009, 106, 19168–19173. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Bajpayee, N.S. Molecular mechanisms of go signaling. Neuro-Signals 2009, 17, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Prather, P.L.; McGinn, T.M.; Claude, P.A.; Liu-Chen, L.Y.; Loh, H.H.; Law, P.Y. Properties of a kappa-opioid receptor expressed in CHO cells: Interaction with multiple G-proteins is not specific for any individual G alpha subunit and is similar to that of other opioid receptors. Brain Res. Mol. Brain Res. 1995, 29, 336–346. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Diaz, J.; Sokoloff, P.; Griffon, N. Interactions of GIPC with dopamine D2, D3 but not D4 receptors define a novel mode of regulation of G protein-coupled receptors. Mol. Biol. Cell 2004, 15, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Seiler, C.Y.; Park, J.G.; Sharma, A.; Hunter, P.; Surapaneni, P.; Sedillo, C.; Field, J.; Algar, R.; Price, A.; Steel, J.; et al. DNASU plasmid and PSI:Biology-Materials repositories: Resources to accelerate biological research. Nucleic Acids Res. 2014, 42, D1253–D1260. [Google Scholar] [CrossRef] [Green Version]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.-P.; Lansu, K.; McCorvy, J.D.; Giguère, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef]
- Harterink, M.; da Silva, M.E.; Will, L.; Turan, J.; Ibrahim, A.; Lang, A.E.; van Battum, E.Y.; Pasterkamp, R.J.; Kapitein, L.C.; Kudryashov, D.; et al. DeActs: Genetically encoded tools for perturbing the actin cytoskeleton in single cells. Nat. Methods 2017, 14, 479–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.; White, K.L.; Doncescu, N.; Didenko, T.; Roth, B.L.; Czaplicki, G.; Stevens, R.C.; Wüthrich, K.; Milon, A. NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 11852–11857. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2019.
- Schrödinger Release 2019-4. In Protein Preparation Wizard; Epik, Schrödinger LLC: New York, NY, USA, 2019.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-4. In Maestro; Schrödinger, LLC: New York, NY, USA, 2019.
- The PyMOL Molecular Graphics System; Version 1.8.0.0; Schrödinger, LLC: New York, NY, USA, 2016.
- Childers, S.R.; Snyder, S.H. Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life Sci. 1978, 23, 759–761. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Land, B.B.; Aita, M.; Xu, M.; Barot, S.K.; Li, S.; Chavkin, C. Stress-induced p38 mitogen-activated protein kinase activation mediates κ-opioid-dependent dysphoria. J. Neurosci. 2007, 27, 11614–11623. [Google Scholar] [CrossRef] [Green Version]
- Stein, C. New concepts in opioid analgesia. Expert Opin. Investig. Drugs 2018, 27, 765–775. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [Green Version]
- Sankararamakrishnan, R.; Weinstein, H. Molecular dynamics simulations predict a tilted orientation for the helical region of dynorphin A(1-17) in dimyristoylphosphatidylcholine bilayers. Biophys. J. 2000, 79, 2331–2344. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorganic Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, G.L.; Canals, M.; Poole, D.P. Biological redundancy of endogenous GPCR ligands in the gut and the potential for endogenous functional selectivity. Front. Pharmacol. 2014, 5, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain J. Neurol. 2006, 129, 1357–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lieb, A.; Thaler, G.; Fogli, B.; Trovato, O.; Posch, M.A.; Kaserer, T.; Zangrandi, L. Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides. Biomedicines 2021, 9, 1882. https://doi.org/10.3390/biomedicines9121882
Lieb A, Thaler G, Fogli B, Trovato O, Posch MA, Kaserer T, Zangrandi L. Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides. Biomedicines. 2021; 9(12):1882. https://doi.org/10.3390/biomedicines9121882
Chicago/Turabian StyleLieb, Andreas, Germana Thaler, Barbara Fogli, Olga Trovato, Mitja Amon Posch, Teresa Kaserer, and Luca Zangrandi. 2021. "Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides" Biomedicines 9, no. 12: 1882. https://doi.org/10.3390/biomedicines9121882
APA StyleLieb, A., Thaler, G., Fogli, B., Trovato, O., Posch, M. A., Kaserer, T., & Zangrandi, L. (2021). Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides. Biomedicines, 9(12), 1882. https://doi.org/10.3390/biomedicines9121882