
Regulation of Oncogenic Targets by Tumor-Suppressive miR-150-3p in Lung Squamous Cell Carcinoma

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human LUSQ Cell Lines and Cell Culture
2.2. RNA Extraction and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.3. Transfection of miRNAs, siRNAs and Plasmid Vectors into LUSQ Cells
2.4. Functional Assays for LUSQ Cells (Cell Proliferation, Migration, Invasion and Cell Cycle)
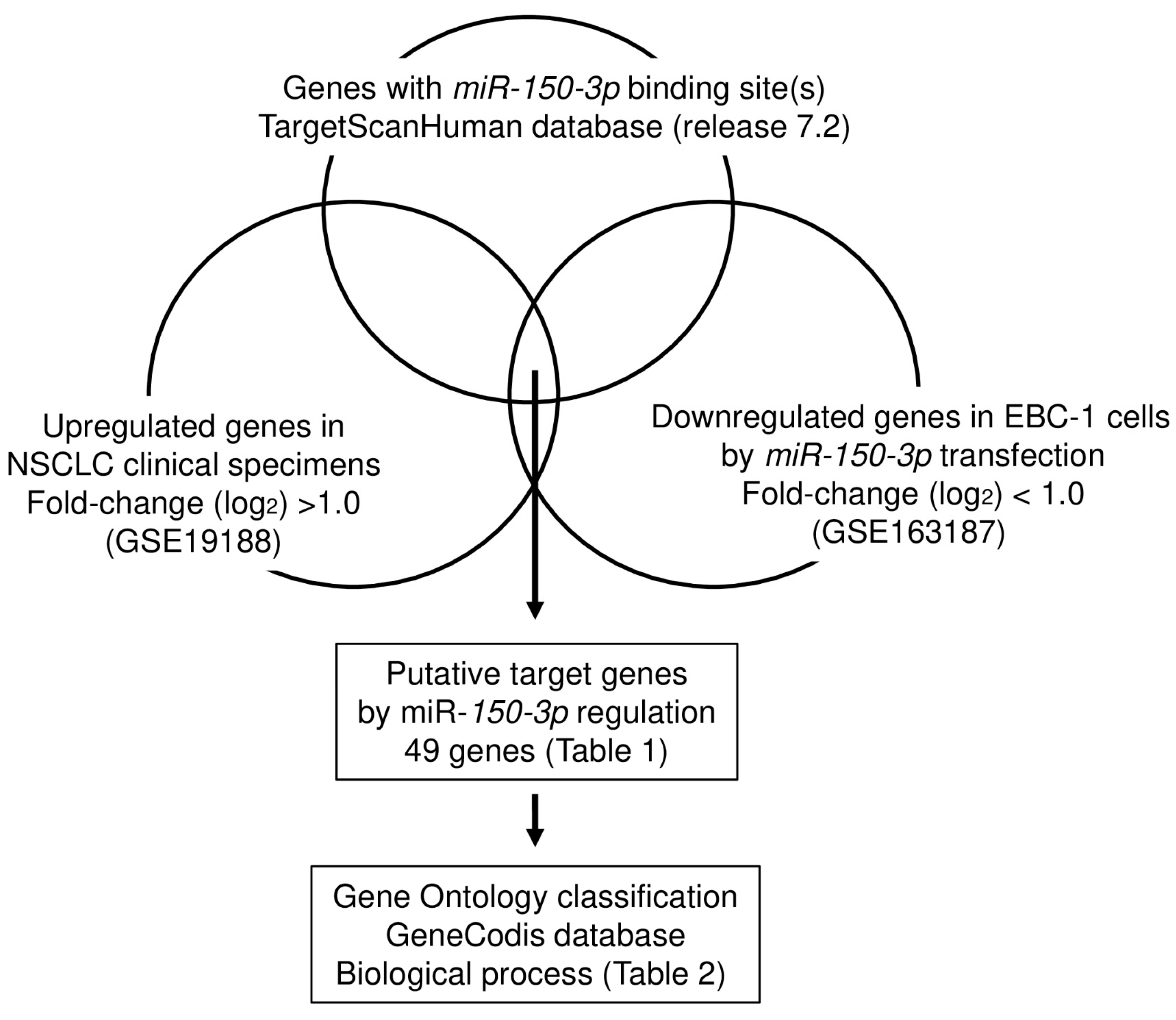
2.5. Identification of Oncogenic Targets by miR-150-3p Regulation in LUSQ Cells
2.6. In Silico Analysis of miRNA and Gene Expression Levels
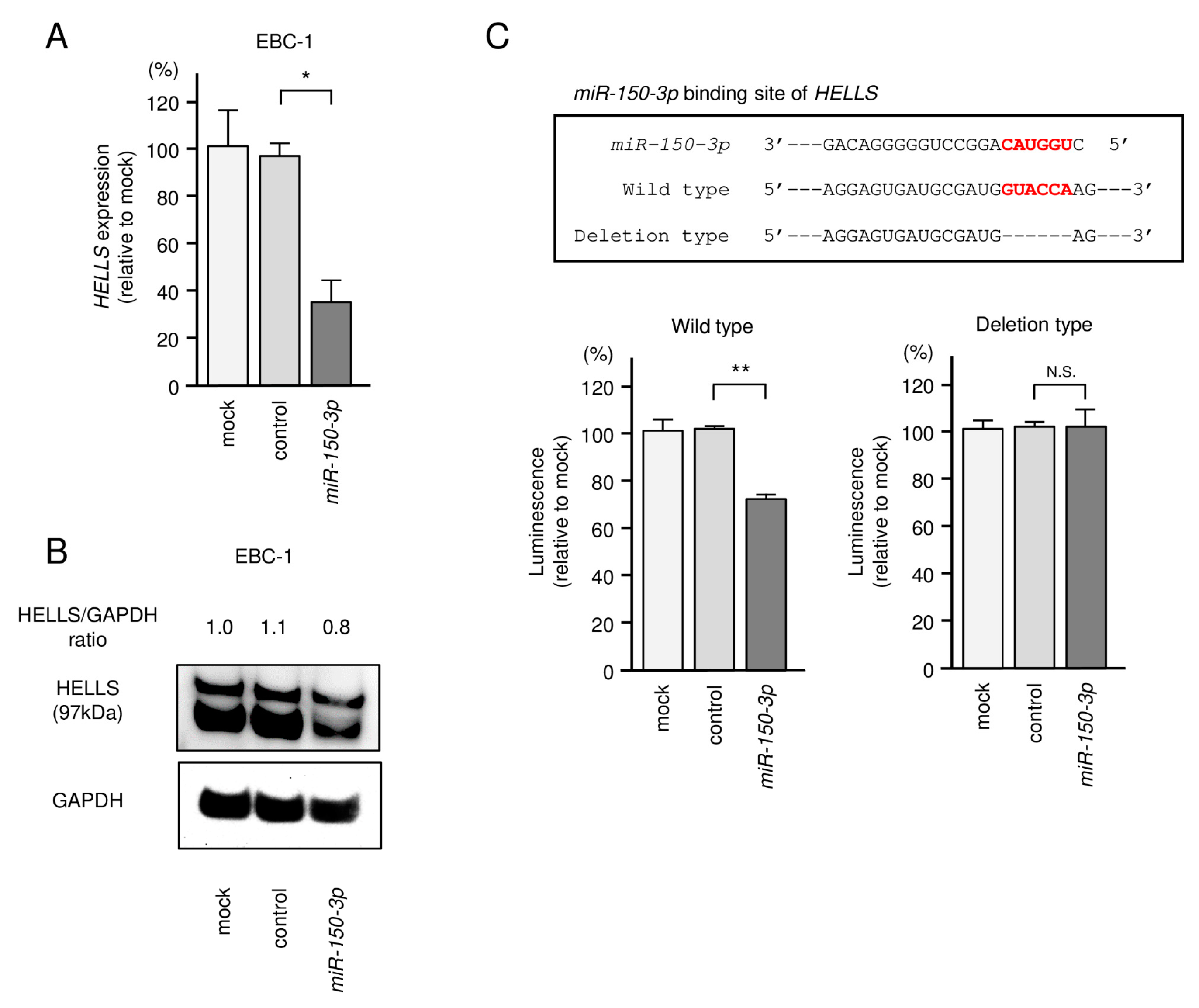
2.7. Plasmid Construction and Dual-Luciferase Reporter Assays
2.8. Western Blotting
2.9. Immunohistochemical Staining
2.10. Statistical Analysis
3. Results
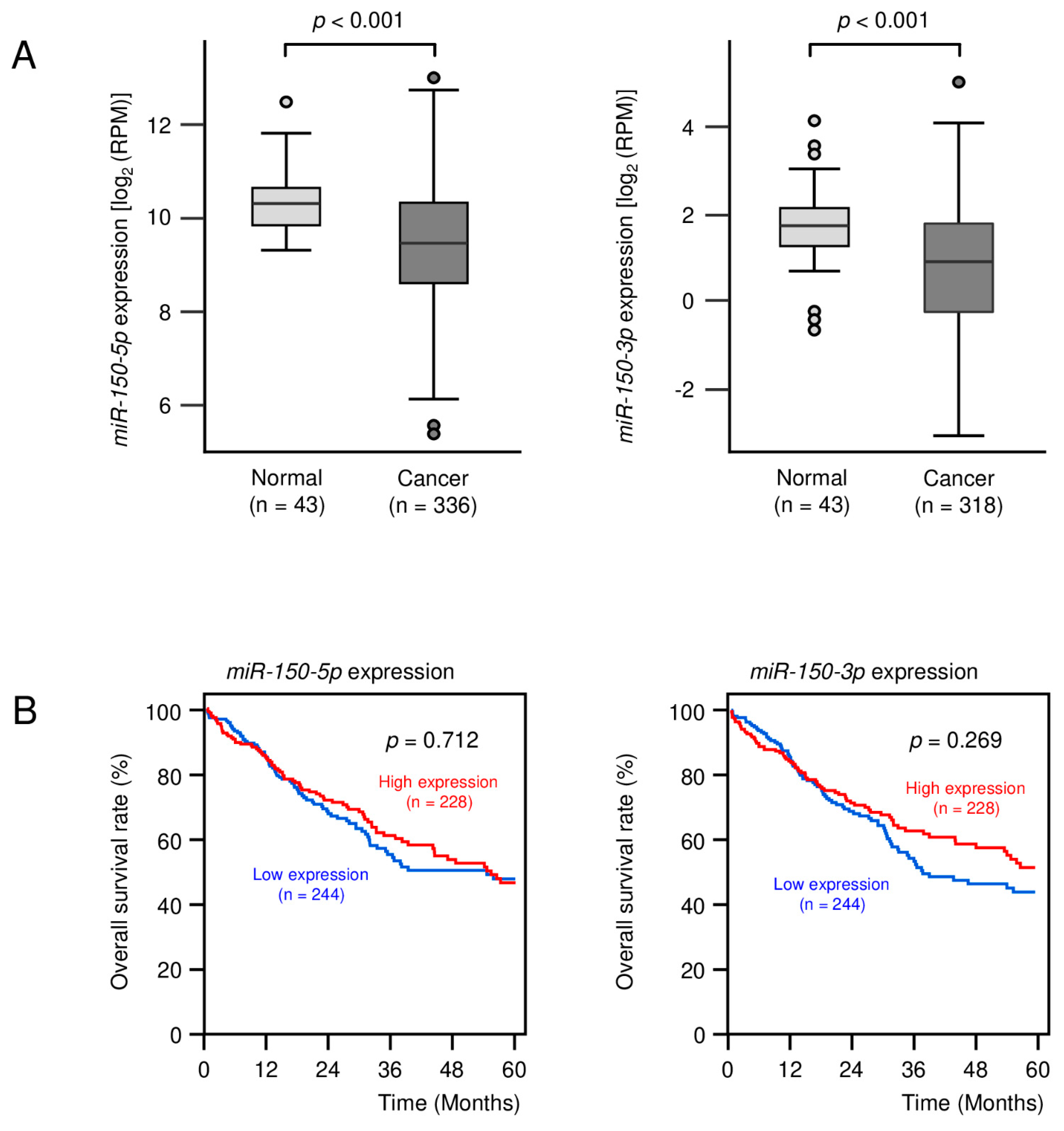
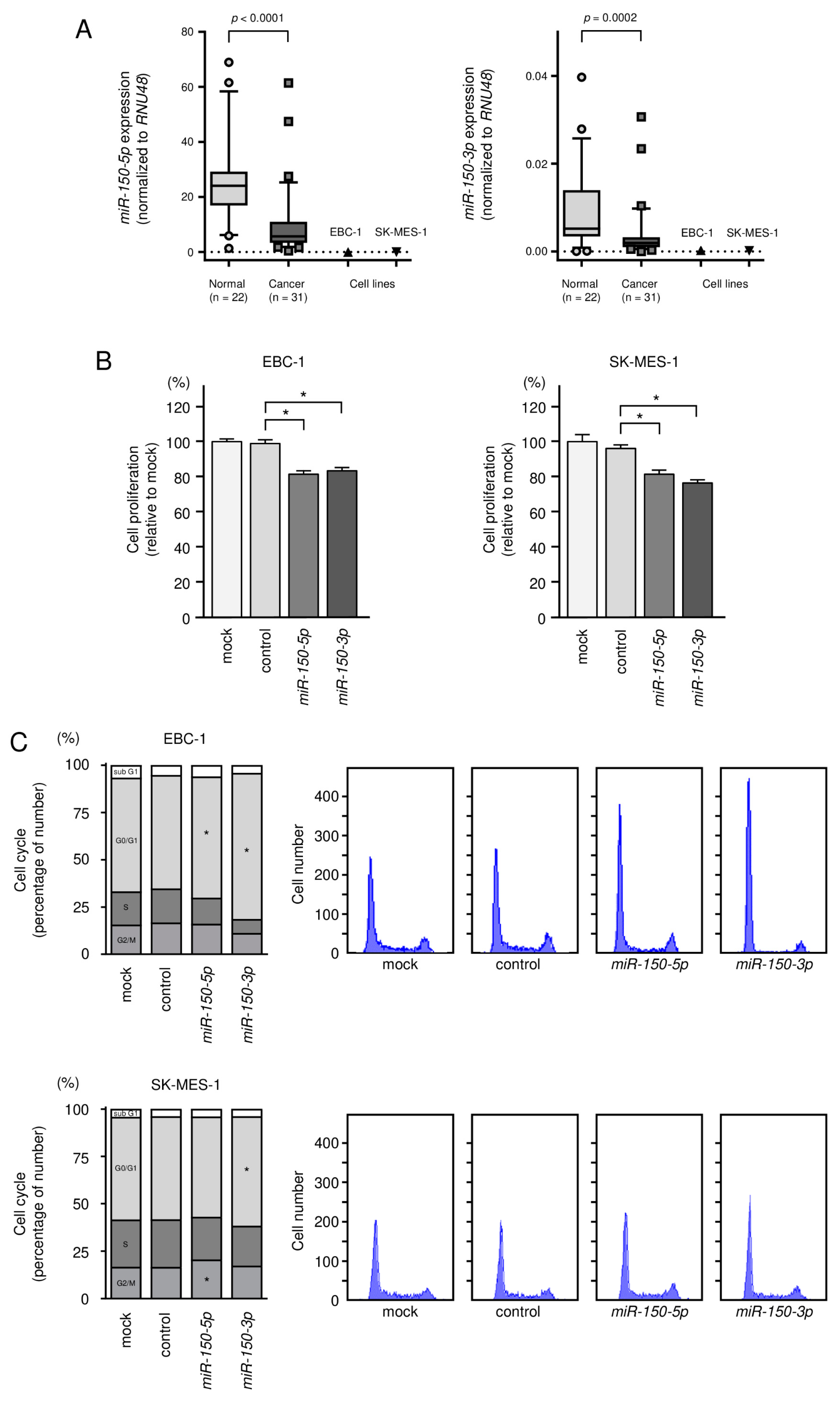
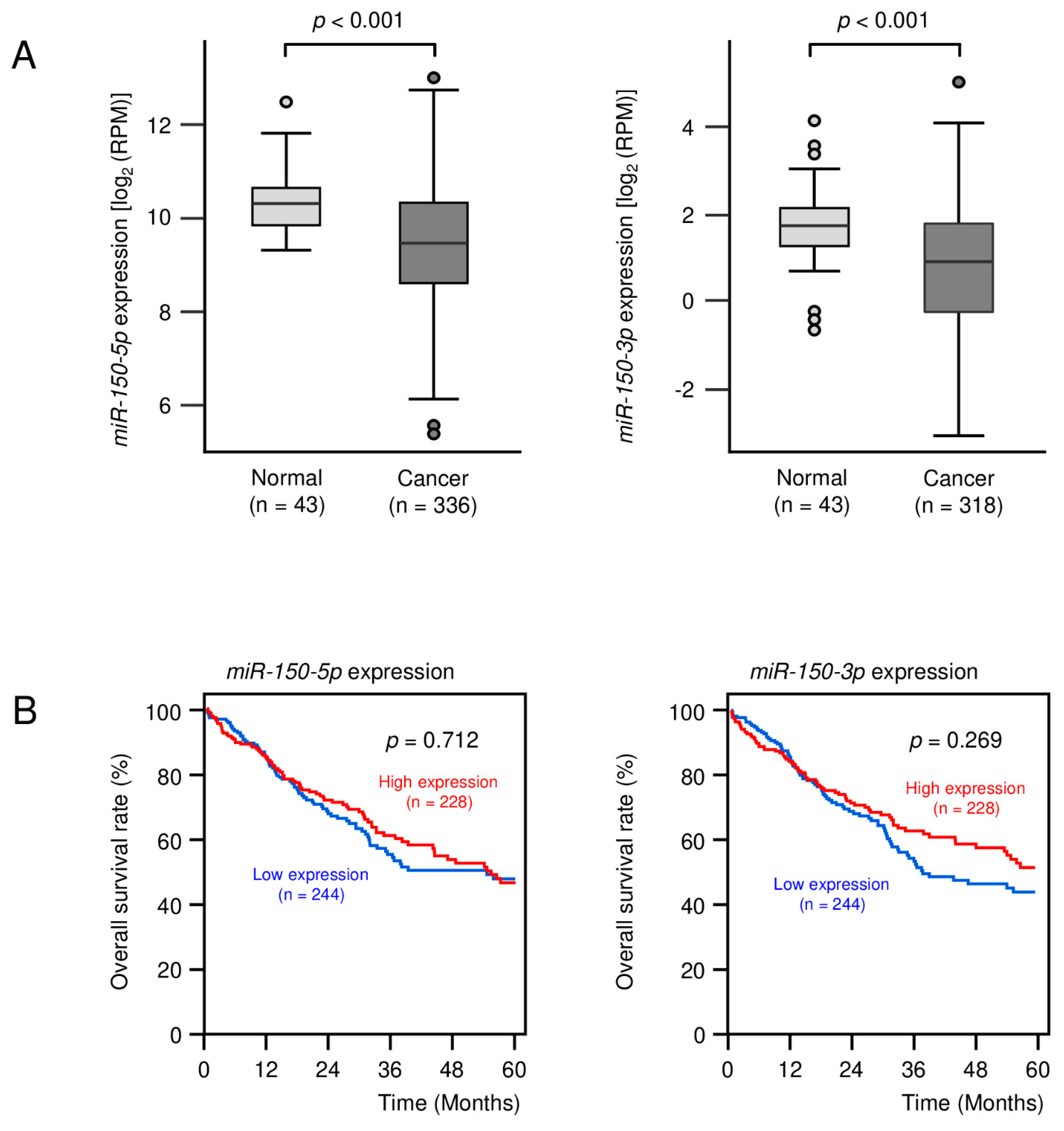
3.1. Expression Levels of miR-150-5p and miR-150-3p in Clinical LUSQ Tissues
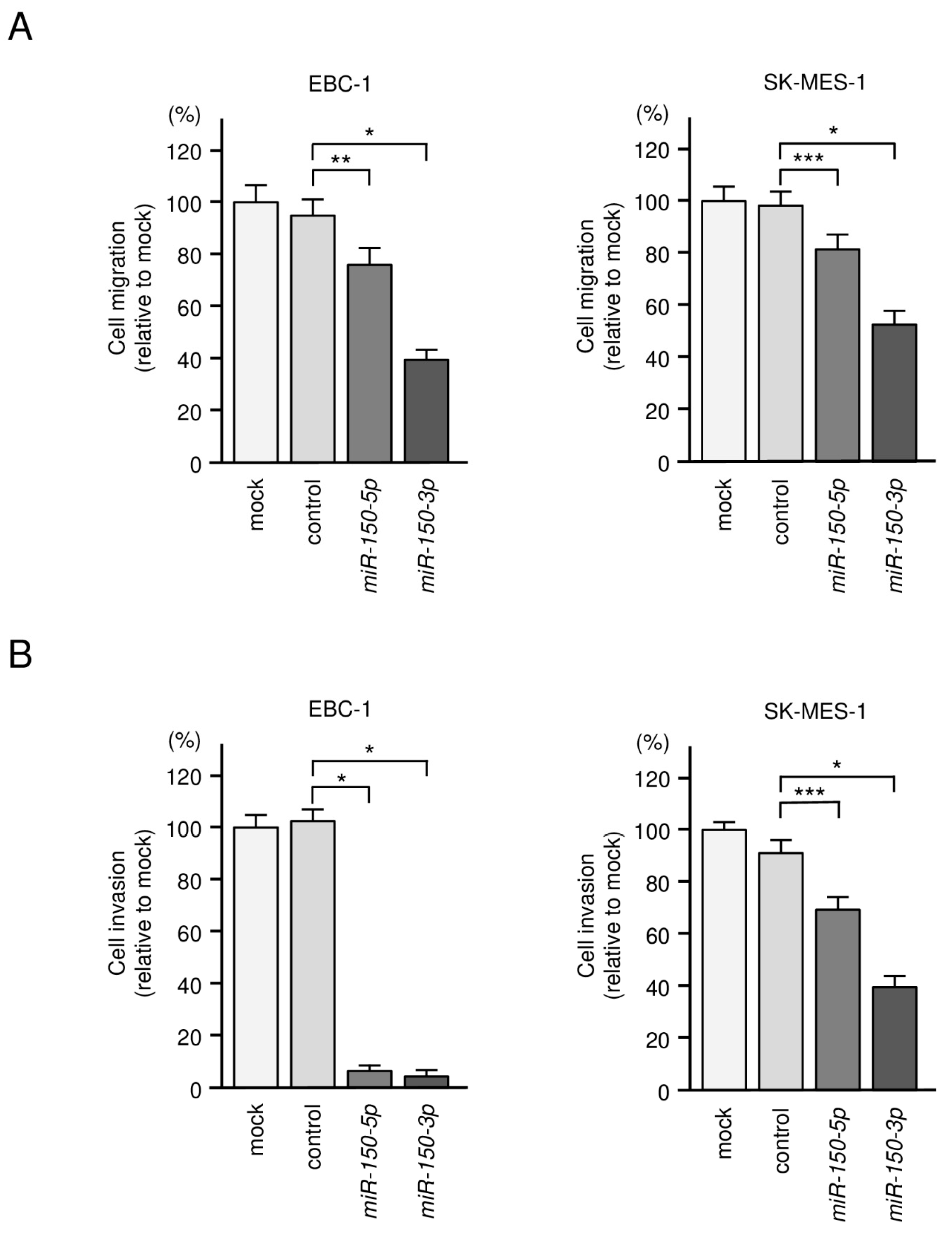
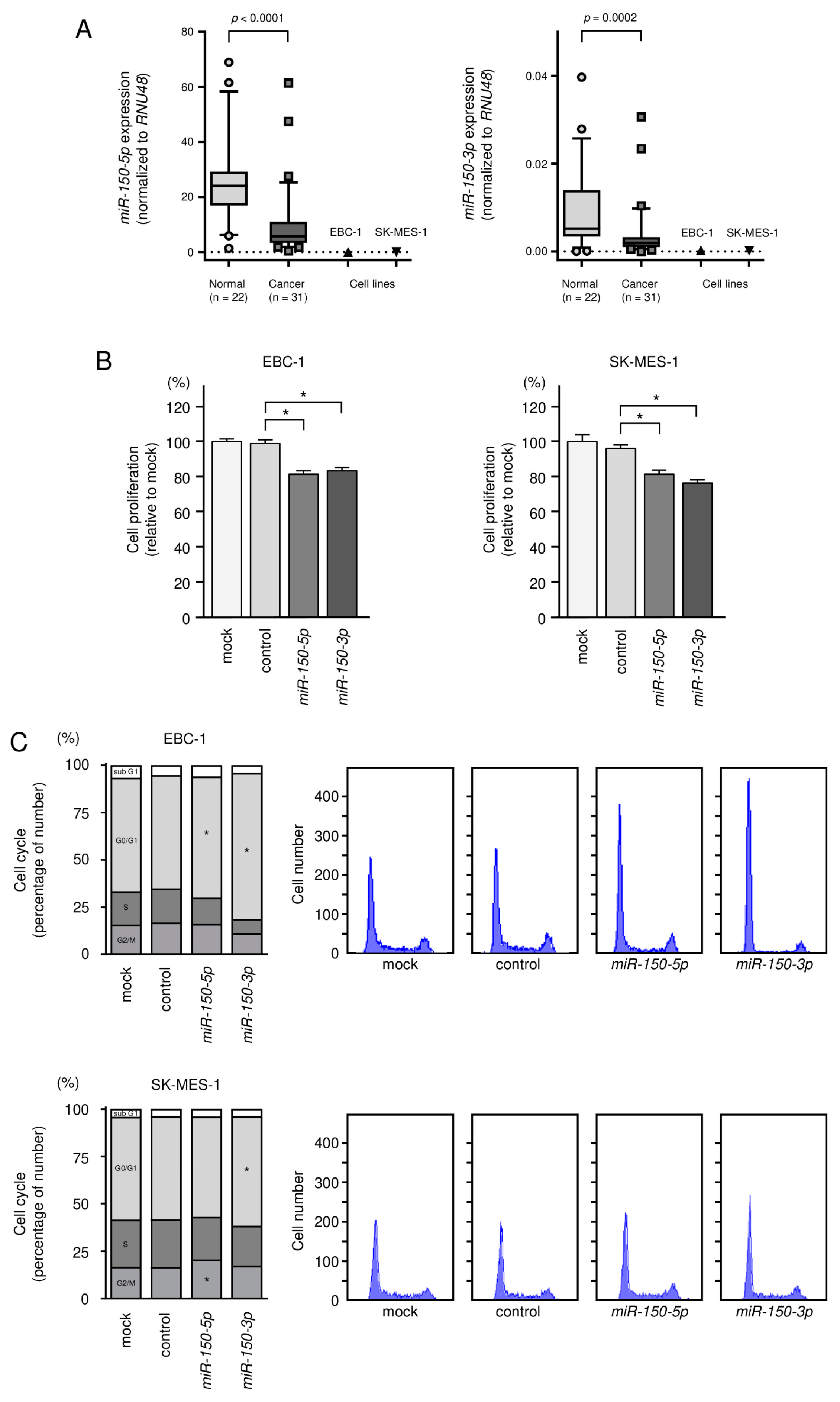
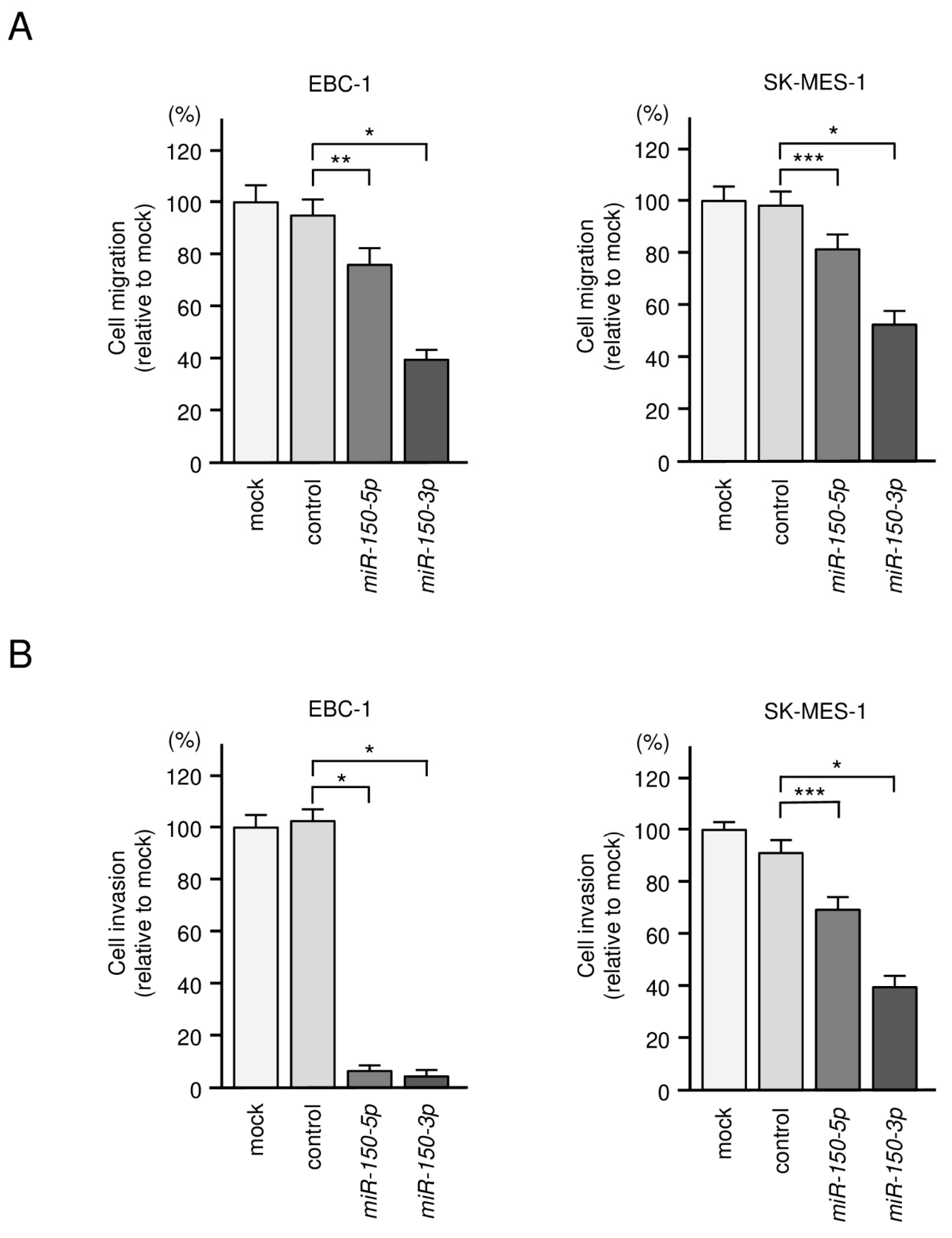
3.2. Tumor-Suppressive Roles of miR-150-5p and miR-150-3p in LUSQ Cells
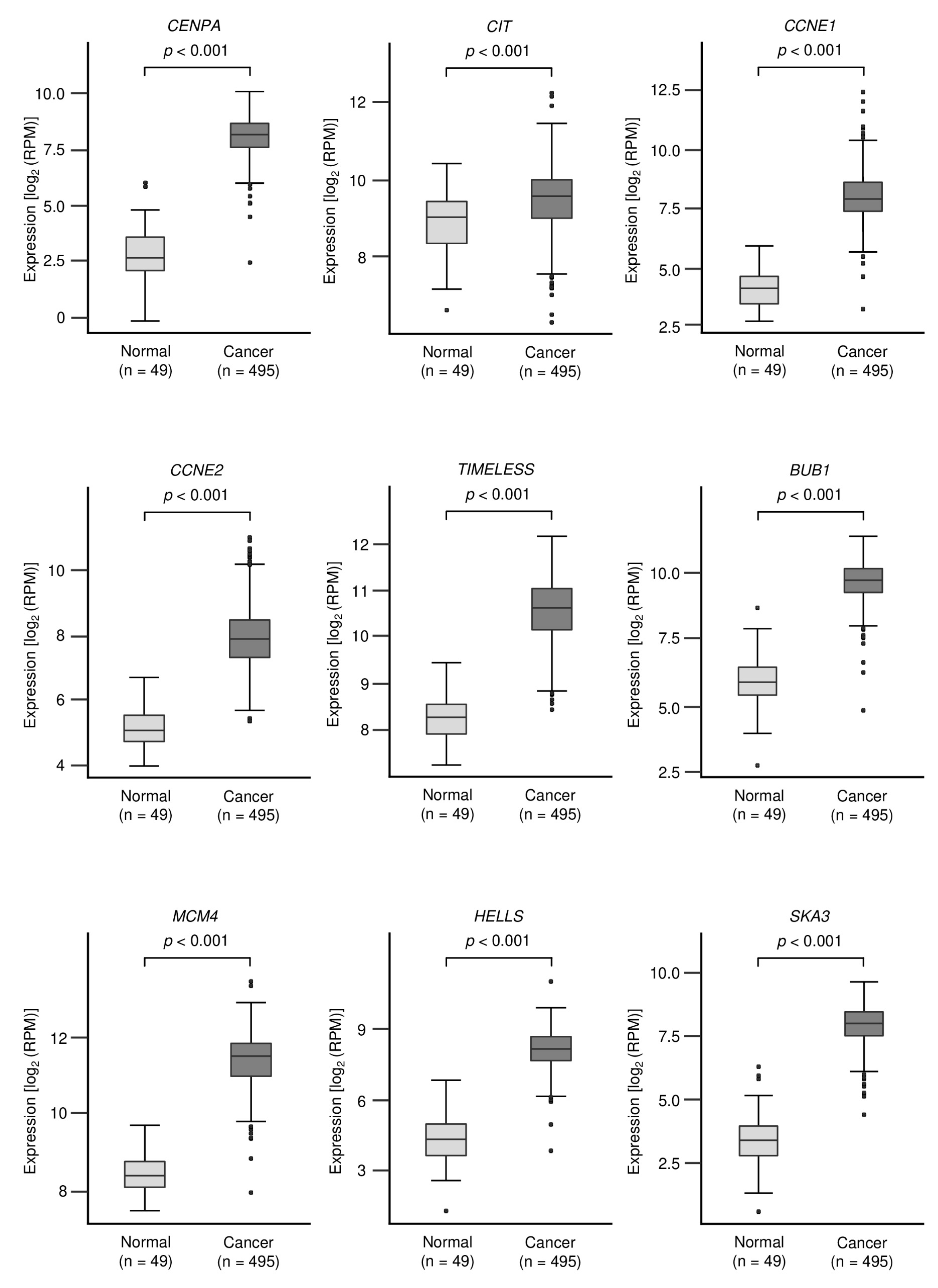
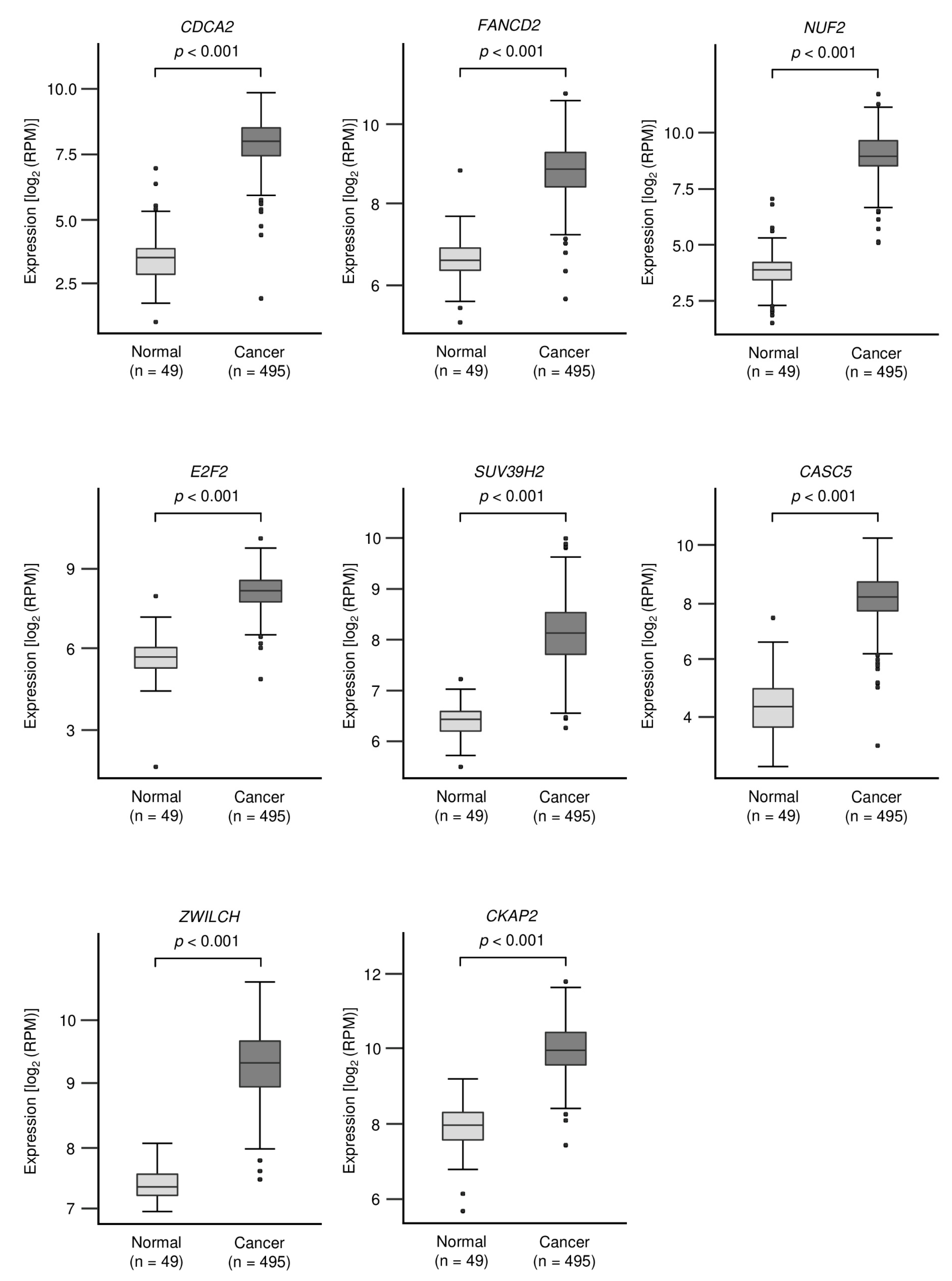
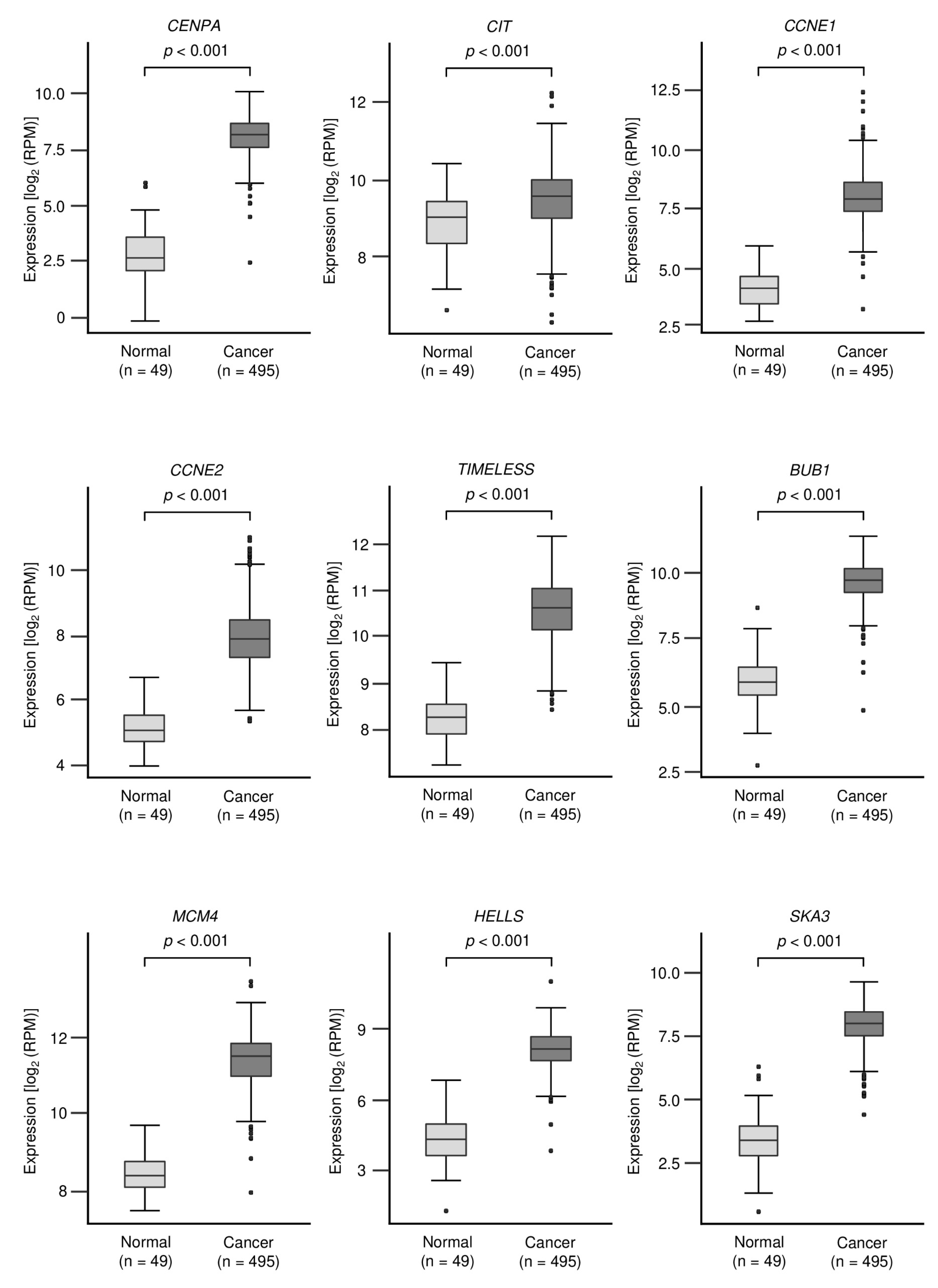
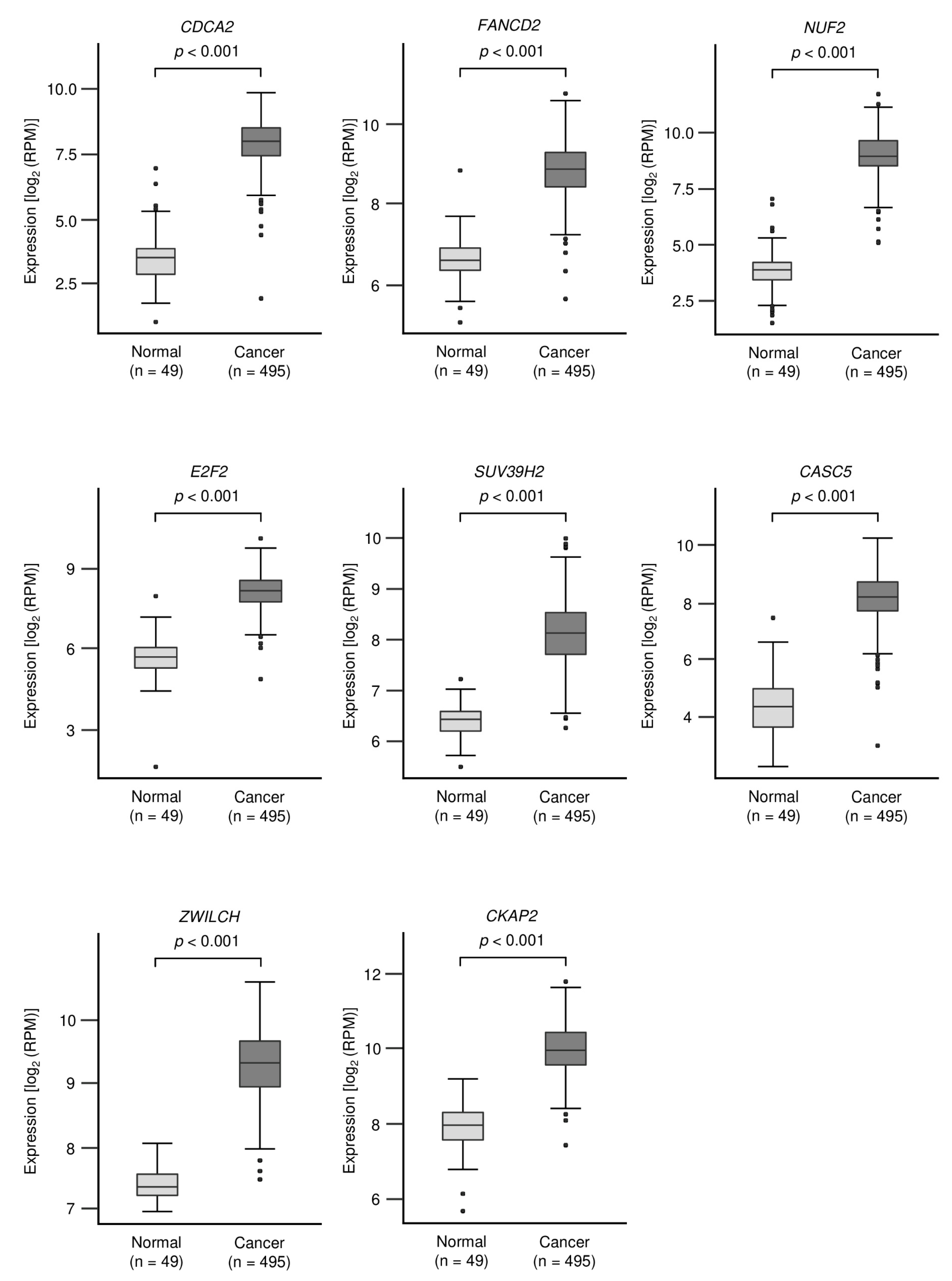
3.3. Identification of Oncogenic Targets Regulated by miR-150-3p in LUSQ Cells
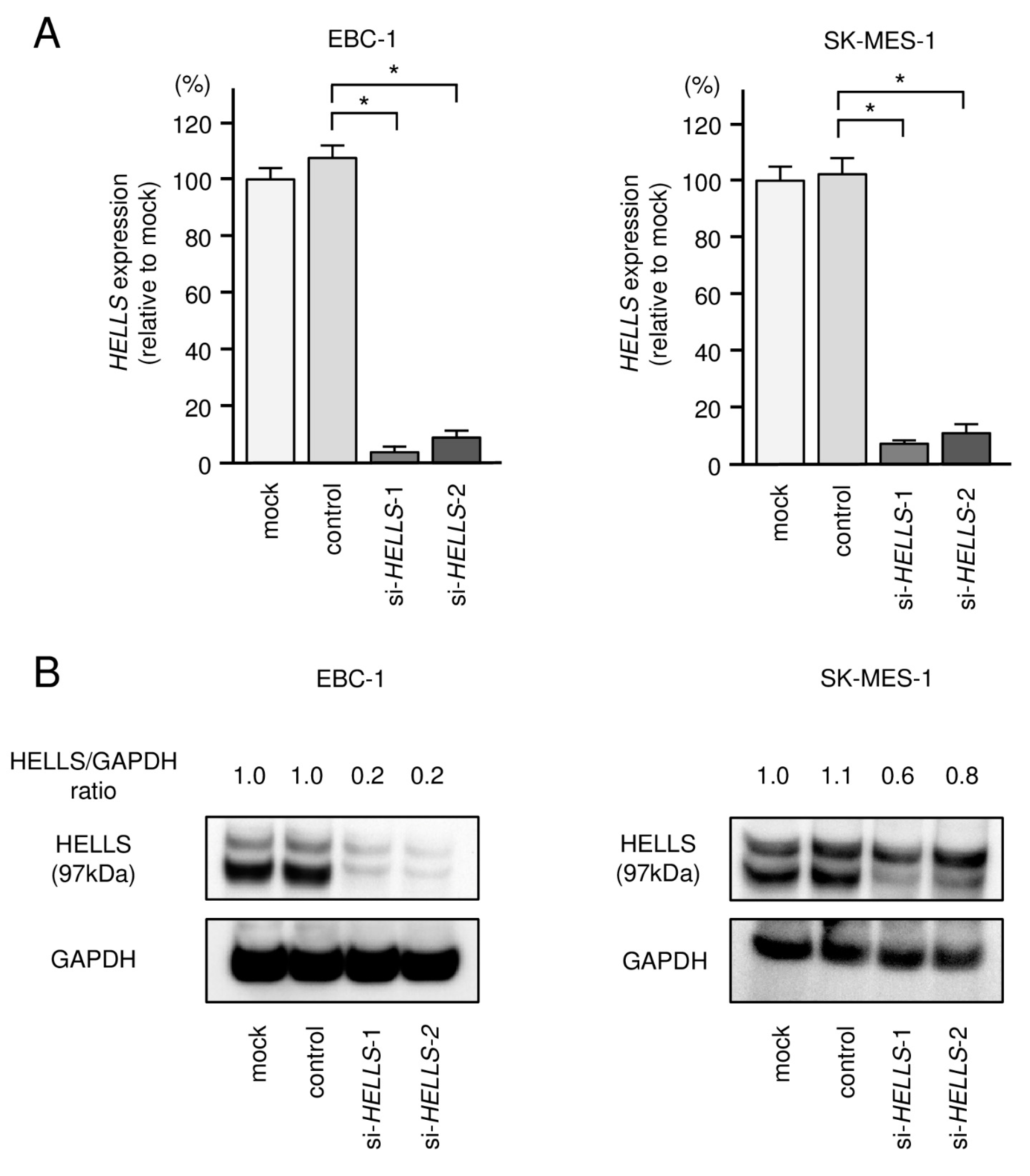
3.4. Direct Regulation of HELLS by miR-150-3p in LUSQ Cells
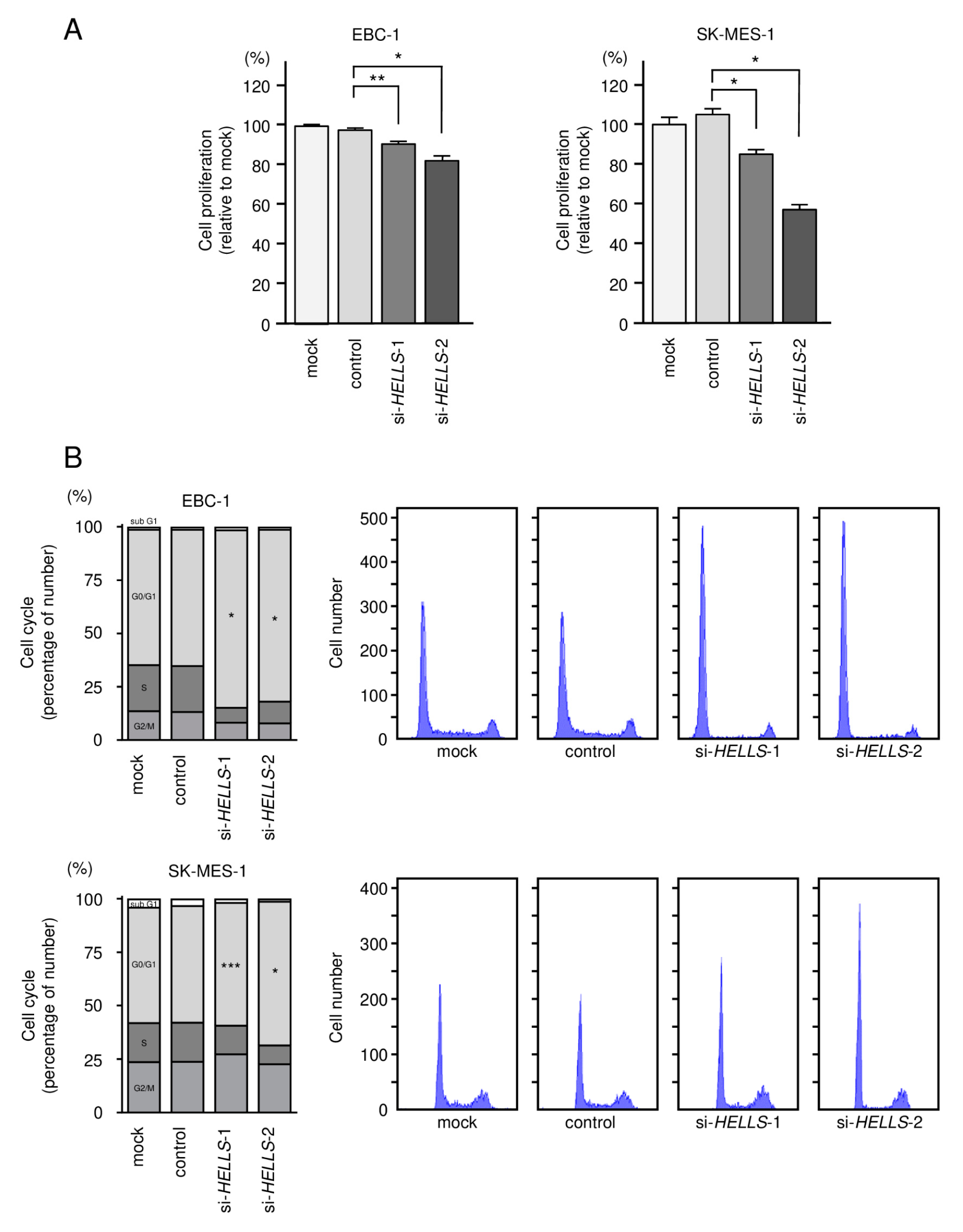
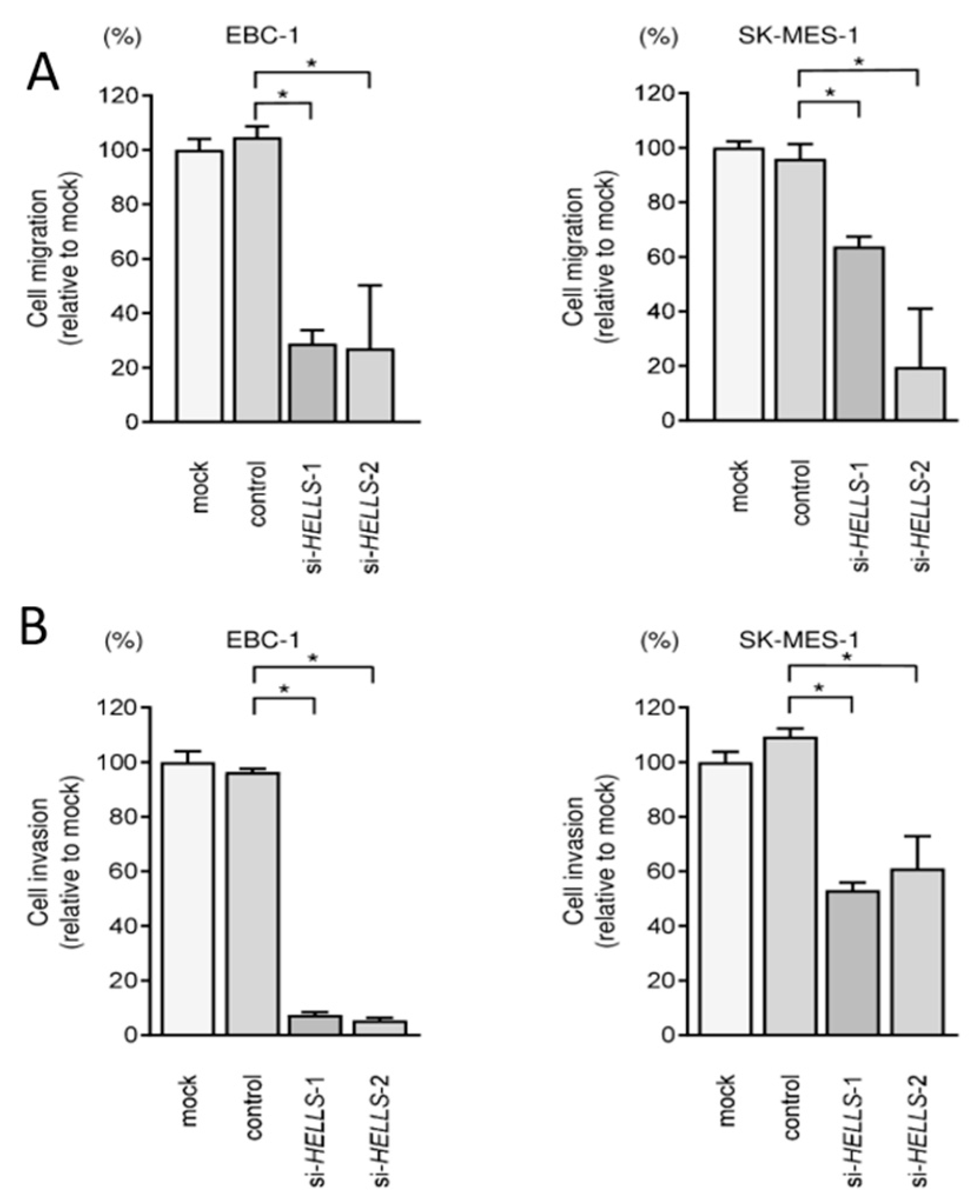
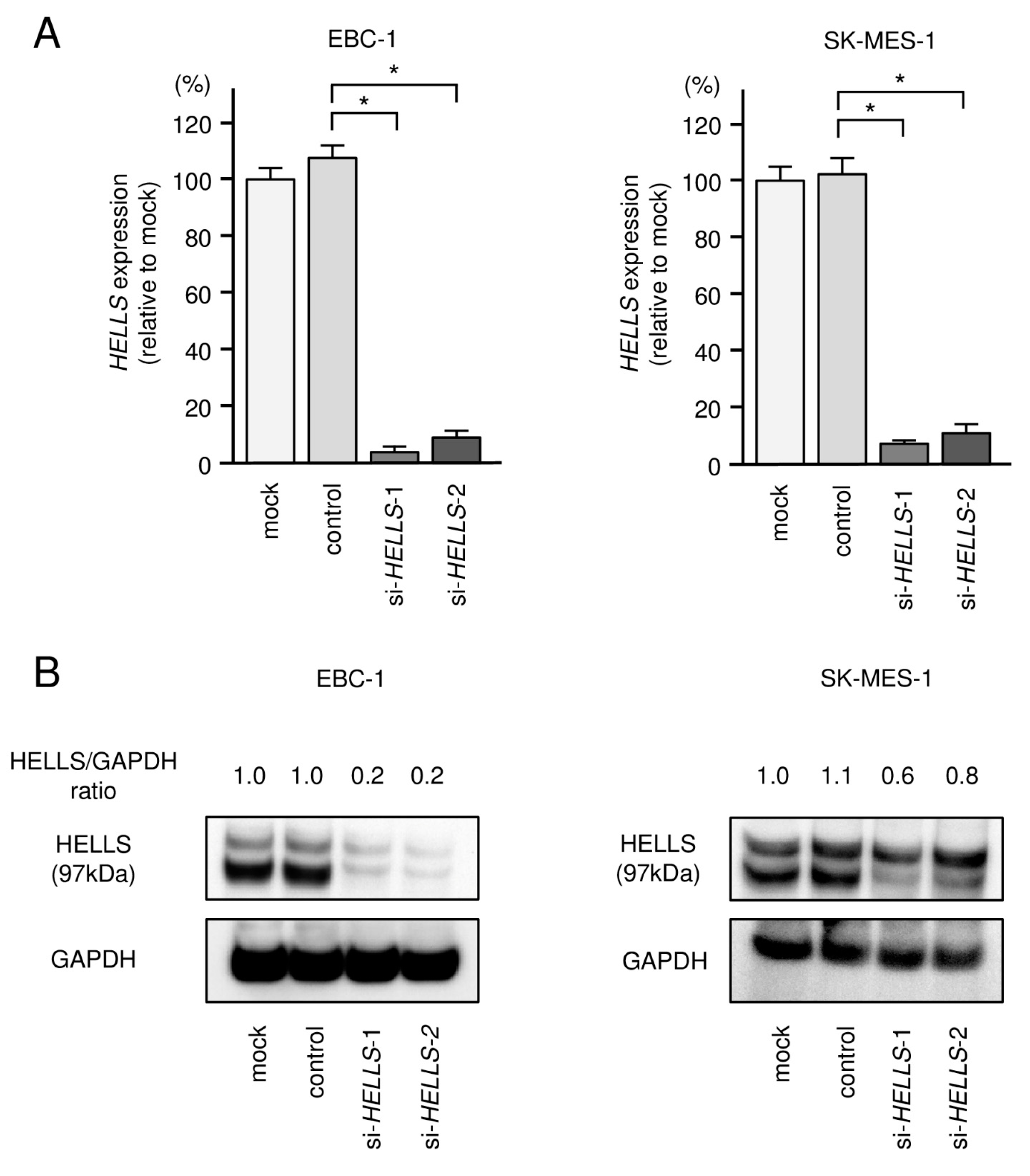
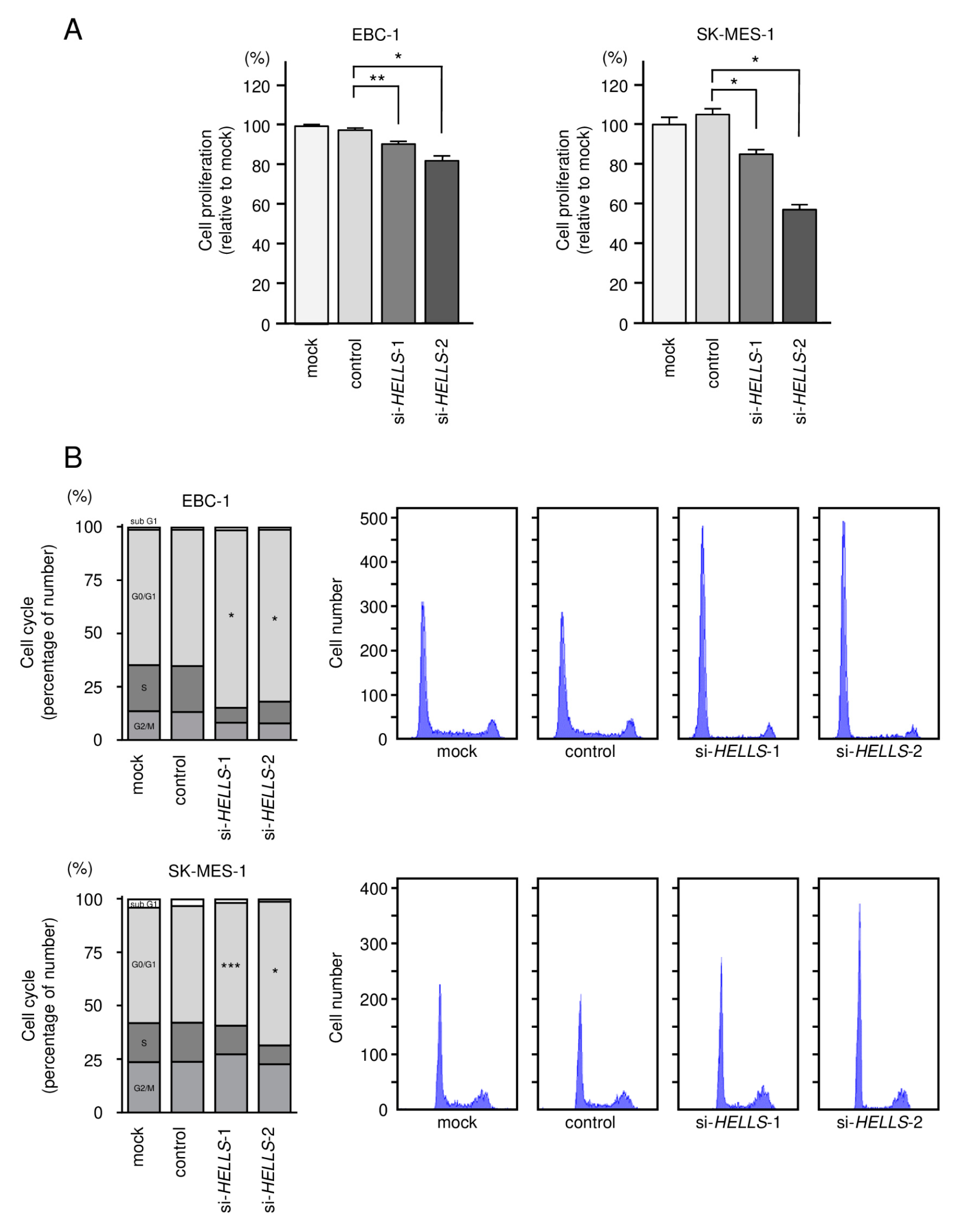
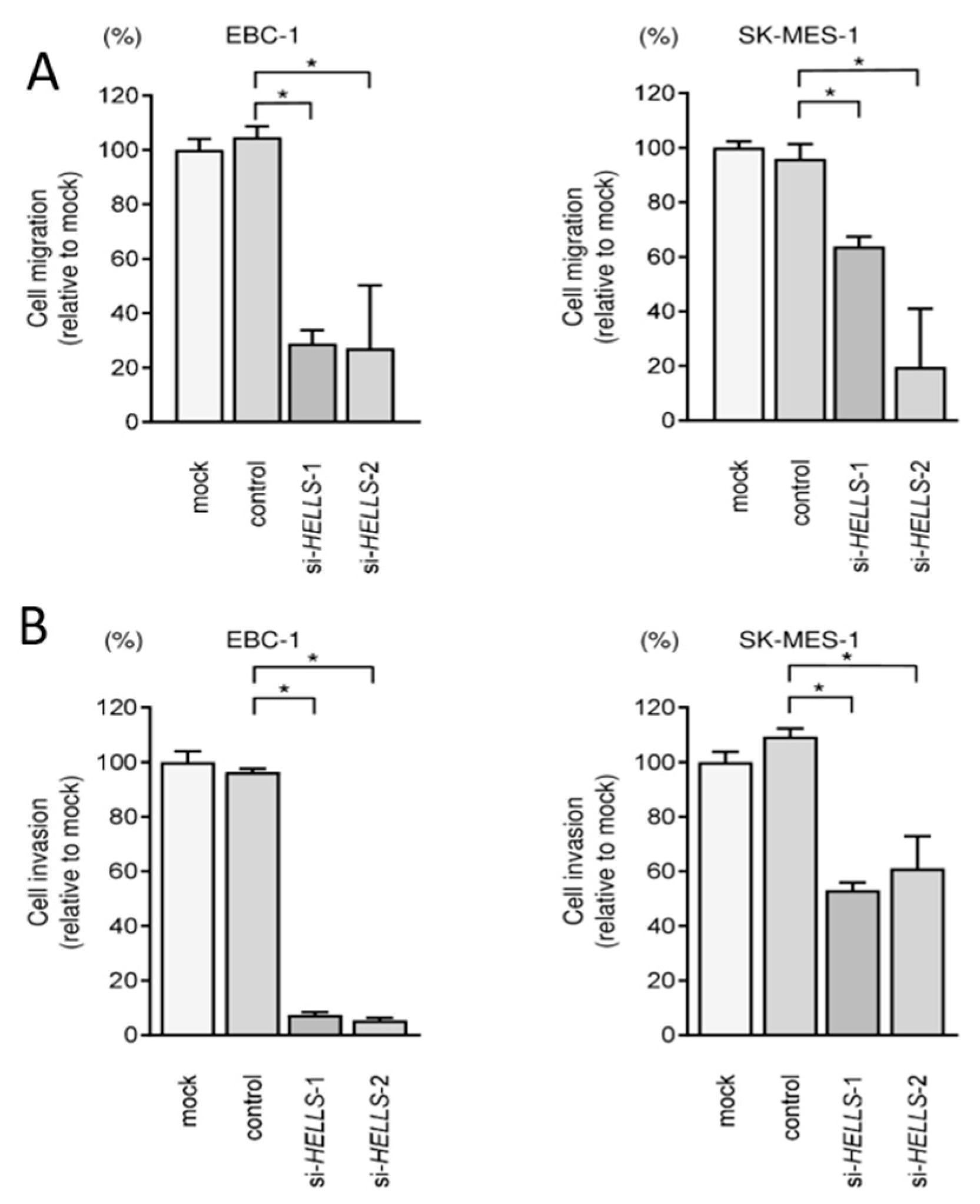
3.5. Effects of Knockdown of HELLS in LUSQ Cells
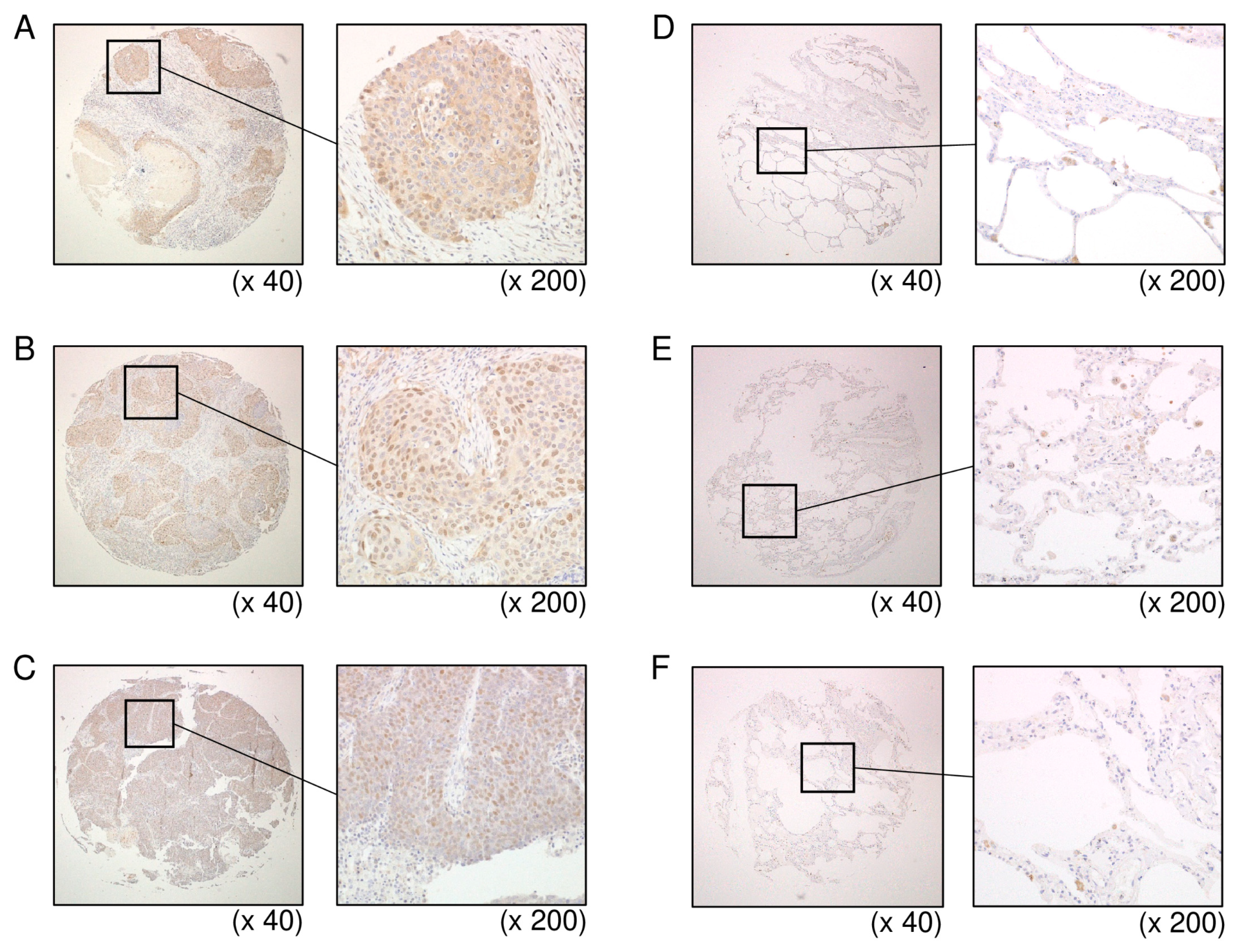
3.6. Overexpression of HELLS in LUSQ Clinical Specimens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Claus, R.; Plass, C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013, 73, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Ann. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [Green Version]
- Ramassone, A.; Pagotto, S.; Veronese, A.; Visone, R. Epigenetics and MicroRNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 459. [Google Scholar] [CrossRef] [Green Version]
- Ma, L. MicroRNA and metastasis. Adv. Cancer Res. 2016, 132, 165–207. [Google Scholar] [CrossRef]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-based nano-strategies as new therapeutic approach in multiple myeloma to overcome disease progression and drug resistance. Int. J. Mol. Sci. 2020, 21, 3084. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The risks of miRNA therapeutics: In a drug target perspective. Drug Des. Devel. Ther. 2021, 15, 721–733. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshizuka, K.; Nohata, N.; Hanazawa, T.; Kikkawa, N.; Arai, T.; Okato, A.; Fukumoto, I.; Katada, K.; Okamoto, Y.; Seki, N. Deep sequencing-based microRNA expression signatures in head and neck squamous cell carcinoma: Dual strands of pre-miR-150 as antitumor miRNAs. Oncotarget 2017, 8, 30288–30304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonemori, K.; Seki, N.; Idichi, T.; Kurahara, H.; Osako, Y.; Koshizuka, K.; Arai, T.; Okato, A.; Kita, Y.; Arigami, T.; et al. The microRNA expression signature of pancreatic ductal adenocarcinoma by RNA sequencing: Anti-tumour functions of the microRNA-216 cluster. Oncotarget 2017, 8, 70097–70115. [Google Scholar] [CrossRef] [Green Version]
- Toda, H.; Kurozumi, S.; Kijima, Y.; Idichi, T.; Shinden, Y.; Yamada, Y.; Arai, T.; Maemura, K.; Fujii, T.; Horiguchi, J.; et al. Molecular pathogenesis of triple-negative breast cancer based on microRNA expression signatures: Antitumor miR-204-5p targets AP1S3. J. Hum. Genet. 2018, 63, 1197–1210. [Google Scholar] [CrossRef]
- Wada, M.; Goto, Y.; Tanaka, T.; Okada, R.; Moriya, S.; Idichi, T.; Noda, M.; Sasaki, K.; Kita, Y.; Kurahara, H.; et al. RNA sequencing-based microRNA expression signature in esophageal squamous cell carcinoma: Oncogenic targets by antitumor miR-143-5p and miR-143-3p regulation. J. Hum. Genet. 2020, 65, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Sanada, H.; Seki, N.; Mizuno, K.; Misono, S.; Uchida, A.; Yamada, Y.; Moriya, S.; Kikkawa, N.; Machida, K.; Kumamoto, T.; et al. Involvement of dual strands of miR-143 (miR-143-5p and miR-143-3p) and their target oncogenes in the molecular pathogenesis of lung adenocarcinoma. Int. J. Mol. Sci. 2019, 20, 4482. [Google Scholar] [CrossRef] [Green Version]
- Misono, S.; Seki, N.; Mizuno, K.; Yamada, Y.; Uchida, A.; Arai, T.; Kumamoto, T.; Sanada, H.; Suetsugu, T.; Inoue, H. Dual strands of the miR-145 duplex (miR-145-5p and miR-145-3p) regulate oncogenes in lung adenocarcinoma pathogenesis. J. Hum. Genet. 2018, 63, 1015–1028. [Google Scholar] [CrossRef]
- Toda, H.; Seki, N.; Kurozumi, S.; Shinden, Y.; Yamada, Y.; Nohata, N.; Moriya, S.; Idichi, T.; Maemura, K.; Fujii, T.; et al. RNA-sequence-based microRNA expression signature in breast cancer: Tumor-suppressive miR-101-5p regulates molecular pathogenesis. Mol. Oncol. 2020, 14, 426–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshizuka, K.; Hanazawa, T.; Kikkawa, N.; Arai, T.; Okato, A.; Kurozumi, A.; Kato, M.; Katada, K.; Okamoto, Y.; Seki, N. Regulation of ITGA3 by the anti-tumor miR-199 family inhibits cancer cell migration and invasion in head and neck cancer. Cancer Sci. 2017, 108, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, T.; Koshizuka, K.; Seki, N.; Mizuno, K.; Okato, A.; Arai, T.; Misono, S.; Uchida, A.; Kumamoto, T.; Inoue, H. Downregulation of matrix metalloproteinase 14 by the antitumor miRNA, miR-150-5p, inhibits the aggressiveness of lung squamous cell carcinoma cells. Int. J. Oncol. 2018, 52, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Misono, S.; Seki, N.; Mizuno, K.; Yamada, Y.; Uchida, A.; Sanada, H.; Moriya, S.; Kikkawa, N.; Kumamoto, T.; Suetsugu, T.; et al. Molecular pathogenesis of gene regulation by the miR-150 duplex: miR-150-3p regulates TNS4 in lung adenocarcinoma. Cancers 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Okato, A.; Yamada, Y.; Sugawara, S.; Kurozumi, A.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Regulation of NCAPG by miR-99a-3p (passenger strand) inhibits cancer cell aggressiveness and is involved in CRPC. Cancer Med. 2018, 7, 1988–2002. [Google Scholar] [CrossRef]
- Lv, Y.; Yang, H.; Ma, X.; Wu, G. Strand-specific miR-28-3p and miR-28-5p have differential effects on nasopharyngeal cancer cells proliferation, apoptosis, migration and invasion. Cancer Cell Int. 2019, 19, 187. [Google Scholar] [CrossRef]
- Liu, X.; Ji, Q.; Zhang, C.; Liu, X.; Liu, Y.; Liu, N.; Sui, H.; Zhou, L.; Wang, S.; Li, Q. miR-30a acts as a tumor suppressor by double-targeting COX-2 and BCL9 in H. pylori gastric cancer models. Sci. Rep. 2017, 7, 7113. [Google Scholar] [CrossRef] [Green Version]
- Okada, R.; Goto, Y.; Yamada, Y.; Kato, M.; Asai, S.; Moriya, S.; Ichikawa, T.; Seki, N. Regulation of oncogenic targets by the tumor-suppressive miR-139 duplex (miR-139-5p and miR-139-3p) in renal cell carcinoma. Biomedicines 2020, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, K.; Hanazawa, T.; Kikkawa, N.; Katada, K.; Okato, A.; Arai, T.; Idichi, T.; Osako, Y.; Okamoto, Y.; Seki, N. Antitumor miR-150-5p and miR-150-3p inhibit cancer cell aggressiveness by targeting SPOCK1 in head and neck squamous cell carcinoma. Auris Nasus Larynx 2018, 45, 854–865. [Google Scholar] [CrossRef]
- Osako, Y.; Seki, N.; Koshizuka, K.; Okato, A.; Idichi, T.; Arai, T.; Omoto, I.; Sasaki, K.; Uchikado, Y.; Kita, Y.; et al. Regulation of SPOCK1 by dual strands of pre-miR-150 inhibit cancer cell migration and invasion in esophageal squamous cell carcinoma. J. Hum. Genet. 2017, 62, 935–944. [Google Scholar] [CrossRef]
- Dai, F.Q.; Li, C.R.; Fan, X.Q.; Tan, L.; Wang, R.T.; Jin, H. miR-150-5p inhibits non-small-cell lung cancer metastasis and recurrence by targeting HMGA2 and β-catenin signaling. Mol. Ther. Nucleic Acids. 2019, 16, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellon, M.; Lepelletier, Y.; Hermine, O.; Nicot, C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood 2009, 113, 4914–4917. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.Y.; Li, Y.J.; Zhang, S.; Li, Z.L.; Yue, Z.; Xie, N.; Xie, S.Y. Regulating A549 cells growth by ASO inhibiting miRNA expression. Mol. Cell. Biochem. 2010, 339, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, W.; Li, J.; Zhang, Y.; Zhang, X.; Xu, Y.; Hu, Y.; Li, Q.; Sun, Q.; Ma, Z. Higher expression of miR-150-5p promotes tumorigenesis by suppressing LKB1 in non-small cell lung cancer. Pathol Res. Pract. 2020, 216, 153145. [Google Scholar] [CrossRef]
- Wu, Q.; Chen, Y.F.; Fu, J.; You, Q.H.; Wang, S.M.; Huang, X.; Feng, X.J.; Zhang, S.H. Short hairpin RNA-mediated down-regulation of CENP-A attenuates the aggressive phenotype of lung adenocarcinoma cells. Cell. Oncol. 2014, 37, 399–407. [Google Scholar] [CrossRef]
- Hu, D.D.; Chen, H.L.; Lou, L.M.; Zhang, H.; Yang, G.L. SKA3 promotes lung adenocarcinoma metastasis through the EGFR-PI3K-Akt axis. Biosci Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yu, J.H.; Lu, Z.H.; Zhang, W. E2F2 induction in related to cell proliferation and poor prognosis in non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 10545–10554. [Google Scholar]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Brümmer, A.; Hausser, J. MicroRNA binding sites in the coding region of mRNAs: Extending the repertoire of post-transcriptional gene regulation. Bioessays 2014, 36, 617–626. [Google Scholar] [CrossRef]
- Lungu, C.; Muegge, K.; Jeltsch, A.; Jurkowska, R.Z. An ATPase-deficient variant of the SNF2 family member HELLS shows altered dynamics at pericentromeric heterochromatin. J. Mol. Biol. 2015, 427, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Law, C.T.; Wei, L.; Tsang, F.H.; Chan, C.Y.; Xu, I.M.; Lai, R.K.; Ho, D.W.; Lee, J.M.; Wong, C.C.; Ng, I.O.; et al. HELLS regulates chromatin remodeling and epigenetic silencing of multiple tumor suppressor genes in human hepatocellular carcinoma. Hepatology 2019, 69, 2013–2030. [Google Scholar] [CrossRef]
- Von Eyss, B.; Maaskola, J.; Memczak, S.; Möllmann, K.; Schuetz, A.; Loddenkemper, C.; Tanh, M.D.; Otto, A.; Muegge, K.; Heinemann, U.; et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012, 31, 972–985. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Yang, L.; Wang, K.; Zhou, Y.; Li, Q.; Kong, F.; Liu, X.; He, J. HELLS, a chromatin remodeler is highly expressed in pancreatic cancer and downregulation of it impairs tumor growth and sensitizes to cisplatin by reexpressing the tumor suppressor TGFBR3. Cancer Med. 2021, 10, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hou, X.; Zhou, Y.; Li, Q.; Kong, F.; Yan, S.; Lei, S.; Xiong, L.; He, J. Downregulation of the helicase lymphoid-specific (HELLS) gene impairs cell proliferation and induces cell cycle arrest in colorectal cancer cells. OncoTargets Ther. 2019, 12, 10153–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseem, A.; Ali, M.; Odell, E.W.; Fortune, F.; Teh, M.T. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral Oncol. 2010, 46, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entrez Gene ID | Gene Symbol | Gene Name | Total Sites | GSE19188 Log2 FC | EBC-1 miR-150-3p Transfectant Log2 FC | TCGA OncoLnc p-Value |
|---|---|---|---|---|---|---|
| 1058 | CENPA | centromere protein A | 1 | 3.49 | −1.56 | 0.097 |
| 83540 | NUF2 | NUF2 component of NDC80 kinetochore complex | 2 | 3.44 | −2.30 | 0.366 |
| 699 | BUB1 | BUB1 mitotic checkpoint serine/threonine kinase | 1 | 3.21 | −1.86 | 0.172 |
| 3070 | HELLS | helicase, lymphoid specific | 1 | 3.15 | −1.75 | 0.002 |
| 4173 | MCM4 | minichromosome maintenance complex component 4 | 1 | 3.13 | −2.07 | 0.315 |
| 150468 | CKAP2L | cytoskeleton associated protein 2 like | 2 | 3.03 | −1.89 | 0.700 |
| 9837 | GINS1 | GINS complex subunit 1 | 1 | 2.99 | −2.29 | 0.509 |
| 84951 | TNS4 | tensin 4 | 1 | 2.56 | −1.05 | 0.908 |
| 157313 | CDCA2 | cell division cycle associated 2 | 1 | 2.39 | −1.53 | 0.538 |
| 29028 | ATAD2 | ATPase family AAA domain containing 2 | 1 | 2.25 | −1.96 | 0.329 |
| 898 | CCNE1 | cyclin E1 | 1 | 2.23 | −1.51 | 0.255 |
| 57082 | CASC5 | cancer susceptibility candidate 5 | 1 | 2.23 | −1.91 | 0.377 |
| 55771 | PRR11 | proline rich 11 | 2 | 2.20 | −1.83 | 0.548 |
| 2146 | EZH2 | enhancer of zeste 2 polycomb repressive complex 2 subunit | 1 | 2.06 | −1.02 | 0.095 |
| 9134 | CCNE2 | cyclin E2 | 1 | 2.04 | −2.73 | 0.175 |
| 221150 | SKA3 | spindle and kinetochore associated complex subunit 3 | 1 | 2.01 | −2.53 | 0.616 |
| 84733 | CBX2 | chromobox 2 | 1 | 1.99 | −1.16 | 0.422 |
| 1870 | E2F2 | E2F transcription factor 2 | 1 | 1.70 | −2.87 | 0.584 |
| 8357 | HIST1H3H | histone cluster 1 H3 family member h | 1 | 1.70 | −2.14 | 0.251 |
| 84798 | C19orf48 | chromosome 19 open reading frame 48 | 1 | 1.68 | −1.35 | 0.159 |
| 8970 | HIST1H2BJ | histone cluster 1 H2B family member j | 1 | 1.66 | −1.08 | 0.451 |
| 8914 | TIMELESS | timeless circadian regulator | 1 | 1.65 | −2.16 | 0.433 |
| 79172 | CENPO | centromere protein O | 1 | 1.62 | −1.48 | 0.259 |
| 26586 | CKAP2 | cytoskeleton associated protein 2 | 1 | 1.54 | −1.20 | 0.454 |
| 94032 | CAMK2N2 | calcium/calmodulin dependent protein kinase II inhibitor 2 | 1 | 1.54 | −2.37 | 0.994 |
| 55055 | ZWILCH | zwilch kinetochore protein | 1 | 1.53 | −2.41 | 0.223 |
| 25886 | POC1A | POC1 centriolar protein A | 1 | 1.43 | −2.10 | 0.084 |
| 79723 | SUV39H2 | suppressor of variegation 3–9 homolog 2 | 1 | 1.39 | −1.58 | 0.261 |
| 81831 | NETO2 | neuropilin and tolloid like 2 | 2 | 1.38 | −1.85 | 0.379 |
| 8358 | HIST1H3B | histone cluster 1 H3 family member b | 1 | 1.36 | −2.24 | 0.127 |
| 5563 | PRKAA2 | protein kinase AMP-activated catalytic subunit alpha 2 | 1 | 1.32 | −1.05 | 0.0382 |
| 54830 | NUP62CL | nucleoporin 62 C-terminal like | 1 | 1.32 | −1.20 | 0.794 |
| 146956 | EME1 | essential meiotic structure-specific endonuclease 1 | 1 | 1.30 | −1.69 | 0.234 |
| 11113 | CIT | citron rho-interacting serine/threonine kinase | 1 | 1.28 | −2.49 | 0.553 |
| 201725 | C4orf46 | chromosome 4 open reading frame 46 | 1 | 1.28 | −2.39 | 0.249 |
| 126433 | FBXO27 | F-box protein 27 | 1 | 1.27 | −2.29 | 0.950 |
| 5738 | PTGFRN | prostaglandin F2 receptor inhibitor | 2 | 1.24 | −2.51 | 0.645 |
| 22824 | HSPA4L | heat shock protein family A (Hsp70) member 4 like | 1 | 1.24 | −1.02 | 0.889 |
| 140707 | BRI3BP | BRI3 binding protein | 1 | 1.23 | −1.87 | 0.829 |
| 80179 | MYO19 | myosin XIX | 1 | 1.19 | −1.50 | 0.333 |
| 2177 | FANCD2 | FA complementation group D2 | 1 | 1.15 | −1.54 | 0.403 |
| 9802 | DAZAP2 | DAZ associated protein 2 | 2 | 1.14 | −1.78 | 0.479 |
| 79071 | ELOVL6 | ELOVL fatty acid elongase 6 | 1 | 1.13 | −1.11 | 0.693 |
| 374655 | ZNF710 | zinc finger protein 710 | 1 | 1.11 | −1.20 | 0.0206 |
| 145282 | MIPOL1 | mirror-image polydactyly 1 | 1 | 1.11 | −1.03 | 0.254 |
| 10849 | CD3EAP | CD3e molecule associated protein | 1 | 1.10 | −1.28 | 0.267 |
| 150223 | YDJC | YdjC chitooligosaccharide deacetylase homolog | 1 | 1.09 | −1.49 | 0.352 |
| 80010 | RMI1 | RecQ mediated genome instability 1 | 1 | 1.01 | −1.26 | 0.621 |
| 26355 | FAM162A | family with sequence similarity 162 member A | 1 | 1.01 | −1.87 | 0.0475 |
| Description | Annotation | Number of Genes | p-Value | Genes |
|---|---|---|---|---|
| cell cycle | GO:0007049 | 17 | 9.64 × 10−12 | CENPA, CIT, CCNE1, CCNE2, TIMELESS, BUB1, MCM4, HELLS, SKA3, CDCA2, FANCD2, NUF2, E2F2, SUV39H2, CASC5, ZWILCH, CKAP2 |
| cell division | GO:0051301 | 12 | 1.36 × 10−8 | CENPA, CIT, CCNE1, CCNE2, TIMELESS, BUB1, HELLS, SKA3, CDCA2, NUF2, CASC5, ZWILCH |
| chromosome segregation | GO:0007059 | 5 | 0.000241047 | BUB1, SKA3, CDCA2, NUF2, CASC5 |
| chromatin organization | GO:0006325 | 7 | 0.00128717 | HIST1H3B, HIST1H3H, PRKAA2, EZH2, CBX2, SUV39H2, ATAD2 |
| DNA replication initiation | GO:0006270 | 3 | 0.00275067 | CCNE1, CCNE2, MCM4 |
| homologous chromosome pairing at meiosis | GO:0007129 | 3 | 0.0027733 | CCNE1, CCNE2, FANCD2 |
| CENP-A containing nucleosome assembly | GO:0034080 | 3 | 0.00566854 | CENPA, CENPO, CASC5 |
| negative regulation of gene expression, epigenetic | GO:0045814 | 3 | 0.00814561 | HIST1H3B, HIST1H3H, EZH2 |
| mitotic cytokinesis | GO:0000281 | 3 | 0.00864655 | CENPA, CIT, CKAP2 |
| regulation of gene silencing | GO:0060968 | 2 | 0.00873016 | HIST1H3B, HIST1H3H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizuno, K.; Tanigawa, K.; Misono, S.; Suetsugu, T.; Sanada, H.; Uchida, A.; Kawano, M.; Machida, K.; Asai, S.; Moriya, S.; et al. Regulation of Oncogenic Targets by Tumor-Suppressive miR-150-3p in Lung Squamous Cell Carcinoma. Biomedicines 2021, 9, 1883. https://doi.org/10.3390/biomedicines9121883
Mizuno K, Tanigawa K, Misono S, Suetsugu T, Sanada H, Uchida A, Kawano M, Machida K, Asai S, Moriya S, et al. Regulation of Oncogenic Targets by Tumor-Suppressive miR-150-3p in Lung Squamous Cell Carcinoma. Biomedicines. 2021; 9(12):1883. https://doi.org/10.3390/biomedicines9121883
Chicago/Turabian StyleMizuno, Keiko, Kengo Tanigawa, Shunsuke Misono, Takayuki Suetsugu, Hiroki Sanada, Akifumi Uchida, Minami Kawano, Kentaro Machida, Shunichi Asai, Shogo Moriya, and et al. 2021. "Regulation of Oncogenic Targets by Tumor-Suppressive miR-150-3p in Lung Squamous Cell Carcinoma" Biomedicines 9, no. 12: 1883. https://doi.org/10.3390/biomedicines9121883
APA StyleMizuno, K., Tanigawa, K., Misono, S., Suetsugu, T., Sanada, H., Uchida, A., Kawano, M., Machida, K., Asai, S., Moriya, S., Inoue, H., & Seki, N. (2021). Regulation of Oncogenic Targets by Tumor-Suppressive miR-150-3p in Lung Squamous Cell Carcinoma. Biomedicines, 9(12), 1883. https://doi.org/10.3390/biomedicines9121883