
Effects of Renal Denervation on the Enhanced Renal Vascular Responsiveness to Angiotensin II in High-Output Heart Failure: Angiotensin II Receptor Binding Assessment and Functional Studies in Ren-2 Transgenic Hypertensive Rats

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Heart Failure Model, Exclusion Criteria, and RDN Technique
2.3. Detailed Experimental Design
2.3.1. Series 1: Effects of RDN on RBF Responsiveness to ANG II
- (a)
- Sham-operated TGR (no ACF) + sham RDN
- (b)
- Sham-operated TGR + RDN
- (c)
- ACF TGR + sham RDN
- (d)
- ACF TGR + RDN
2.3.2. Series 2: Effects of RDN on Kidney ANG II Receptor Characterization Using Ligand Binding Studies
2.4. Preparation of Crude Membrane Fraction
2.5. ANG II Receptors Binding Assay
2.6. Statistical Analysis
2.7. Statement of Ethics
3. Results
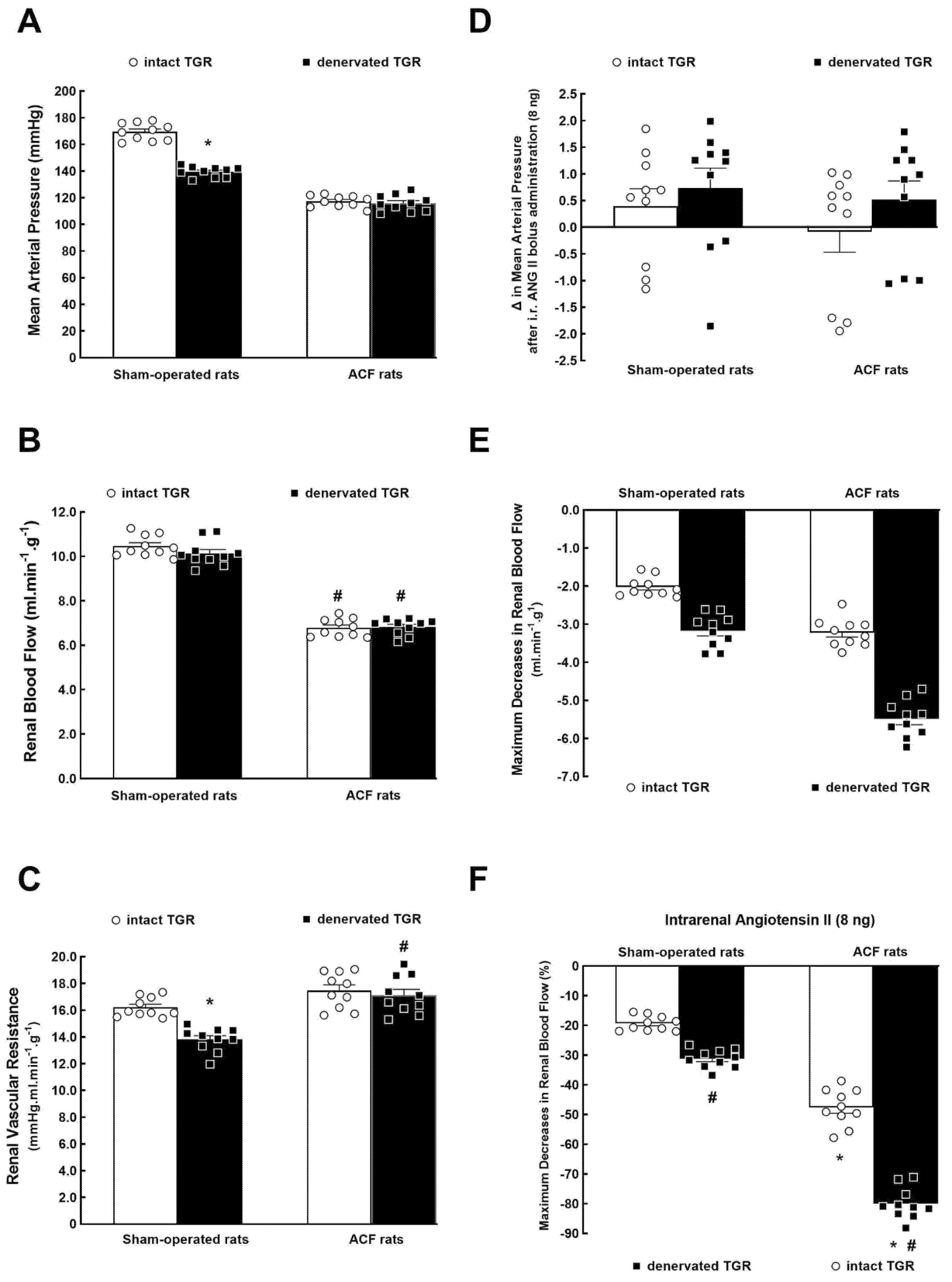
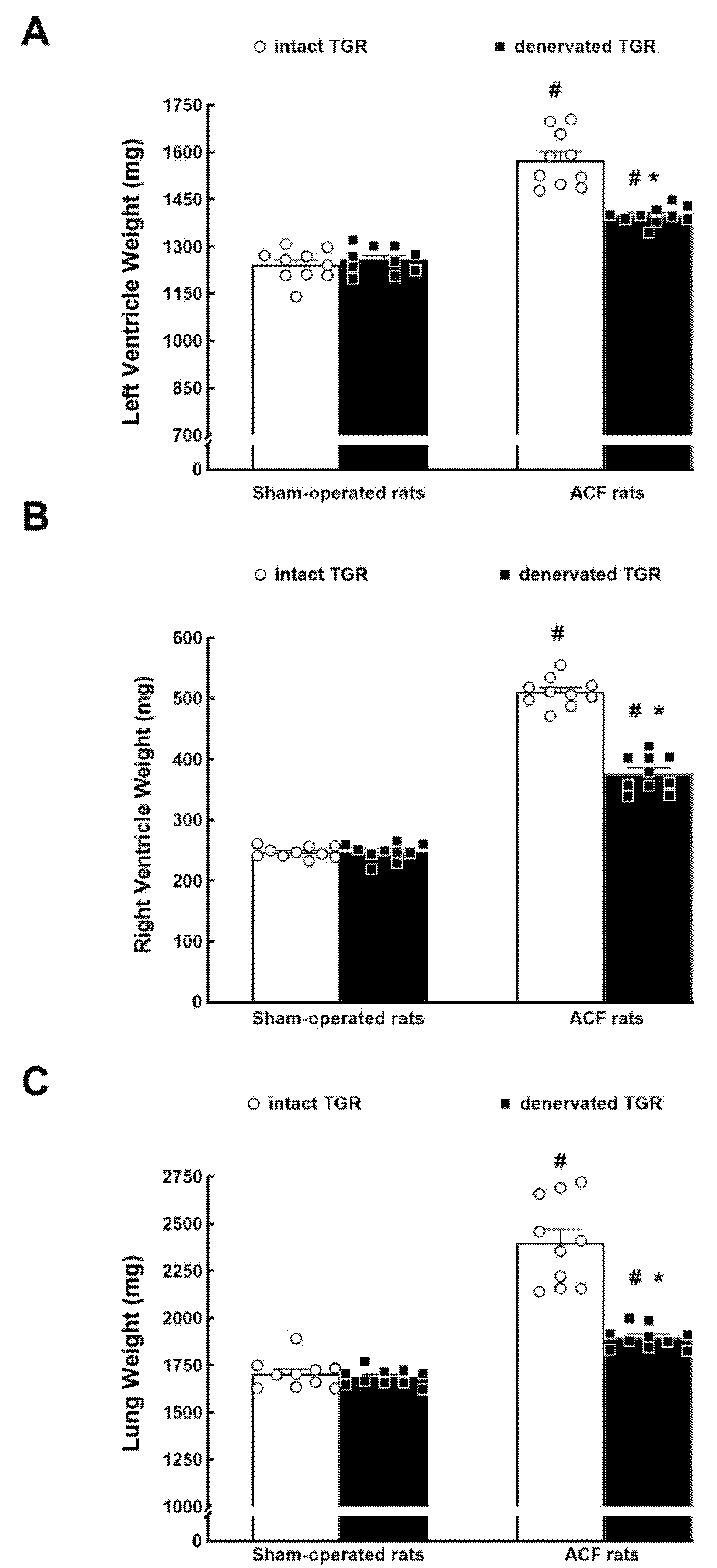
3.1. Series 1: Effects of RDN on Renal Vascular Responsiveness to ANG II
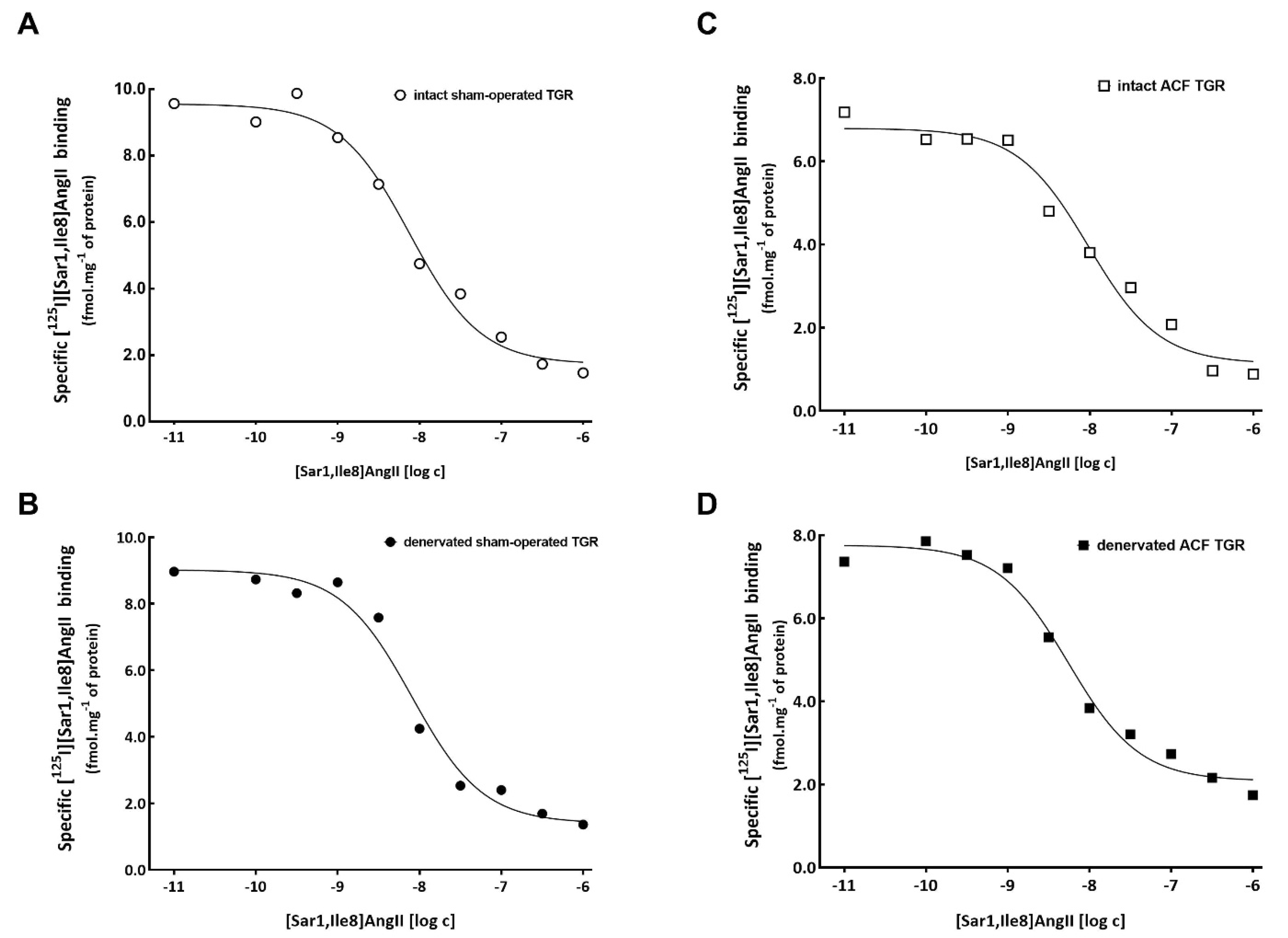
3.2. Series 2: Effects of RDN on Kidney ANG II Receptors Characterization Using Ligand Binding Studies
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2K1C | two-kidney: one clip Goldblatt hypertension |
| ACF | aorto-caval fistula |
| ANG II | angiotensin II |
| AT1 | angiotensin II type 1 receptor |
| AT2 | angiotensin II type 2 receptor |
| CKD | chronic kidney disease |
| FHH | Fawn-hooded hypertensive rat |
| GFR | glomerular filtration rate |
| HanSD | Hannover-Sprague transgene-negative rats |
| HF | heart failure |
| LV | left ventricle |
| MAP | mean arterial pressure |
| MI | myocardial infarction |
| NE | norepinephrine |
| RAS | renin-angiotensin system |
| RBF | renal blood flow |
| RDN | renal denervation |
| RSNA | renal sympathetic nerve activity |
| RV | right ventricle |
| RVR | renal vascular resistance |
| SNS | sympathetic nervous system |
| TGR | Ren-2 transgenic rat |
References
- Rangawwami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezeu, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal syndrome: Classification, pathophysiology, diagnosis, and treatment strategies. Circulation 2019, 139, e840–e878. [Google Scholar]
- Yogasundaram, H.; Chappell, M.C.; Braam, B.; Oudit, G.Y. Cardiorenal syndrome and heart failure-challenges and opportunities. Can. J. Cardiol. 2019, 35, 1208–1219. [Google Scholar] [CrossRef]
- Khayyat-Kholghi, M.; Oparil, S.; Davis, B.R.; Tereshchenko, L.G. Worsening kidney function is the major mechanism of heart failure in hypertension. The ALLHAT study. JACC Heart Fail. 2021, 9, 100–111. [Google Scholar] [CrossRef]
- Weber, M.A.; Mahfoud, F.; Schmieder, R.E.; Kandzari, D.E.; Tsioufis, K.P.; Twonsend, R.R.; Kario, K.; Bohm, M.; Sharp, A.S.P.; Davies, J.E.; et al. Renal denervation for treating hypertension: Current scientific and clinical evidence. JACC Cardiovasc. Interv. 2019, 12, 1095–1105. [Google Scholar] [CrossRef]
- Schmieder, R.E. Renal denervation: Where do we stand and what is the relevance to the nephrologist? Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef]
- Oluwaseun, A.; Ralston, W.F.; Johnson, K.C.; Ketron, L.L.; Womack, C.R.; Ibebuogu, U.N. Renal sympathetic denervation: A comprehenshive review. Curr. Probl. Cardiol. 2021, 46, 100598. [Google Scholar]
- Schmieder, R.E.; Mahfoud, F.; Mancia, G.; Azizi, M.; Bohm, M.; Dimitriadis, K.; Kario, K.; Kroon, A.A.; Lobo, M.D.; Ott, C.; et al. European Society of Hypertension position paper on renal denervation 2021. J. Hypertens. 2021, 39, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Kopp, U.C. Neural control of renal function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef]
- Osborn, J.W.; Tyshynsky, R.; Vulchanova, L. Function of renal nerves in kidney physiology an pathophysiology. Annu. Rev. Physiol. 2021, 83, 429–450. [Google Scholar] [CrossRef]
- Antoine, S.; Vaidya, G.; Imam, H.; Villarreal, D. Pathophysiologic mechanisms in heart failure: Role of the sympathetic nervous system. Am. J. Med. Sci. 2017, 353, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Florea, V.G.; Cohn, J.N. The autonomic nervous system and heart failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.E.; Lefer, D.J. Renal denervation to treat heart failure. Annu. Rev. Physiol. 2021, 83, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Rodinova, K.; Hindermann, M.; Hilgers, K.; Ot, C.; Schmieder, R.E.; Schiffer, M.; Amann, K.; Veelken, R.; Ditting, T. ANG II receptor blockade and renal denervation: Different interventions with comparable effects? Kidney Blood Press Res. 2021, 46, 331–341. [Google Scholar] [CrossRef]
- Kresoja, K.J.; Rommel, K.-P.; Fengler, K.; von Roeder, M.; Besler, C.; Lucke, C.; Gutberlet, M.; Desch, S.; Thiele, H.; Bohm, M.; et al. Renal sympathetic denervation in patients with heart failure with preserved ejection fraction. Circ. Heart Fail. 2021, 14, e007421. [Google Scholar] [CrossRef] [PubMed]
- Fudim, M.; Sobotka, P.A.; Piccini, J.P.; Patel, M.R. Renal denervation for patients with heart failure. Making a full circle. Circ. Heart Fail. 2021, 14, e008301. [Google Scholar] [CrossRef] [PubMed]
- Honetschlagerová, Z.; Škaroupková, P.; Kikerlová, S.; Husková, Z.; Maxová, H.; Melenovský, V.; Kompanowska-Jezierska, E.; Sadowski, J.; Gawrys, O.; Kujal, P.; et al. Effects of renal sympathetic denervation on the course of congestive heart failure combined with chronic kidney disease: Insight from studies with fawn-hooded hypertensive rats with volume overload induced using aorto-caval fistula. Clin. Exp. Hypertens. 2021, 43, 522–535. [Google Scholar] [CrossRef]
- Honetschlagerová, Z.; Škaroupková, P.; Kikerlová, S.; Vaňourková, Z.; Husková, Z.; Melenovský, V.; Kompanowska-Jezierska, E.; Sadowski, J.; Červenka, L. Renal sympathetic denervation attenuates congestive heart failure in angiotensin II-dependent hypertension: Studies with Ren-2 transgenic hypertensive rats with aortocaval fistula. Kidney Blood Press Res. 2021, 46, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Červenka, L.; Melenovský, V.; Husková, Z.; Škaroupková, P.; Nishiyama, A.; Sadowski, J. Inhibition of soluble epoxide hydrolase counteracts the development of renal dysfunction and progression of congestive heart failure in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Clin. Exp. Pharmacol. Physiol. 2015, 42, 795–807. [Google Scholar] [CrossRef]
- Kala, P.; Sedláková, L.; Škaroupková, P.; Kopkan, L.; Vaňourková, Z.; Táborský, M.; Nishiyama, A.; Hwang, S.H.; Hammock, B.D.; Sadowski, J.; et al. Effects of angiotensin-converting enzyme blockade, alone or combined with blockade of soluble epoxide hydrolase, on the course of congestive heart failure and occurrence of renal dysfunction in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Physiol. Res. 2018, 67, 401–415. [Google Scholar] [CrossRef]
- Kratky, V.; Kopkan, L.; Kikerlova, S.; Huskova, Z.; Taborsky, M.; Sadowski, J.; Kolar, F.; Cervenka, L. The role of renal vascular reactivity in the development of renal dysfunction in compensated and decompensated congestive heart failure. Kidney Blood Press Res. 2018, 43, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Krátký, V.; Kikerlová, S.; Husková, Z.; Sadowski, J.; Kolář, F.; Červenka, L. Enhanced renal vascular responsiveness to angiotensin II and norepinephrine: A unique feature of female rats with congestive heart failure. Kidney Blood Press Res. 2019, 44, 1128–1141. [Google Scholar] [CrossRef]
- Vacková, Š.; Kikerlová, S.; Melenovský, V.; Kolář, F.; Imig, J.D.; Kompanovska-Jezierska, E.; Sadowski, J.; Červenka, L. Altered renal vascular responsiveness in rats with angiotensin II-dependent hypertension and congestive heart failure. Kidney Blood Press Res. 2019, 44, 792–809. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Inscho, E.W.; Imig, J.D.; Michel, R.E.; Navar, L.G. Role of renal nerves in afferent arteriolar reactivity in angiotensin II-induced hypertension. Hypertension 1997, 29, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Osborn, J.W.; Foss, J.D. Renal nerves and long-term control of arterial pressure. Compr. Physiol. 2017, 7, 263–320. [Google Scholar] [PubMed]
- Li, X.C.; Widdop, R.E. AT2 receptor mediated vasodilatation is unmasked by AT1 receptor blockade in conscious SHR. Br. J. Pharmacol. 2004, 142, 821–830. [Google Scholar] [CrossRef]
- Carrey, R.M. Update on angiotensin AT2 receptors. Curr. Opin. Nehprol. Hypertens. 2017, 26, 91–96. [Google Scholar] [CrossRef][Green Version]
- Pandey, A.; Gaikwad, A.B. AT2 receptor agonist Compound 21: A silver lining for diabetic nephropathy. Eur. J. Pharmacol. 2017, 815, 251–257. [Google Scholar] [CrossRef]
- Kratky, V.; Vanourkova, Z.; Sykora, M.; Szeiffova Bacova, B.; Hruskova, Z.; Kikerlova, S.; Huskova, Z.; Kopkan, L. AT1 receptor blocker, but not an ACE inhibitor, prevents kidneys form hypoperfusion during congestive heart failure in normotensive and hypertensive rats. Sci. Rep. 2021, 11, 4271. [Google Scholar] [CrossRef]
- Bello-Reuss, E.; Colindres, R.E.; Pastoriza-Monuz, E.; Mueller, R.A.; Gottschalk, C.W. Effect of acute unilateral renal denervation in the rat. J. Clin. Investig. 1975, 56, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Kline, R.L.; Mercer, P.F. Functional reinnervation and development of supersensitivity to NE after renal denervation in rats. Am. J. Physiol. 1980, 238, R353–R358. [Google Scholar] [CrossRef] [PubMed]
- Krayacich, J.; Kline, R.L.; Mercer, P.F. Supersensitivity to NE alters renal function of chronically denervated rat kidneys. Am. J. Physiol. 1987, 252, F856–F864. [Google Scholar] [CrossRef] [PubMed]
- Chatziantoniou, C.; Daniels, F.H.; Arendhosrt, W.J. Exaggerated renal vascular reactivity to angiotensin and tromboxane in young genetically hypertensive rats. Am. J. Physiol. 1990, 259, F372–F382. [Google Scholar]
- Jacinto, S.M.; Mullins, J.J.; Mitchell, K.D. Enhanced renal vascular responsiveness to angiotensin II in hypertensive ren-2 transgenic rats. Am. J. Physiol. 1999, 276, F315–F322. [Google Scholar] [CrossRef]
- Kopkan, L.; Kramer, H.J.; Husková, Z.; Vaňourková, Z.; Škaroupková, P.; Thumová, M.; Červenka, L. The role of intrarenal angiotensin II in the development of hypertension in Ren-2 transgenic rats. J. Hypertens. 2005, 23, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Čertíková Chábová, V.; Walkowska, A.; Kompanowska-Jezierska, E.; Sadowski, J.; Kujal, P.; Verenrova, Z.; Vaňourková, Z.; Kopkan, L.; Kramer, H.J.; Falck, J.R.; et al. Combined inhibition of 20-hydroxyeicosatrienoic acid formation and epoxyeicosatrienoic dgradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin. Sci. 2010, 118, 617–632. [Google Scholar] [CrossRef]
- Amiri, F.; Garcia, R. Differential regulation of renal glomerular and preglomerular vascular angiotensin II receptors. Am. J. Physiol. 1996, 270, E810–E815. [Google Scholar] [CrossRef]
- Amiri, F.; Garcia, R. Renal angiotensin II receptor regulation in two-kidney, one-clip hypertensive rats. Effect of ACE inhibition. Hypertension 1997, 30, 337–344. [Google Scholar] [CrossRef]
- Chatziantoniou, C.; Arendshorst, W.J. Angiotensin receptor sites in renal vasculature of rats developing genetic hypertension. Am. J. Physiol. 1993, 265, F853–F862. [Google Scholar] [CrossRef] [PubMed]
- Mento, P.F.; Pica, M.E.; Hilepo, J.; Hirsch, L.; Wilkes, B.M. Increased expression of glomerular AT1 receptors in rats with myocardial infarction. Am. J. Physiol. 1998, 275, H1247–H1253. [Google Scholar] [CrossRef]
- Licea, H.; Walters, M.R.; Navar, L.G. Renal nuclear angiotensin II receptors in normal and hypertensive rats. Acta Physiol. Hung. 2002, 89, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Bernard, L.M.; Zhou, J.; Kobori, H.; Ohishi, M.; Navar, L.G. Intrarenal AT1 receptor and ACE binding in ANG II-induced hypertensive rats. Am. J. Physiol. 2002, 281, F19–F25. [Google Scholar] [CrossRef]
- Oliver-Dussault, C.; Ascah, A.; Marcil, M.; Matas, J.; Picard, S.; Pibarot, B.; Burelle, Y.; Deschepper, C.F. Early predictors of cardiac decompensation in experimental volume overload. Mol. Cell Biochem. 2010, 338, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Goltsmna, I.; Karram, T.; Winaver, J.; Horrman, A. Aortocaval fistula in rat: A unique model of volume-overload congestive heart failure and cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 729497. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An updated on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Harrison-Bernard, L.M.; Imig, J.D.; Cervenka, L.; Mitchell, K.D. Renal responses to AT1 receptor blockade. Am. J. Hypertens. 2000, 13, 45S–54S. [Google Scholar] [CrossRef][Green Version]
- Pieruzzi, F.; Abassi, Z.A.; Keiser, H.R. Expression of renin-angiotensin system components in the heart, kidneys, and lungs of rats with experimental heart failure. Circulation 1995, 92, 3105–3112. [Google Scholar] [CrossRef]
- Harrison-Bernard, L.M.; El-Dahr, S.S.; O´Leary, D.F.; Navar, L.G. Regulation of angiotensin II type 1 receptor mRNA and protein in angiotensin II-induced hypertension. Hypertension 1999, 33, 340–346. [Google Scholar] [CrossRef]
- Červenka, L.; Horáček, V.; Vaněčková, I.; Hubáček, J.A.; Oliverio, M.I.; Coffman, T.M.; Navar, L.G. Essentials role of AT1A receptor in the development of 2K1C Hypertension. Hypertension 2002, 40, 735–741. [Google Scholar] [CrossRef][Green Version]
- Červenka, L.; Vaněčková, I.; Malý, J.; Horáček, V.; El-Dahr, S.S. Genetic inactivation of the B2 receptor mice worsens two-kidney, one-clip hypertension: Role of NO and the AT2 receptor. J. Hypertens. 2003, 21, 1531–1538. [Google Scholar] [CrossRef]
- Červenka, L.; Vaněčková, I.; Husková, Z.; Vaňourková, Z.; Erbanová, M.; Thumová, M.; Škaroupková, P.; Opočenský, M.; Malý, J.; Čertíková Chábová, V.; et al. Pivotal role of angiotensin II receptor subtype 1A in the development of two-kidney, one-clip hypertension: Study in angiotensin II receptor subtype 1A knockout mice. J. Hypertens. 2008, 26, 1379–1389. [Google Scholar] [CrossRef]
- Clayton, S.C.; Haack, K.K.A.; Zucker, I.H. Renal denervation modulates angiotensin receptor expression in the renal cortex of rabbits with chronic heart failure. Am. J. Physiol. 2011, 300, F31–F39. [Google Scholar] [CrossRef]
- Mendelsohn, F.A.; Dunbar, M.; Allen, A.; Chou, S.T.; Milan, M.A.; Aguilera, G.; Catt, K.J. Angiotensin II receptors in the kidney. Fed. Proc. 1986, 45, 1420–1425. [Google Scholar] [PubMed]
- Kala, P.; Miklovič, M.; Jíchová, Š.; Škaroupková, P.; Vaňourková, Z.; Maxová, H.; Gawrys, O.; Kompanowska-Jezierska, E.; Sadowski, J.; Imig, J.D.; et al. Effects of epoxyeicoatrienoci acid-enhancing therapy on the course of congestive heart failure in angiotensin II-dependent rat hypertension: From mRNA analysis towards functional in vivo evaluation. Biomedicines 2021, 9, 1053. [Google Scholar] [CrossRef]
- Trendelenburg, U. Mechanisms of supersensitivity and subsensitivity to sympathomimetic amines. Pharmacol. Rev. 1966, 18, 629–640. [Google Scholar]
- Sadowski, J.; Portalska, E. Denervated and intact kidney responses to norepinephrine infusion in conscious dogs. J. Auton. Nerv. Syst. 1982, 6, 373–379. [Google Scholar] [CrossRef]
- Szenasi, G.; Bencsath, P.; Takacs, L. Supersensitivity of the renal tubule to catecholamines in chronically denervated canine kidney. Pflugers. Arch. 1986, 406, 57–59. [Google Scholar] [CrossRef]
- Lohmeier, T.; Reinhart, G.A.; Mizelle, L.; Han, M.; Dean, M.M. Renal denervation supersensitivity revisited. Am. J. Physiol. 1998, 275, R1239–R1246. [Google Scholar] [CrossRef]
- Ramcharda, R.; Barrett, C.J.; Guild, S.-J.; Malpes, S. Is the chronically denervated kidney supersensitive to catecholamines? Am. J. Physiol. 2002, 282, R603–R610. [Google Scholar]
- Booth, L.C.; de Silva, R.A.U.; Pontes, R.B.; Yano, S.T.; Hood, S.G.; Lankadeva, Y.R.; Kosaka, J.; Eikelis, N.; Lambert, G.W.; Schlaich, M.P.; et al. Renal, cardiac, and autonomic effects of cathether-based renal denervation in ovine heart failure. Hypertension 2021, 78, 706–715. [Google Scholar] [CrossRef]
- Singh, R.R.; McArdle, Z.M.; Booth, L.C.; May, C.N.; Moritz, K.M.; Schlaich, M.P.; Denton, K.M. Increase in bioavailability of nitric oxide after renal denervation improves kidney function in sheep with hypertensive kidney disease. Hypertension 2021, 77, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, X.; Katsurada, K.; Patel, K.P. Renal denervation improves sodium excretion in rats with chronic heart failure: Effects on expression of renal ENaC and AQP2. Am. J. Physiol. 2019, 317, H958–H968. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, X.; Rao, U.S.; Patel, K.P. Increased renal ENaC subunits and sodium retention in rats with chronic heart failure. Am. J. Physiol. 2011, 300, F641–F649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, X.; Sharma, N.M.; Li, Y.; Pliquett, R.U.; Patel, K.P. Urinary proteolyitic activation of renal epithelial Na+ channels in chronic heart failure. Hypertension 2016, 67, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Giebisch, G.H. A long affair with renal tubules. Annu Rev. Physiol. 2011, 73, 1–28. [Google Scholar] [CrossRef]
- Wang, H.-J.; Wang, W.; Cornish, K.G.; Rozanski, G.J.; Zucker, I.H. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 2014, 64, 745–755. [Google Scholar] [CrossRef]
- Lopes, N.R.; Milanez, M.I.O.; Martins, B.S.; Veiga, C.A.; Ferreira, G.R.; Gomes, G.N.; Girardi, A.C.; Carvalho, P.M.; Noqueira, F.N.; Campos, R.R.; et al. Afferent innervation of the ischemic kidney contributes to renal dysfunction in renovascular hypertensive rats. Pfluger. Arch.-Eur. J. Physiol. 2020, 472, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Sata, Y.; Burke, L.S.; Eikelis, N.; Watson, A.M.D.; Gueguen, C.; Jackoson, K.L.; Lambert, G.W.; Lim, K.; Denton, K.M.; Schlaich, M.P.; et al. Renal deafferentation prevents progression of hypertension and changes to sympathetic reflexes in a rabbit model of chronic kidney disease. Hypertension 2021, 78, 1310–1321. [Google Scholar] [CrossRef]
- Foss, J.D.; Wainford, R.D.; Engeland, W.C.; Fink, G.D.; Osborn, J.W. A novel method of selective ablation of afferent renal nerves by periaxonal application of capsaicin. Am. J. Physiol. 2015, 308, R112–R122. [Google Scholar] [CrossRef]
- Cheng, H.F.; Becker, B.N.; Burns, K.D.; Harris, R.C. Angiotensin II upregulates type-1 angiotensin II receptors in renal proximal tubule. J. Clin. Investig. 1995, 95, 2012–2019. [Google Scholar] [CrossRef]
- Douglas, J.G. Angiotensin receptor subtypes of the kidney cortex. Am. J. Physiol. 1987, 253, F1–F7. [Google Scholar] [CrossRef]
- Mendelsohn, F.A.O.; Millan, M.; Quirion, R.; Aguilera, G.; Chou, S.-T.; Catt, K.J. Localization of angiotensin II receptors in rat and monkey kidney by in vitro autoradiography. Kidney Int. 1987, 31 (Suppl. 20), S40–S44. [Google Scholar]
- Zhou, J.; Song, K.; Harris, P.J.; Mendelsohn, F.A.O. In vitro autoradiography revelas predominantly AT1 angiotensin II receptors in rat kidney. Renal Physiol. Biochem. 1992, 15, 231–239. [Google Scholar]
- Miyata, N.; Park, F.; Li, X.F.; Cowley, A.W., Jr. Distribution of angiotensin AT1 and AT2 receptors subtypes in the rat kidney. Am. J. Physiol. 1999, 277, F437–F446. [Google Scholar] [CrossRef]
- Husková, Z.; Kramer, H.J.; Vaňourková, Z.; Červenka, L. Effect of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J. Hypertens. 2006, 24, 517–522. [Google Scholar] [CrossRef]
- Husková, Z.; Kramer, H.J.; Thumová, M.; Vaňourková, Z.; Burgelová, M.; Teplan, V.; Malý, J.; Červenka, L. Effects of anaesthesia on plasma and kidney ANG II levels in normotensive and ANG II-dependent hypertensive rats. Kidney Blood Press Res. 2006, 29, 74–83. [Google Scholar] [CrossRef]
- Mullins, J.J.; Peters, J.; Ganten, D. Fulminant hypertension in transgenic rats harboring the mouse Ren-2 renin gene. Nature 1990, 344, 541–544. [Google Scholar] [CrossRef]
- Langheririch, M.; Lee, M.A.; Bohm, M.; Pinto, Y.M.; Ganten, D.; Paul, M. The hypertensive Ren-2 transgenic rat TGR(mRen2)27 in hypertension research. Characterizatics and functional aspects. Am. J. Hypertens. 1996, 9, 506–512. [Google Scholar] [CrossRef]
- Peters, J.; Hilgers, K.F.; Maser-Gluth, C.; Kreutz, R. Role of the circulating renin-angiotensin system in the pathogenesis of hypertension in transgenic rats TGR(mRen2)27. Clin. Exp. Hypertens. 1996, 18, 933–948. [Google Scholar] [CrossRef]
- Rong, P.; Campbell, D.J.; Skinner, S.L. Hypertension in the (mRen2)27 rat is not explained by enhanced kinetics of transgenic Ren-2 renin. Hypertension 2003, 42, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Husková, Z.; Kramer, H.J.; Vaňourková, Z.; Thumová, M.; Malý, J.; Opočenský, M.; Škaroupková, P.; Vernerová, Z.; Červenka, L. Effects of dietary salt load and salt depletion on the course of hypertension and angiotensin II levels in male and female heterozygous Ren-2 transgenic rats. Kidney Blood Press Res. 2007, 30, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.A.; Bohm, M.; Bader, M.; Ganten, U.; Ganten, D. Physiological characterization of the hypertensive transgenic rat TGR(mRen2)27. Am. J. Physiol. 1996, 270, E919–E929. [Google Scholar] [CrossRef]
- Mitchell, K.D.; Mullins, J.J. ANG II dependence of tubuloglomerular feedback responsiveness in hypertensive ren-2 transgenic rats. Am. J. Physiol. 1995, 268, F821–F828. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.D.; Jacinto, S.M.; Mullins, J.J. Proximal tubular fluid, kidney, and plasma levels of angitotensin II in hypertensive ren-2 transgenic rats. Am. J. Physiol. 1997, 273, F246–F253. [Google Scholar]
- Dube, P.; Weber, K.T. Congestive heart failure: Pathophysiologic consequences of neurohormonal activation and the potential for recovery: Part I. Am. J. Med. Sci. 2011, 342, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; Felker, G.M. Mechanisms and models in heart failure. A translation approach. Cir. Res. 2021, 128, 1435–1450. [Google Scholar] [CrossRef]
- Roger, V.L. Epidemiology of heart failure. A contemporary perspective. Cir. Res. 2021, 128, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cheng, Z.; Zhang, M.; Zhu, P.; Gu, Y. Impact of aortocaval shunt flow on cardiac and renal function in unilateral nephrectomized rats. Sci. Rep. 2016, 6, 27493. [Google Scholar] [CrossRef]
- Fiksen-Olson, M.J.; Strick, D.M.; Hawley, H.; Romero, J.C. Renal effects of angiotensin II inhibition during increases in renal venous pressure. Hypertension 1992, 19 (Suppl. II), II137–II141. [Google Scholar] [CrossRef]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef]
- Riehle, C.; Bauersachs, J. Small animals models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sham-Operated TGR + Intact | Sham-Operated TGR + RDN | ACF TGR + Intact | ACF TGR + RDN | |
|---|---|---|---|---|
| Bmax (fmol mg−1 of protein) | 22 ± 4 | 19 ± 3 | 18 ± 3 | 17 ± 4 |
| KD (nM) | 3.5 ± 0.8 | 4.2 ± 0.9 | 3.2 ± 0.9 | 3.9 ± 0.8 |
| Sham-Operated TGR + Intact | Sham-Operated TGR + RDN | ACF TGR + Intact | ACF TGR + RDN | |
|---|---|---|---|---|
| % of specific [125I][Sar1,Ile8]Ang II binding | 98 ± 7 | 93 ± 5 | 103 ± 11 | 92 ± 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honetschlägerová, Z.; Hejnová, L.; Novotný, J.; Marek, A.; Červenka, L. Effects of Renal Denervation on the Enhanced Renal Vascular Responsiveness to Angiotensin II in High-Output Heart Failure: Angiotensin II Receptor Binding Assessment and Functional Studies in Ren-2 Transgenic Hypertensive Rats. Biomedicines 2021, 9, 1803. https://doi.org/10.3390/biomedicines9121803
Honetschlägerová Z, Hejnová L, Novotný J, Marek A, Červenka L. Effects of Renal Denervation on the Enhanced Renal Vascular Responsiveness to Angiotensin II in High-Output Heart Failure: Angiotensin II Receptor Binding Assessment and Functional Studies in Ren-2 Transgenic Hypertensive Rats. Biomedicines. 2021; 9(12):1803. https://doi.org/10.3390/biomedicines9121803
Chicago/Turabian StyleHonetschlägerová, Zuzana, Lucie Hejnová, Jiří Novotný, Aleš Marek, and Luděk Červenka. 2021. "Effects of Renal Denervation on the Enhanced Renal Vascular Responsiveness to Angiotensin II in High-Output Heart Failure: Angiotensin II Receptor Binding Assessment and Functional Studies in Ren-2 Transgenic Hypertensive Rats" Biomedicines 9, no. 12: 1803. https://doi.org/10.3390/biomedicines9121803
APA StyleHonetschlägerová, Z., Hejnová, L., Novotný, J., Marek, A., & Červenka, L. (2021). Effects of Renal Denervation on the Enhanced Renal Vascular Responsiveness to Angiotensin II in High-Output Heart Failure: Angiotensin II Receptor Binding Assessment and Functional Studies in Ren-2 Transgenic Hypertensive Rats. Biomedicines, 9(12), 1803. https://doi.org/10.3390/biomedicines9121803