
TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter

 ,
,  and
and
Abstract
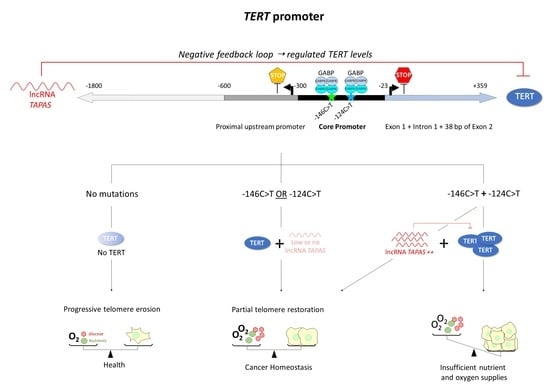
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Construction of TERT Promoter-Luciferase Reporter Expression Vectors
2.3. Transfection of Cells
2.4. RNA Extraction and RT-qPCR
2.5. Statistical Analyses
3. Results
3.1. Regulation of Sense and Antisense Transcription from the TERT Promoter
3.1.1. Regulation of Sense Expression from the TERT Promoter
3.1.2. Regulation of Antisense Expression from the TERT Promoter
3.2. TERT Promoter Mutations −146C>T and −124C>T Increase TERT Transcription in an Additive Fashion
3.3. TERT Promoter Mutations Increase Antisense Transcription from the TERT Promoter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Velasco-Herrera Mdel, C.; Hayward, N.K.; Adams, D.J. Telomere-regulating genes and the telomere interactome in familial cancers. Mol. Cancer Res. 2015, 13, 211–222. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Leao, R.; Apolonio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–10118. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.E.; Wright, W.E.; Shay, J.W. Regulation of telomerase activity in immortal cell lines. Mol. Cell. Biol. 1996, 16, 2932–2939. [Google Scholar] [CrossRef]
- Yasumoto, S.; Kunimura, C.; Kikuchi, K.; Tahara, H.; Ohji, H.; Yamamoto, H.; Ide, T.; Utakoji, T. Telomerase activity in normal human epithelial cells. Oncogene 1996, 13, 433–439. [Google Scholar]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Renaud, S.; Bosman, F.T.; Benhattar, J. Implication of the exon region in the regulation of the human telomerase reverse transcriptase gene promoter. Biochem. Biophys. Res. Commun. 2003, 300, 47–54. [Google Scholar] [CrossRef]
- Renaud, S.; Loukinov, D.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005, 33, 6850–6860. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Abdullaev, Z.; Guilleret, I.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007, 35, 1245–1256. [Google Scholar] [CrossRef]
- Rowland, T.J.; Bonham, A.J.; Cech, T.R. Allele-specific proximal promoter hypomethylation of the telomerase reverse transcriptase gene (TERT) associates with TERT expression in multiple cancers. Mol. Oncol. 2020, 14, 2358–2374. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef]
- Malhotra, S.; Freeberg, M.A.; Winans, S.J.; Taylor, J.; Beemon, K.L. A Novel Long Non-Coding RNA in the hTERT Promoter Region Regulates hTERT Expression. Noncoding RNA 2017, 4, 1. [Google Scholar] [CrossRef]
- Hafezi, F.; Perez Bercoff, D. The Solo Play of TERT Promoter Mutations. Cells 2020, 9, 749. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, G.; Shan, Y.; Hartmann, C.; von Deimling, A.; Xing, M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle 2013, 12, 1637–1638. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Han, S.; Meng, L.; Li, Z.; Zhang, X.; Wu, A. TERT promoter mutations lead to high transcriptional activity under hypoxia and temozolomide treatment and predict poor prognosis in gliomas. PLoS ONE 2014, 9, e100297. [Google Scholar] [CrossRef]
- Park, C.K.; Lee, S.H.; Kim, J.Y.; Kim, J.E.; Kim, T.M.; Lee, S.T.; Choi, S.H.; Park, S.H.; Kim, I.H. Expression level of hTERT is regulated by somatic mutation and common single nucleotide polymorphism at promoter region in glioblastoma. Oncotarget 2014, 5, 3399–3407. [Google Scholar] [CrossRef][Green Version]
- Huang, F.W.; Bielski, C.M.; Rinne, M.L.; Hahn, W.C.; Sellers, W.R.; Stegmeier, F.; Garraway, L.A.; Kryukov, G.V. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis 2015, 4, e176. [Google Scholar] [CrossRef]
- Spiegl-Kreinecker, S.; Lotsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro Oncol. 2015, 17, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef]
- Arita, H.; Narita, Y.; Takami, H.; Fukushima, S.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Shibui, S.; Ichimura, K. TERT promoter mutations rather than methylation are the main mechanism for TERT upregulation in adult gliomas. Acta Neuropathol. 2013, 126, 939–941. [Google Scholar] [CrossRef]
- Johanns, T.M.; Fu, Y.; Kobayashi, D.K.; Mei, Y.; Dunn, I.F.; Mao, D.D.; Kim, A.H.; Dunn, G.P. High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016, 33, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S.; Wang, Z.; He, X.J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.Y.; Wang, W.; Jiang, X.T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro Oncol. 2015, 17, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Yamasaki, K.; Matsushita, Y.; Nakamura, T.; Shimokawa, A.; Takami, H.; Tanaka, S.; Mukasa, A.; Shirahata, M.; Shimizu, S.; et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol. Commun. 2016, 4, 79. [Google Scholar] [CrossRef]
- Yang, P.; Cai, J.; Yan, W.; Zhang, W.; Wang, Y.; Chen, B.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. Classification based on mutations of TERT promoter and IDH characterizes subtypes in grade II/III gliomas. Neuro Oncol. 2016, 18, 1099–1108. [Google Scholar] [CrossRef]
- Mosrati, M.A.; Malmstrom, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.L.; Bratthall, C.; Strandeus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663–16673. [Google Scholar] [CrossRef]
- Batista, R.; Cruvinel-Carloni, A.; Vinagre, J.; Peixoto, J.; Catarino, T.A.; Campanella, N.C.; Menezes, W.; Becker, A.P.; de Almeida, G.C.; Matsushita, M.M.; et al. The prognostic impact of TERT promoter mutations in glioblastomas is modified by the rs2853669 single nucleotide polymorphism. Int. J. Cancer 2016, 139, 414–423. [Google Scholar] [CrossRef]
- Williams, E.A.; Miller, J.J.; Tummala, S.S.; Penson, T.; Iafrate, A.J.; Juratli, T.A.; Cahill, D.P. TERT promoter wild-type glioblastomas show distinct clinical features and frequent PI3K pathway mutations. Acta Neuropathol. Commun. 2018, 6, 106. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Moller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Schilling, B.; Scholz, S.; Sucker, A.; Song, M.; Susskind, D.; Grabellus, F.; Zimmer, L.; Hillen, U.; et al. TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br. J. Cancer 2013, 109, 497–501. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Garcia-Casado, Z.; Requena, C.; Traves, V.; Becker, J.; Soufir, N.; Hemminki, K.; Kumar, R. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat. Commun. 2014, 5, 3401. [Google Scholar] [CrossRef]
- Populo, H.; Boaventura, P.; Vinagre, J.; Batista, R.; Mendes, A.; Caldas, R.; Pardal, J.; Azevedo, F.; Honavar, M.; Guimaraes, I.; et al. TERT promoter mutations in skin cancer: The effects of sun exposure and X-irradiation. J. Investig. Dermatol. 2014, 134, 2251–2257. [Google Scholar] [CrossRef]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Ofner, R.; Ritter, C.; Heidenreich, B.; Kumar, R.; Ugurel, S.; Schrama, D.; Becker, J.C. Distribution of TERT promoter mutations in primary and metastatic melanomas in Austrian patients. J. Cancer Res. Clin. Oncol. 2017, 143, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Andres-Lencina, J.J.; Rachakonda, S.; Garcia-Casado, Z.; Srinivas, N.; Skorokhod, A.; Requena, C.; Soriano, V.; Kumar, R.; Nagore, E. TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int. J. Cancer 2018, 144, 1027–1036. [Google Scholar] [CrossRef]
- Nguyen, D.; Taheri, D.; Springer, S.; Cowan, M.; Guner, G.; Mendoza Rodriguez, M.A.; Wang, Y.; Kinde, I.; Vanden Bussche, C.J.; Olson, M.T.; et al. High prevalence of TERT promoter mutations in micropapillary urothelial carcinoma. Virchows Arch. 2016, 469, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Cowan, M.; Springer, S.; Nguyen, D.; Taheri, D.; Guner, G.; Rodriguez, M.A.; Wang, Y.; Kinde, I.; VandenBussche, C.J.; Olson, M.T.; et al. High prevalence of TERT promoter mutations in primary squamous cell carcinoma of the urinary bladder. Mod. Pathol. 2016, 29, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Allory, Y.; Beukers, W.; Sagrera, A.; Flandez, M.; Marques, M.; Marquez, M.; van der Keur, K.A.; Dyrskjot, L.; Lurkin, I.; Vermeij, M.; et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: High frequency across stages, detection in urine, and lack of association with outcome. Eur. Urol. 2014, 65, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jeng, Y.M.; Chang, C.N.; Lee, H.J.; Hsu, H.C.; Lai, P.L.; Yuan, R.H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Liu, T.; Wang, N.; Cao, J.; Sofiadis, A.; Dinets, A.; Zedenius, J.; Larsson, C.; Xu, D. The age- and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene 2014, 33, 4978–4984. [Google Scholar] [CrossRef]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metab. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef]
- Muzza, M.; Colombo, C.; Rossi, S.; Tosi, D.; Cirello, V.; Perrino, M.; De Leo, S.; Magnani, E.; Pignatti, E.; Vigo, B.; et al. Telomerase in differentiated thyroid cancer: Promoter mutations, expression and localization. Mol. Cell. Endocrinol. 2015, 399, 288–295. [Google Scholar] [CrossRef]
- George, J.R.; Henderson, Y.C.; Williams, M.D.; Roberts, D.B.; Hei, H.; Lai, S.Y.; Clayman, G.L. Association of TERT Promoter Mutation, But Not BRAF Mutation, With Increased Mortality in PTC. J. Clin. Endocrinol. Metab. 2015, 100, E1550–E1559. [Google Scholar] [CrossRef]
- Shi, X.; Liu, R.; Qu, S.; Zhu, G.; Bishop, J.; Liu, X.; Sun, H.; Shan, Z.; Wang, E.; Luo, Y.; et al. Association of TERT promoter mutation 1,295,228 C>T with BRAF V600E mutation, older patient age, and distant metastasis in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2015, 100, E632–E637. [Google Scholar] [CrossRef]
- Bae, J.S.; Kim, Y.; Jeon, S.; Kim, S.H.; Kim, T.J.; Lee, S.; Kim, M.H.; Lim, D.J.; Lee, Y.S.; Jung, C.K. Clinical utility of TERT promoter mutations and ALK rearrangement in thyroid cancer patients with a high prevalence of the BRAF V600E mutation. Diagn. Pathol. 2016, 11, 21. [Google Scholar] [CrossRef]
- Song, Y.S.; Lim, J.A.; Choi, H.; Won, J.K.; Moon, J.H.; Cho, S.W.; Lee, K.E.; Park, Y.J.; Yi, K.H.; Park, D.J.; et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016, 122, 1370–1379. [Google Scholar] [CrossRef]
- Vinothkumar, V.; Arunkumar, G.; Revathidevi, S.; Arun, K.; Manikandan, M.; Rao, A.K.; Rajkumar, K.S.; Ajay, C.; Rajaraman, R.; Ramani, R.; et al. TERT promoter hot spot mutations are frequent in Indian cervical and oral squamous cell carcinomas. Tumour Biol. 2016, 37, 7907–7913. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.; Pezzuto, F.; Greggi, S.; Ionna, F.; Losito, S.; Botti, G.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Distinct profiles of TERT promoter mutations and telomerase expression in head and neck cancer and cervical carcinoma. Int. J. Cancer 2018, 143, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Xavier-Magalhaes, A.; Woods, W.S.; Nguyen, K.T.; Amen, A.M.; Hayes, J.L.; Fellmann, C.; Gapinske, M.; McKinney, A.M.; Hong, C.; et al. Disruption of the beta1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell 2018, 34, 513–528.e8. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.C.; Brown, T.A.; McKnight, S.L. Convergence of Ets- and notch-related structural motifs in a heteromeric DNA binding complex. Science 1991, 253, 762–768. [Google Scholar] [CrossRef]
- Oikawa, T.; Yamada, T. Molecular biology of the Ets family of transcription factors. Gene 2003, 303, 11–34. [Google Scholar] [CrossRef]
- La Marco, K.; Thompson, C.C.; Byers, B.P.; Walton, E.M.; McKnight, S.L. Identification of Ets- and notch-related subunits in GA binding protein. Science 1991, 253, 789–792. [Google Scholar] [CrossRef]
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 2015, 29, 2219–2224. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Gaspar, T.B.; Sa, A.; Lopes, J.M.; Sobrinho-Simoes, M.; Soares, P.; Vinagre, J. Telomere Maintenance Mechanisms in Cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- De Wilde, J.; Kooter, J.M.; Overmeer, R.M.; Claassen-Kramer, D.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer 2010, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Hosen, I.; Rachakonda, P.S.; Heidenreich, B.; de Verdier, P.J.; Ryk, C.; Steineck, G.; Hemminki, K.; Kumar, R. Mutations in TERT promoter and FGFR3 and telomere length in bladder cancer. Int. J. Cancer 2015, 137, 1621–1629. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef]
- Mukherjee, S.; Firpo, E.J.; Wang, Y.; Roberts, J.M. Separation of telomerase functions by reverse genetics. Proc. Natl. Acad. Sci. USA 2011, 108, E1363–E1371. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef]
- Li, C.; Wu, S.; Wang, H.; Bi, X.; Yang, Z.; Du, Y.; He, L.; Cai, Z.; Wang, J.; Fan, Z. The C228T mutation of TERT promoter frequently occurs in bladder cancer stem cells and contributes to tumorigenesis of bladder cancer. Oncotarget 2015, 6, 19542–19551. [Google Scholar] [CrossRef]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Bjorkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.M. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget 2016, 7, 11364–11379. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.S. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-kappaB-dependent transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tergaonkar, V. Noncanonical functions of telomerase: Implications in telomerase-targeted cancer therapies. Cancer Res. 2014, 74, 1639–1644. [Google Scholar] [CrossRef]
- Pestana, A.; Vinagre, J.; Sobrinho-Simoes, M.; Soares, P. TERT biology and function in cancer: Beyond immortalisation. J. Mol. Endocrinol. 2017, 58, R129–R146. [Google Scholar] [CrossRef]
- Rachakonda, S.; Kong, H.; Srinivas, N.; Garcia-Casado, Z.; Requena, C.; Fallah, M.; Heidenreich, B.; Planelles, D.; Traves, V.; Schadendorf, D.; et al. Telomere length, telomerase reverse transcriptase promoter mutations, and melanoma risk. Genes Chromosomes Cancer 2018, 57, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife 2015, 4, e07918. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafezi, F.; Jaxel, L.; Lemaire, M.; Turner, J.D.; Perez-Bercoff, D. TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter. Biomedicines 2021, 9, 1773. https://doi.org/10.3390/biomedicines9121773
Hafezi F, Jaxel L, Lemaire M, Turner JD, Perez-Bercoff D. TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter. Biomedicines. 2021; 9(12):1773. https://doi.org/10.3390/biomedicines9121773
Chicago/Turabian StyleHafezi, François, Lisa Jaxel, Morgane Lemaire, Jonathan D. Turner, and Danielle Perez-Bercoff. 2021. "TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter" Biomedicines 9, no. 12: 1773. https://doi.org/10.3390/biomedicines9121773
APA StyleHafezi, F., Jaxel, L., Lemaire, M., Turner, J. D., & Perez-Bercoff, D. (2021). TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter. Biomedicines, 9(12), 1773. https://doi.org/10.3390/biomedicines9121773