
A Combined Spectroscopic and In Silico Approach to Evaluate the Interaction of Human Frataxin with Mitochondrial Superoxide Dismutase


 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
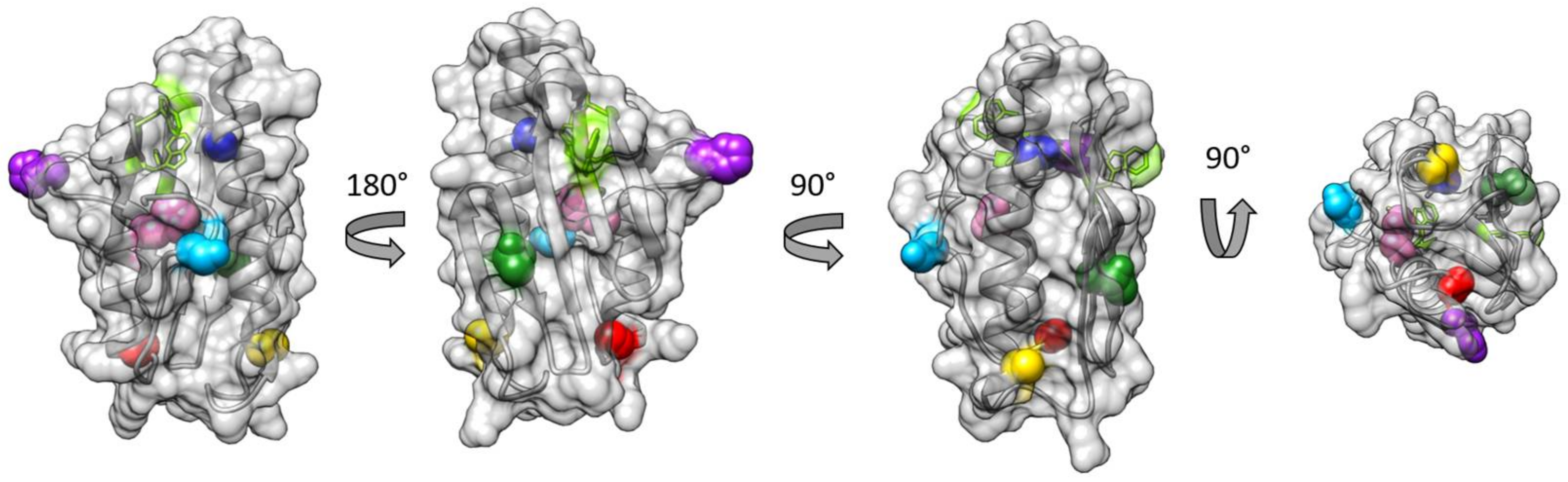
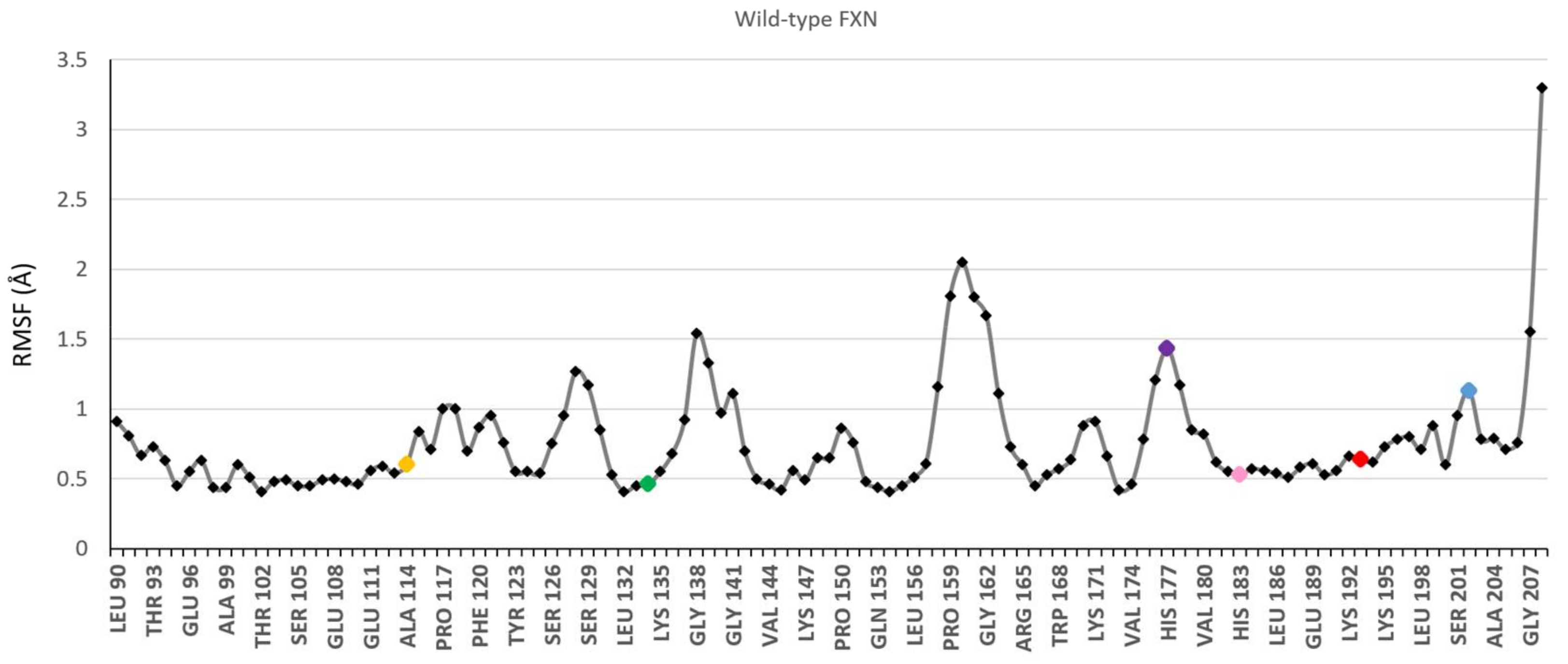
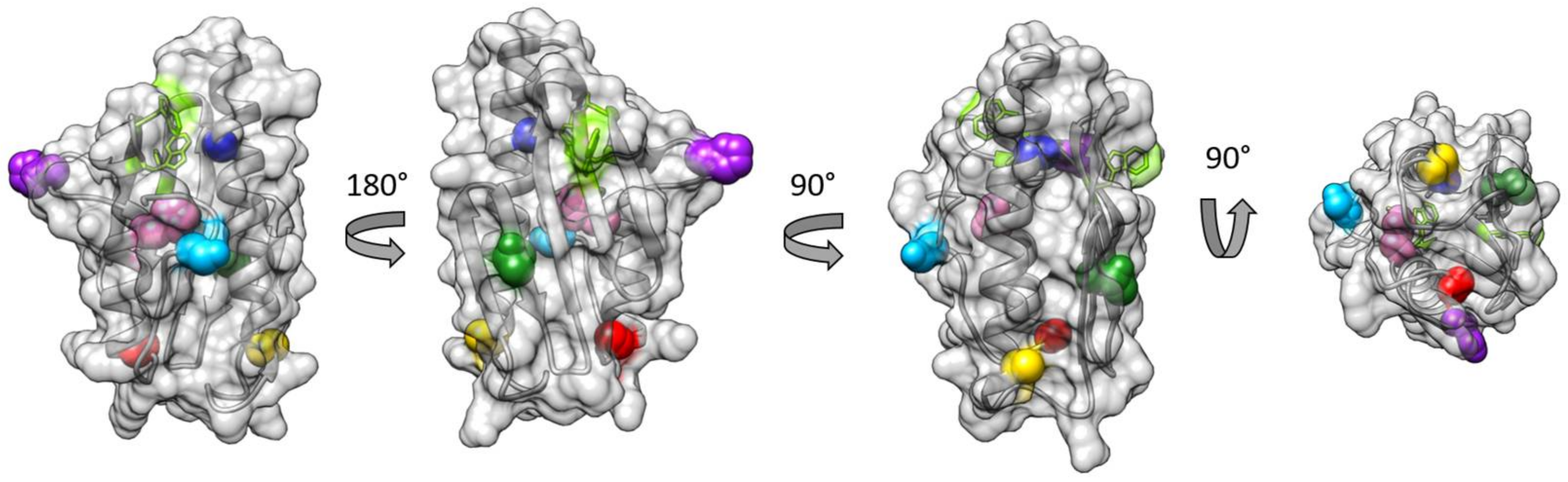
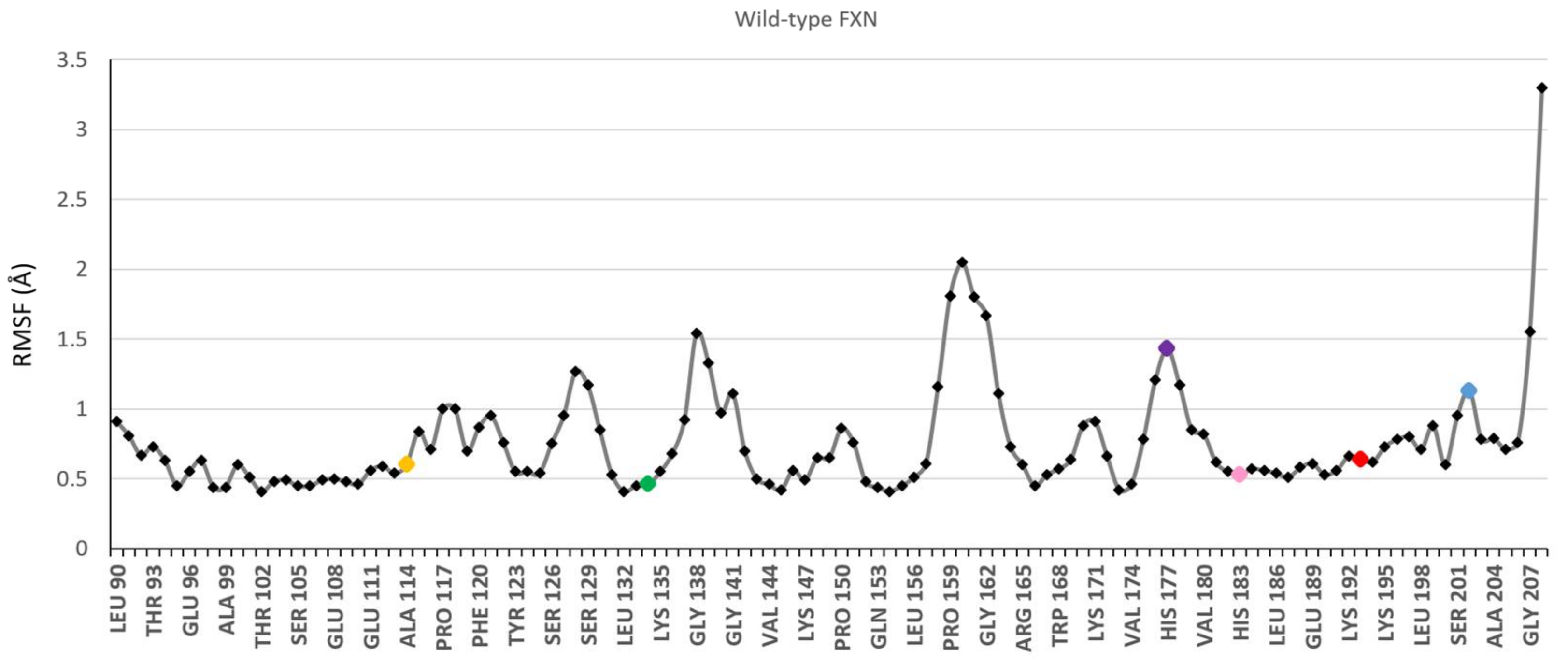
3.1. Choice of Labelling Sites and Molecular Dynamics of FXN
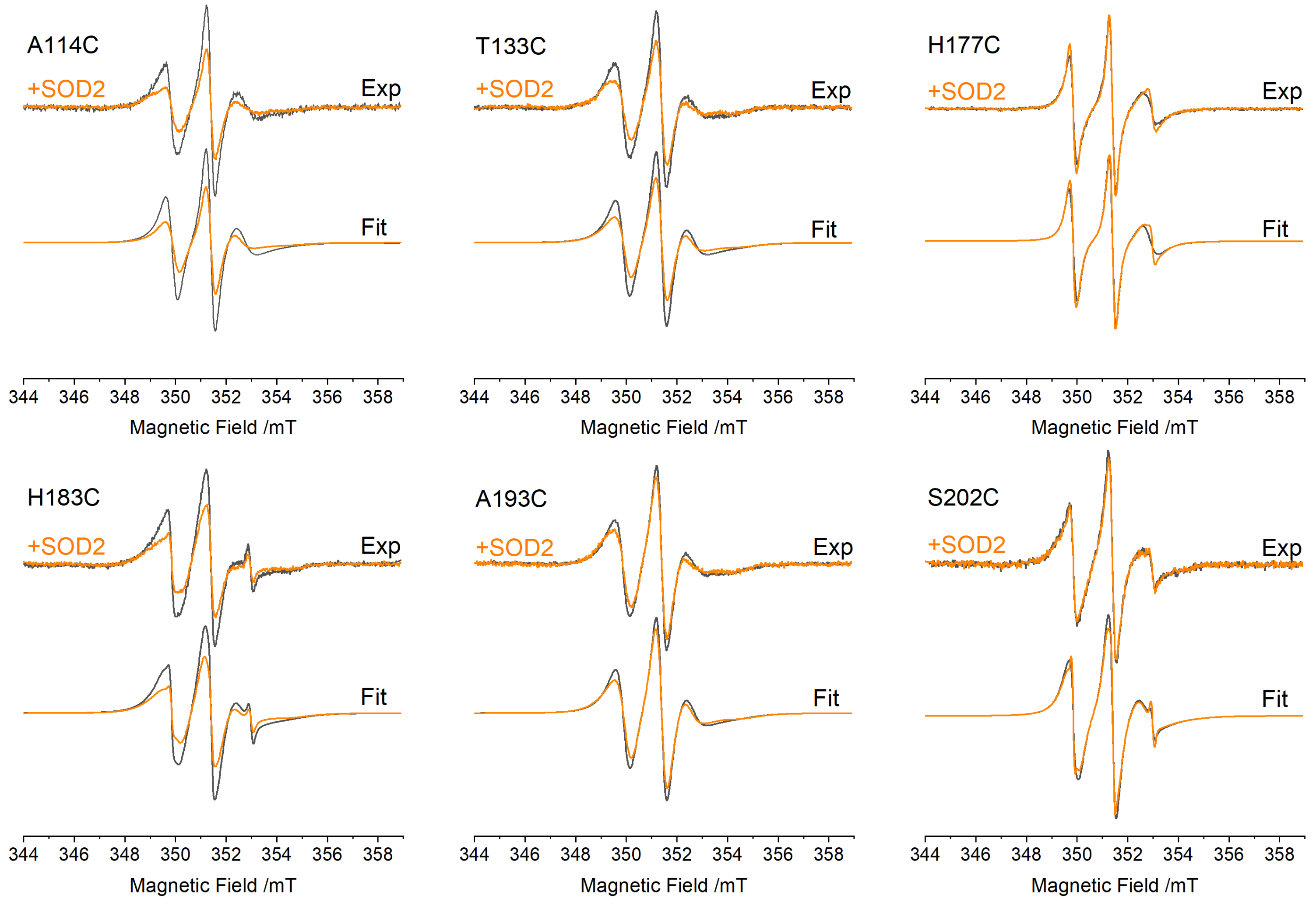
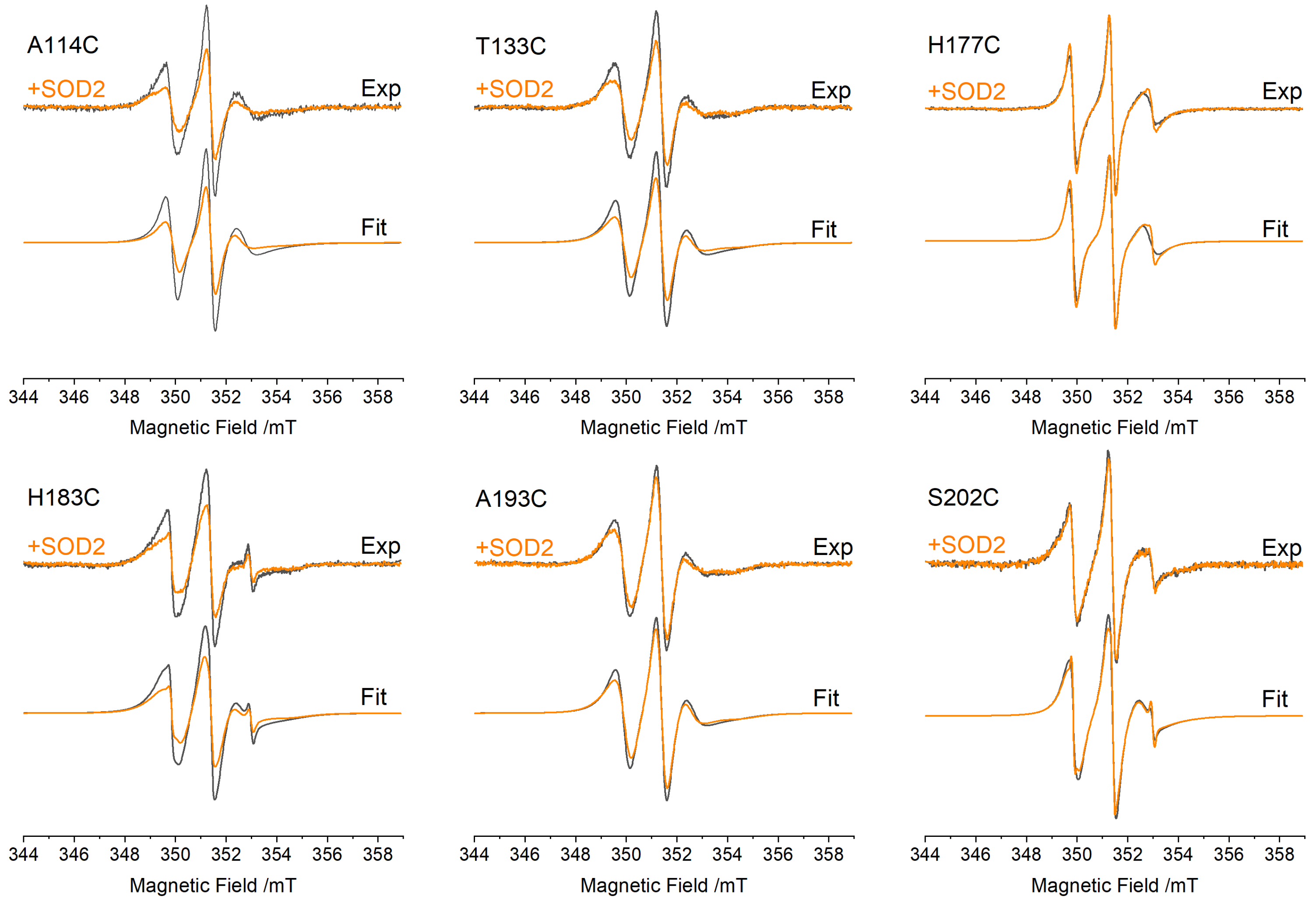
3.2. EPR Spectra of FXN Interacting with SOD2
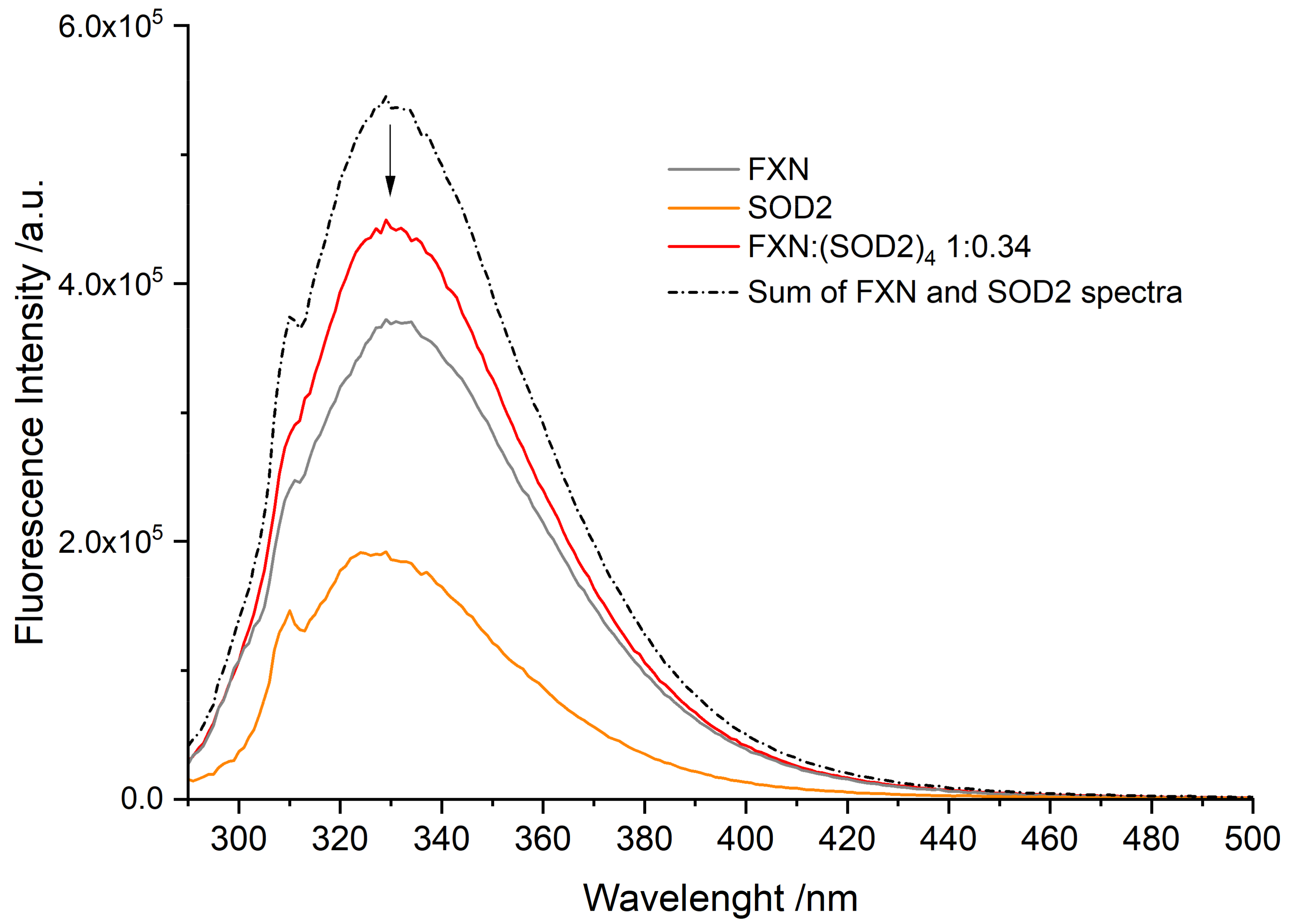
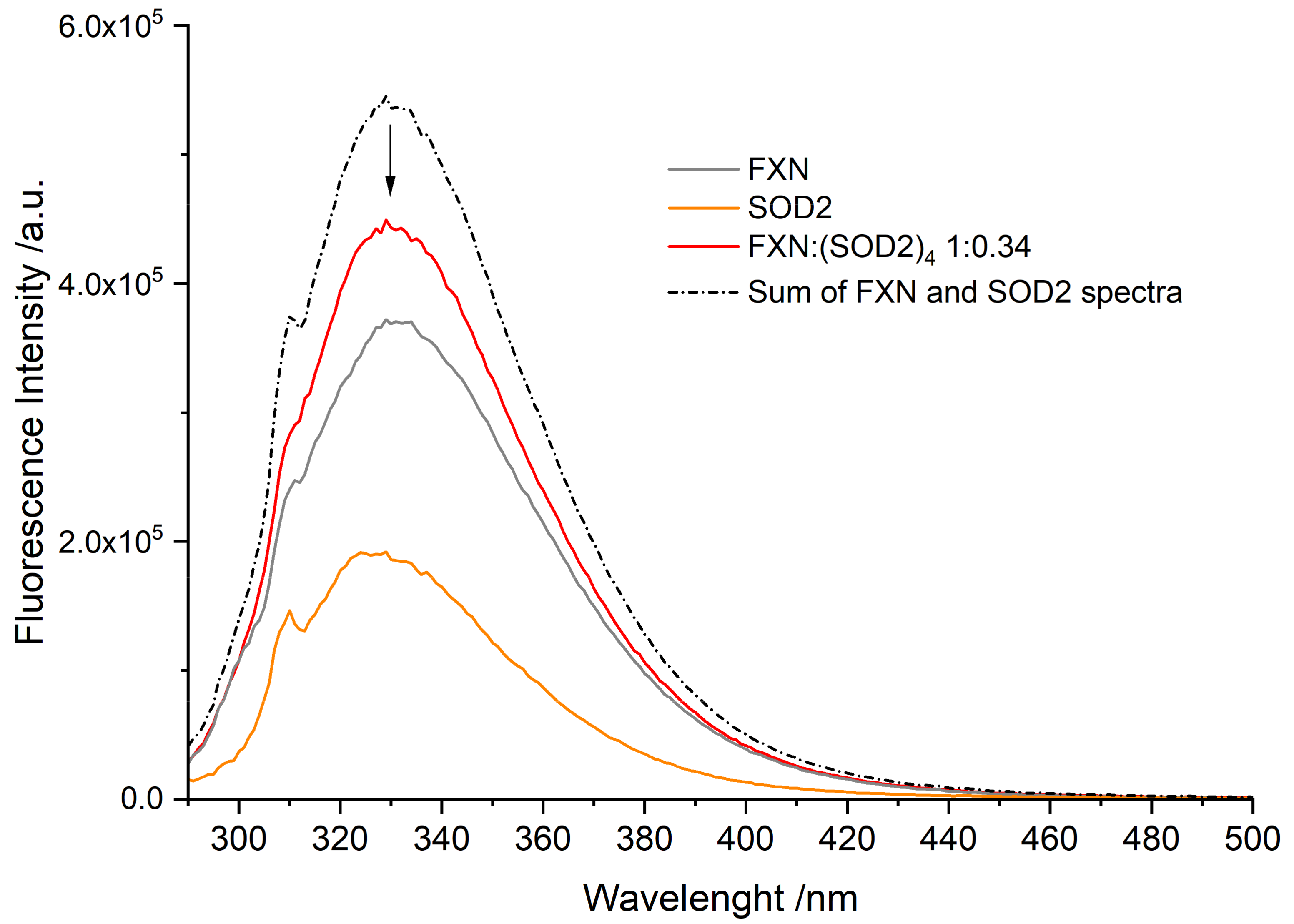
3.3. Fluorescence Spectra of FXN Interacting with SOD2
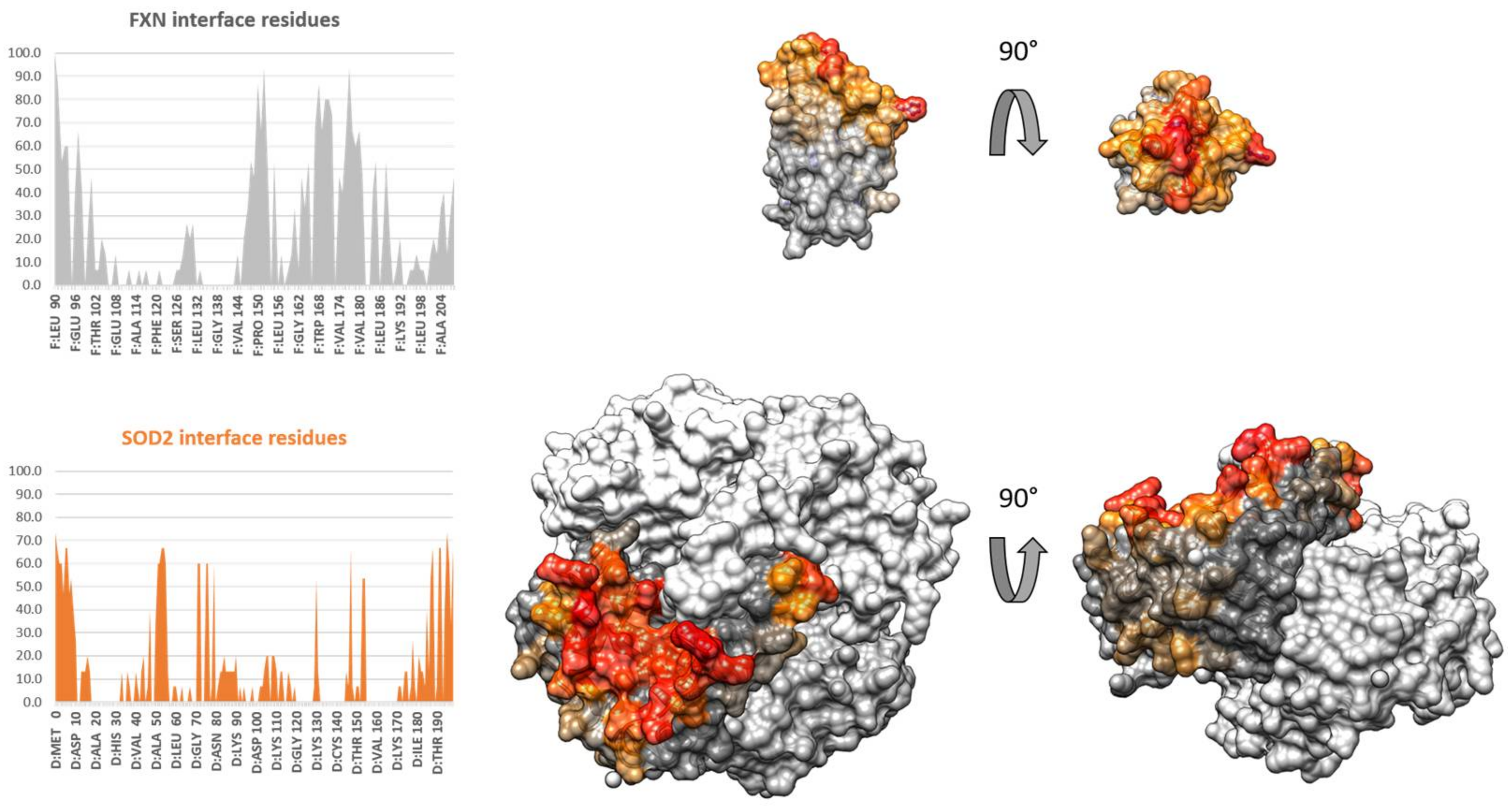
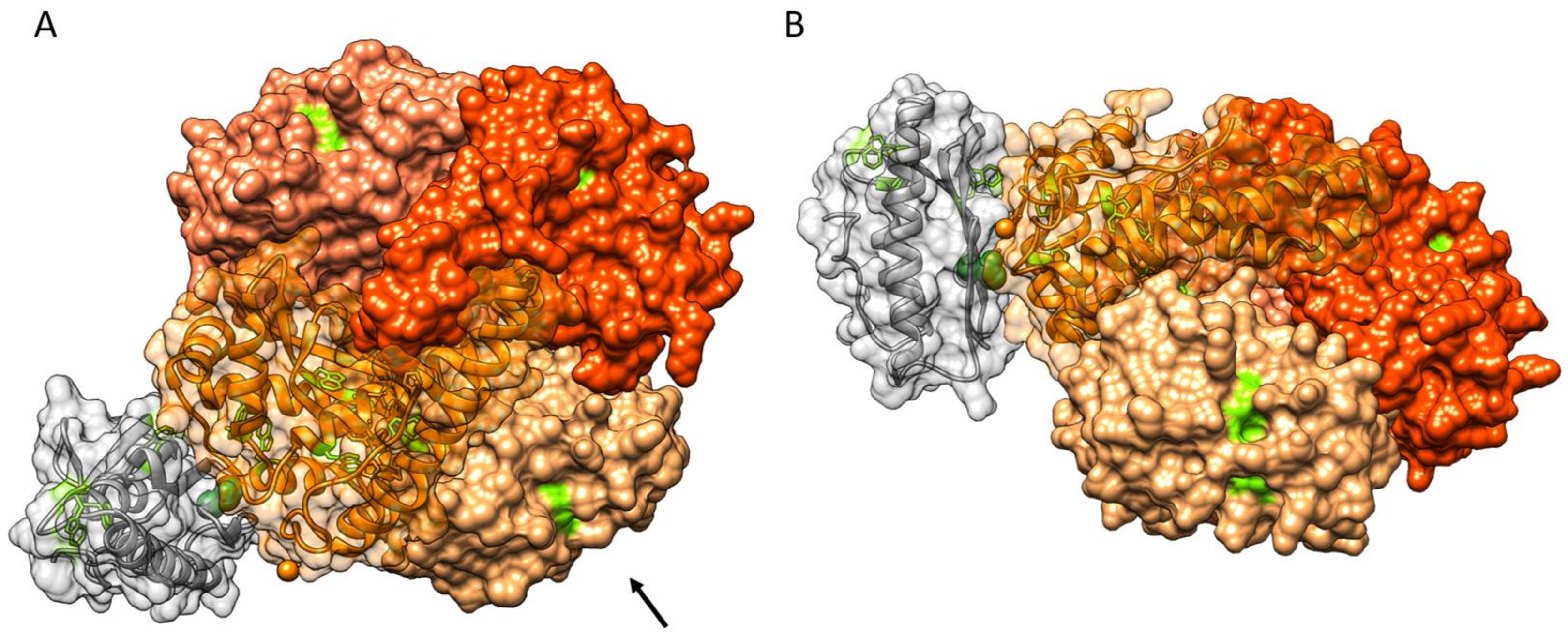
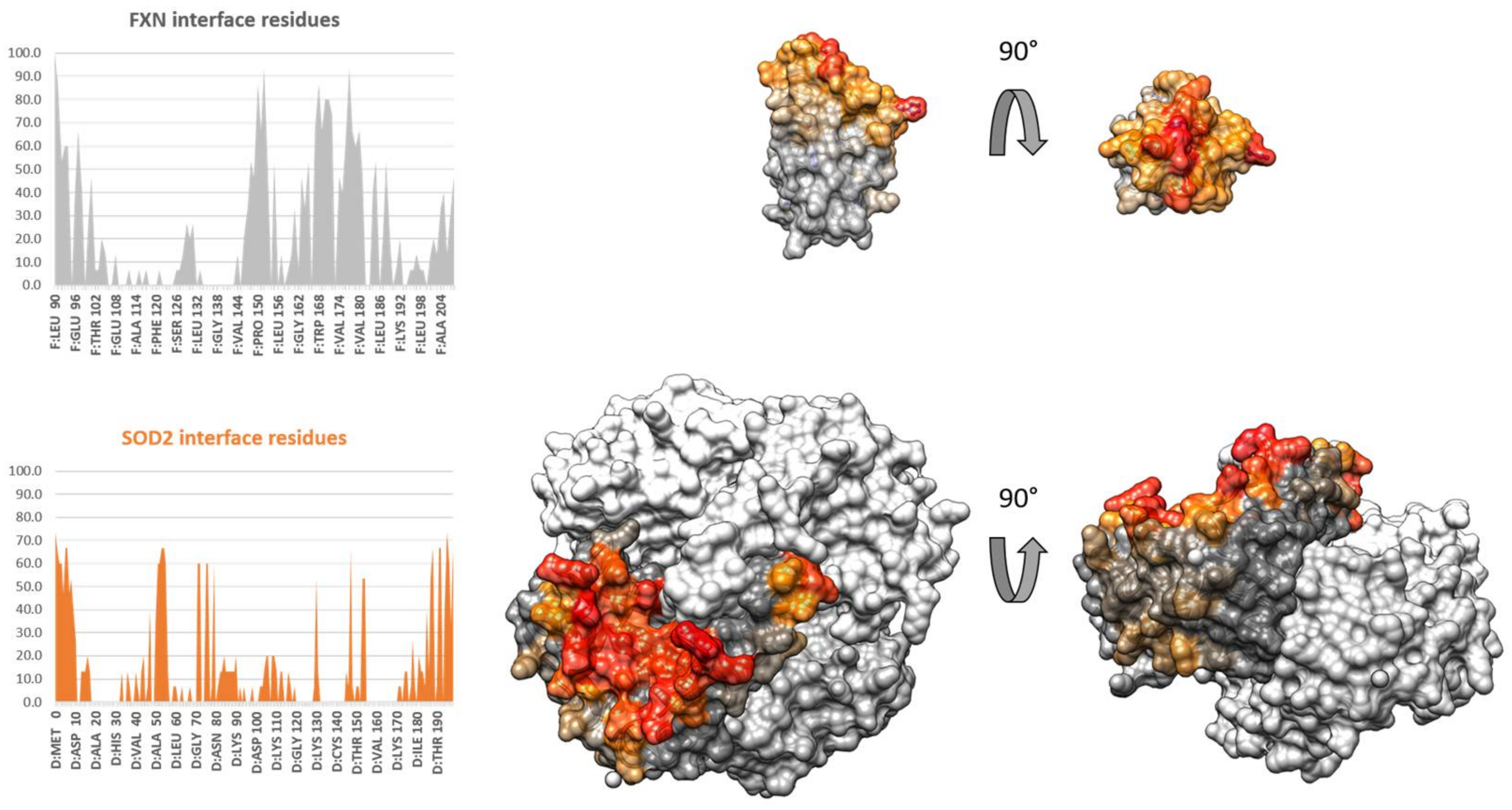
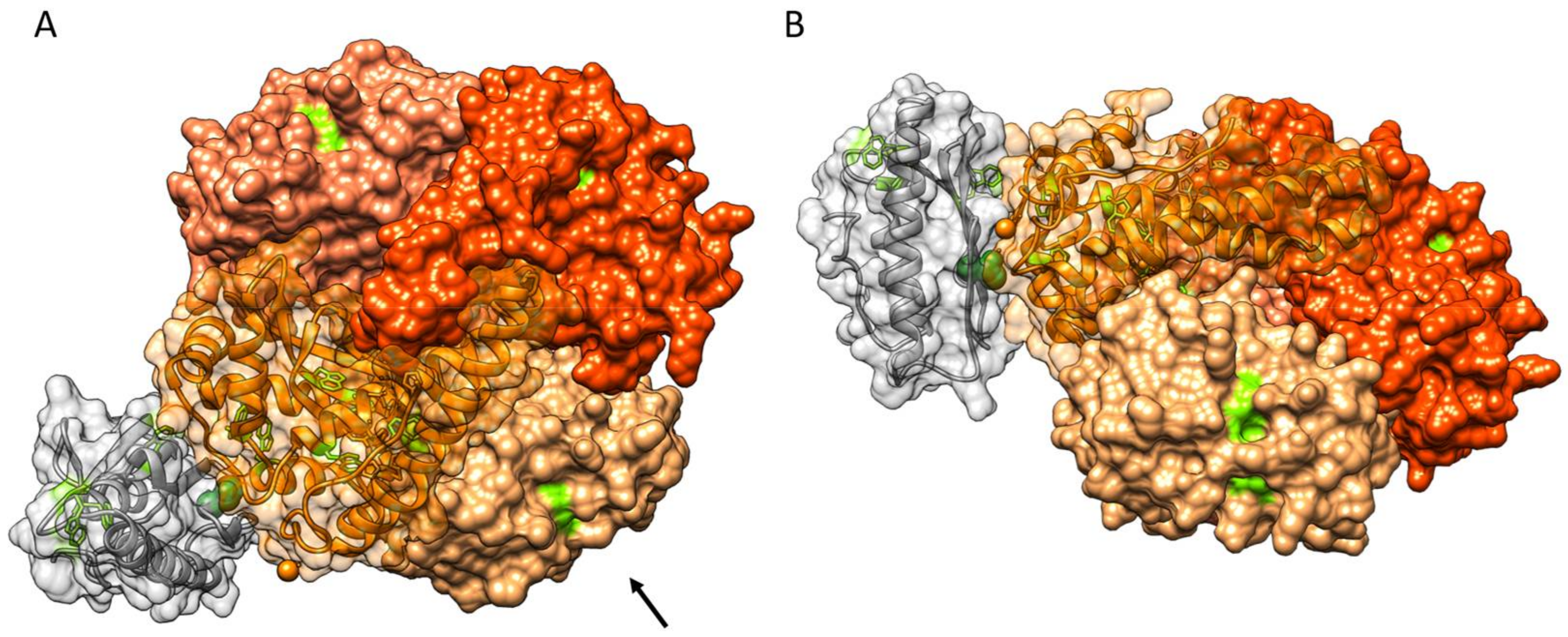
3.4. Protein–Protein Docking of FXN and SOD2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- De Castro, M.; García-Planells, J.; Monrós, E.; Cañizares, J.; Vázquez-Manrique, R.; Vílchez, J.J.; Urtasun, M.; Lucas, M.; Navarro, G.; Izquierdo, G.; et al. Genotype and phenotype analysis of Friedreich’s ataxia compound heterozygous patients. Hum. Genet. 2000, 106, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Gellera, C.; Castellotti, B.; Mariotti, C.; Mineri, R.; Seveso, V.; DiDonato, S.; Taroni, F. Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics 2007, 8, 289–299. [Google Scholar] [CrossRef]
- Chiabrando, D.; Bertino, F.; Tolosano, E. Hereditary ataxia: A focus on heme metabolism and fe-s cluster biogenesis. Int. J. Mol. Sci. 2020, 21, 3760. [Google Scholar] [CrossRef]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schols, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Burk, K.; Matson, W.; Dichgans, J.; Beal, M.F. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Koutnikova, H.; Campuzano, V.; Koenig, M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 1998, 7, 1485–1489. [Google Scholar] [CrossRef]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Schmucker, S.; Argentini, M.; Carelle-Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- Huynen, M.A.; Snel, B.; Bork, P.; Gibson, T.J. The phylogenetic distribution of frataxin indicates a role in iron-sulfur cluster protein assembly. Hum. Mol. Genet. 2001, 10, 2463–2468. [Google Scholar] [CrossRef] [Green Version]
- Musco, G.; Stier, G.; Kolmerer, B.; Adinolfi, S.; Martin, S.; Frenkiel, T.; Gibson, T.; Pastore, A. Towards a structural understanding of Friedreich’s ataxia: Solution structure of frataxin. Structure 2000, 8, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Gentry, L.E.; Thacker, M.A.; Doughty, R.; Timkovich, R.; Busenlehner, L.S. His86 from the N-terminus of frataxin coordinates iron and is required for Fe-S cluster synthesis. Biochemistry 2013, 52, 6085–6096. [Google Scholar] [CrossRef]
- Sirano, D.P.; Shigeta, R.; Chi, Y.I.; Ristow, M.; Shoelson, S.E. Crystal structure of human frataxin. J. Biol. Chem. 2000, 275, 30753–30756. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Patraa, S.; Barondeaua, D.P. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. USA 2019, 116, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Srour, B.; Gervason, S.; Monfort, B.; D’Autréaux, B. Mechanism of Iron–Sulfur Cluster Assembly: In the Intimacy of Iron and Sulfur Encounter. Inorganics 2020, 8, 55. [Google Scholar] [CrossRef]
- Campbell, C.J.; Pall, A.E.; Naik, A.R.; Thompson, L.N.; Stemmler, T.L. Molecular Details of the Frataxin–Scaffold Interaction during Mitochondrial Fe–S Cluster Assembly. Int. J. Mol. Sci. 2021, 22, 6006. [Google Scholar] [CrossRef]
- Alsina, D.; Purroy, R.; Ros, J.; Tamarit, J. Iron in friedreich ataxia: A central role in the pathophysiology or an epiphenomenon? Pharmaceuticals 2018, 11, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Grazia Cotticelli, M.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a novel therapeutic target for Friedreich’s ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Manrique, R.P.; González-Cabo, P.; Ros, S.; Aziz, H.; Baylis, H.A.; Palau, F. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J. 2006, 20, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Runko, A.P.; Griswold, A.J.; Min, K.-T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008, 582, 715–719. [Google Scholar] [CrossRef]
- Busi, M.V.; Maliandi, M.V.; Valdez, H.; Clemente, M.; Zabaleta, E.J.; Araya, A.; Gomez-Casati, D.F. Deficiency of Arabidopsis thaliana frataxin alters activity of mitochondrial Fe-S proteins and induces oxidative stress. Plant J. 2006, 48, 873–882. [Google Scholar] [CrossRef]
- Uceda, A.B.; Donoso, J.; Frau, J.; Vilanova, B.; Adrover, M. Frataxins Emerge as New Players of the Intracellular Antioxidant Machinery. Antioxidants 2021, 10, 315. [Google Scholar] [CrossRef]
- Han, T.H.L.; Camadro, J.-M.M.; Barbault, F.; Santos, R.; El Hage Chahine, J.-M.M.; Ha-Duong, N.-T.T. In Vitro interaction between yeast frataxin and superoxide dismutases: Influence of mitochondrial metals. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 883–892. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Doni, D.; Passerini, L.; Audran, G.; Marque, S.R.A.; Schulz, M.; Santos, J.; Costantini, P.; Bortolus, M.; Carbonera, D. Effects of Fe2+/Fe3+ Binding to Human Frataxin and Its D122Y Variant, as Revealed by Site-Directed Spin Labeling (SDSL) EPR Complemented by Fluorescence and Circular Dichroism Spectroscopies. Int. J. Mol. Sci. 2020, 21, 9619. [Google Scholar] [CrossRef]
- Roman, E.A.; Faraj, S.E.; Gallo, M.; Salvay, A.G.; Ferreiro, D.U.; Santos, J. Protein Stability and Dynamics Modulation: The Case of Human Frataxin. PLoS ONE 2012, 7, e45743. [Google Scholar] [CrossRef] [Green Version]
- Budil, D.E.; Sanghyuk, L.; Saxena, S.; Freed, J.H. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified levenberg-marquardt algorithm. J. Magn. Reson.-Ser. A 1996, 120, 155–189. [Google Scholar] [CrossRef] [Green Version]
- MultiComponent. Available online: http://www.biochemistry.ucla.edu/Faculty/Hubbell/ (accessed on 23 November 2021).
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Genet. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Chermak, E.; Petta, A.; Serra, L.; Vangone, A.; Scarano, V.; Cavallo, L.; Oliva, R. CONSRANK: A server for the analysis, comparison and ranking of docking models based on inter-residue contacts. Bioinformatics 2015, 31, 1481–1483. [Google Scholar] [CrossRef]
- Oliva, R.; Vangone, A.; Cavallo, L. Ranking multiple docking solutions based on the conservation of inter-residue contacts. Proteins Struct. Funct. Bioinforma. 2013, 81, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Cavallo, L.; Oliva, R. Using a consensus approach based on the conservation of inter-residue contacts to rank CAPRI models. Proteins Struct. Funct. Bioinforma. 2013, 81, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering. Methods in Molecular Biology; Bornscheuer, U., Höhne, M., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1685, pp. 43–67. ISBN 978-1-4939-7366-8. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Bellanda, M.; Maso, L.; Doni, D.; Bortolus, M.; De Rosa, E.; Lunardi, F.; Alfonsi, A.; Noguera, M.E.; Herrera, M.G.; Santos, J.; et al. Exploring iron-binding to human frataxin and to selected Friedreich ataxia mutants by means of NMR and EPR spectroscopies. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 140254. [Google Scholar] [CrossRef]
- Huang, J.; Dizin, E.; Cowan, J.A. Mapping iron binding sites on human frataxin: Implications for cluster assembly on the ISU Fe-S cluster scaffold protein. J. Biol. Inorg. Chem. 2008, 13, 825–836. [Google Scholar] [CrossRef]
- Azadmanesh, J.; Trickel, S.R.; Borgstahl, G.E.O. Substrate-analog binding and electrostatic surfaces of human manganese superoxide dismutase. J. Struct. Biol. 2017, 199, 68–75. [Google Scholar] [CrossRef]
- Doni, D.; Rigoni, G.; Palumbo, E.; Baschiera, E.; Peruzzo, R.; De Rosa, E.; Caicci, F.; Passerini, L.; Bettio, D.; Russo, A.; et al. The displacement of frataxin from the mitochondrial cristae correlates with abnormal respiratory supercomplexes formation and bioenergetic defects in cells of Friedreich ataxia patients. FASEB J. 2021, 35, e21362. [Google Scholar] [CrossRef]
- Gong, H.; Li, J.; Xu, A.; Tang, Y.; Ji, W.; Gao, R.; Wang, S.; Yu, L.; Tian, C.; Li, J.; et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science 2018, 362, eaat8923. [Google Scholar] [CrossRef] [Green Version]
- Suthammarak, W.; Somerlot, B.H.; Opheim, E.; Sedensky, M.; Morgan, P.G. Novel interactions between mitochondrial superoxide dismutases and the electron transport chain. Aging Cell 2013, 12, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Jiralerspong, S.; Ge, B.; Hudson, T.J.; Pandolfo, M. Manganese superoxide dismutase induction by iron is impaired in Friedreich ataxia cells. FEBS Lett. 2001, 509, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Chantrel-Groussard, K. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef]
- Irazusta, V.; Cabiscol, E.; Reverter-Branchat, G.; Ros, J.; Tamarit, J. Manganese Is the Link between Frataxin and Iron-Sulfur Deficiency in the Yeast Model of Friedreich Ataxia. J. Biol. Chem. 2006, 281, 12227–12232. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Cobine, P.A.; Molik, S.; Naranuntarat, A.; Lill, R.; Winge, D.R.; Culotta, V.C. The effects of mitochondrial iron homeostasis on cofactor specificity of superoxide dismutase 2. EMBO J. 2006, 25, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Irazusta, V.; Obis, E.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free Radic. Biol. Med. 2010, 48, 411–420. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | τC−SOD/ns | τC+SOD/ns | ΔτC (τC+SOD − τC−SOD)/ns |
|---|---|---|---|
| A114C | 2.52 ± 0.06 | 2.96 ± 0.06 | 0.44 ± 0.09 |
| T133C | 2.70 ± 0.06 | 3.10 ± 0.07 | 0.40 ± 0.095 |
| H177C | 1.13 ± 0.03 | 1.24 ± 0.03 | 0.11 ± 0.04 |
| H183C | 2.77 ± 0.06 | 3.33 ± 0.06 | 0.56 ± 0.10 |
| A193C | 2.77 ± 0.06 | 3.18 ± 0.06 | 0.41 ± 0.10 |
| S202C | 2.30 ± 0.05 | 2.47 ± 0.05 | 0.16 ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doni, D.; Meggiolaro, M.; Santos, J.; Audran, G.; Marque, S.R.A.; Costantini, P.; Bortolus, M.; Carbonera, D. A Combined Spectroscopic and In Silico Approach to Evaluate the Interaction of Human Frataxin with Mitochondrial Superoxide Dismutase. Biomedicines 2021, 9, 1763. https://doi.org/10.3390/biomedicines9121763
Doni D, Meggiolaro M, Santos J, Audran G, Marque SRA, Costantini P, Bortolus M, Carbonera D. A Combined Spectroscopic and In Silico Approach to Evaluate the Interaction of Human Frataxin with Mitochondrial Superoxide Dismutase. Biomedicines. 2021; 9(12):1763. https://doi.org/10.3390/biomedicines9121763
Chicago/Turabian StyleDoni, Davide, Marta Meggiolaro, Javier Santos, Gérard Audran, Sylvain R. A. Marque, Paola Costantini, Marco Bortolus, and Donatella Carbonera. 2021. "A Combined Spectroscopic and In Silico Approach to Evaluate the Interaction of Human Frataxin with Mitochondrial Superoxide Dismutase" Biomedicines 9, no. 12: 1763. https://doi.org/10.3390/biomedicines9121763
APA StyleDoni, D., Meggiolaro, M., Santos, J., Audran, G., Marque, S. R. A., Costantini, P., Bortolus, M., & Carbonera, D. (2021). A Combined Spectroscopic and In Silico Approach to Evaluate the Interaction of Human Frataxin with Mitochondrial Superoxide Dismutase. Biomedicines, 9(12), 1763. https://doi.org/10.3390/biomedicines9121763