
A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation


Abstract
1. Introduction
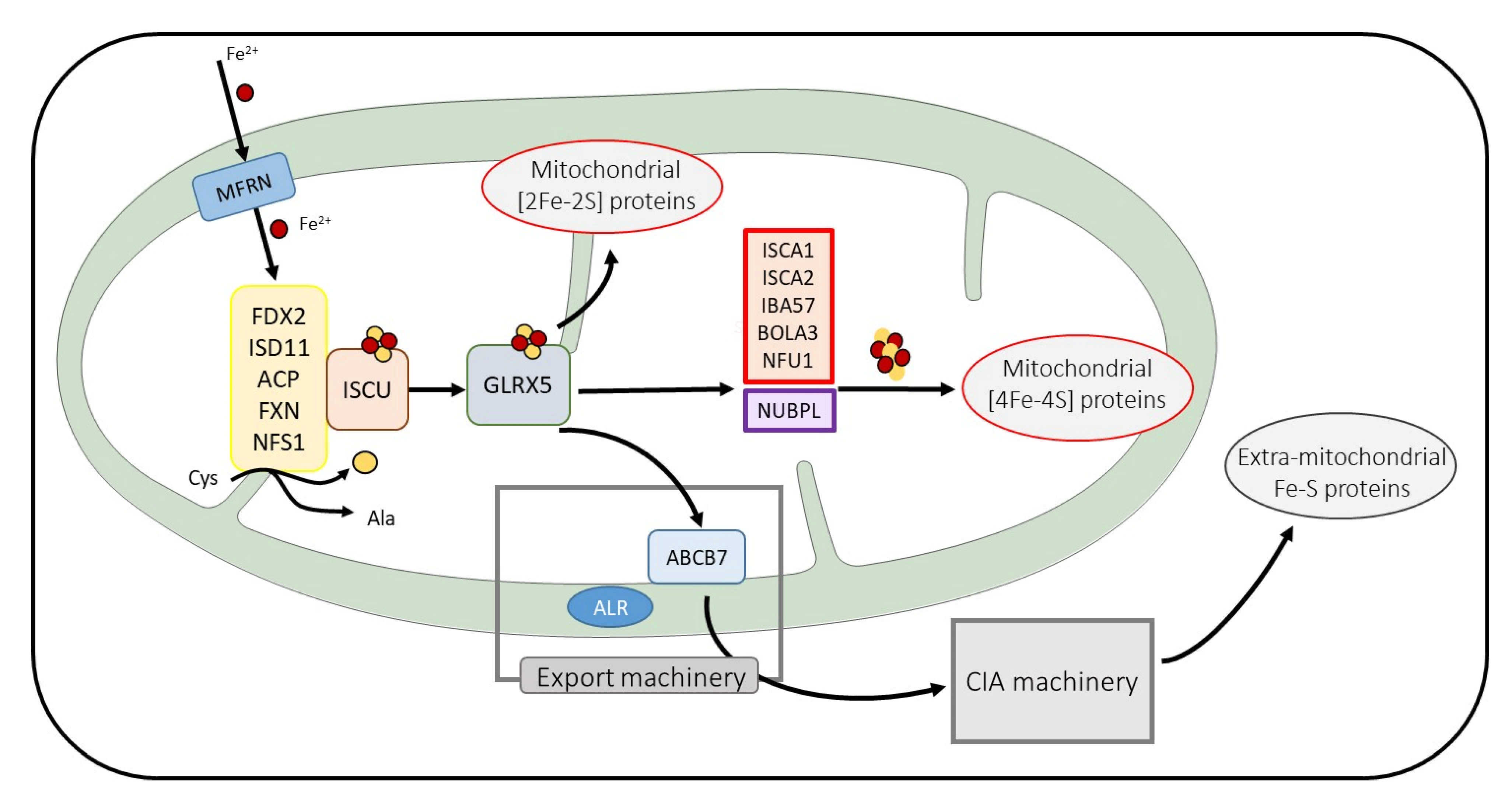
2. Fe-S Proteins: Essential Proteins with Elaborated Maturation Pathways
3. MMDS Type 1 (MMDS1), Mutation in NFU1
4. MMDS Type 2, Mutations in BOLA3
5. MMDS Type 3 (MMDS3), Mutations in IBA57
6. MMDS Type 4, Mutations in ISCA2
7. MMDS Type 5, Mutations in ISCA1
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, Function, and Formation of Biological Iron-Sulfur Clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and Functions of Mammalian Iron-Sulfur Proteins in the Regulation of Iron Homeostasis and Pivotal Metabolic Pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic Concepts of Iron-Sulfur Protein Biogenesis in Biology. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Wachnowsky, C.; Fidai, I.; Cowan, J.A. Iron-Sulfur Cluster Biosynthesis and Trafficking-Impact on Human Disease Conditions. Metallomics 2018, 10, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein Networks in the Maturation of Human Iron-Sulfur Proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef]
- Beinert, H. Iron-Sulfur Proteins: Ancient Structures, Still Full of Surprises. J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J. Iron-Sulfur Protein Folds, Iron-Sulfur Chemistry, and Evolution. J. Biol. Inorg. Chem. 2008, 13, 157–170. [Google Scholar] [CrossRef]
- Pain, D.; Dancis, A.; Bielas, J.; Suzuki, C. Roles of Fe–S Proteins: From Cofactor Synthesis to Iron Homeostasis to Protein Synthesis This Review Comes from a Themed Issue on Molecular and Genetic Bases of Disease. Curr. Opin. Genet. Dev. 2016, 38, 45–51. [Google Scholar] [CrossRef]
- Rouault, T.A. The Indispensable Role of Mammalian Iron Sulfur Proteins in Function and Regulation of Multiple Diverse Metabolic Pathways. Biometals 2019, 32, 343–353. [Google Scholar] [CrossRef]
- Lauble, H.; Stout, C.D. Steric and Conformational Features of the Aconitase Mechanism. Proteins 1995, 22, 1–11. [Google Scholar] [CrossRef]
- Broderick, W.E.; Broderick, J.B. Radical SAM Enzymes: Surprises along the Path to Understanding Mechanism. J. Biol. Inorg. Chem. 2019, 24, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Ferecatu, I.; Gonçalves, S.; Golinelli-Cohen, M.-P.; Clémancey, M.; Martelli, A.; Riquier, S.; Guittet, E.; Latour, J.-M.; Puccio, H.; Drapier, J.-C.; et al. The Diabetes Drug Target MitoNEET Governs a Novel Trafficking Pathway to Rebuild an Fe-S Cluster into Cytosolic Aconitase/Iron Regulatory Protein 1. J. Biol. Chem. 2014, 289, 28070–28086. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Sipos, K.; Lange, H.; Fekete, Z.; Bedekovics, T.; Janáky, T.; Bassler, J.; Aguilar Netz, D.J.; Balk, J.; Rotte, C.; et al. Biogenesis of Cytosolic Ribosomes Requires the Essential Iron-Sulphur Protein Rli1p and Mitochondria. EMBO J. 2005, 24, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Fuss, J.O.; Tsai, C.L.; Ishida, J.P.; Tainer, J.A. Emerging Critical Roles of Fe-S Clusters in DNA Replication and Repair. Biochim. Biophys. Acta 2015, 1853, 1253–1271. [Google Scholar] [CrossRef]
- Fugate, C.J.; Jarrett, J.T. Biotin Synthase: Insights into Radical-Mediated Carbon-Sulfur Bond Formation. Biochim. Biophys. Acta 2009, 1824, 1213–1222. [Google Scholar] [CrossRef]
- Crack, J.C.; Green, J.; Thomson, A.J.; le Brun, N.E. Iron-Sulfur Clusters as Biological Sensors: The Chemistry of Reactions with Molecular Oxygen and Nitric Oxide. Acc. Chem. Res. 2014, 47, 3196–3205. [Google Scholar] [CrossRef]
- Golinelli-Cohen, M.-P.; Bouton, C. Fe-S Proteins Acting as Redox Switch: New Key Actors of Cellular Adaptive Responses. Curr. Chem. Biol. 2017, 11, 1–19. [Google Scholar] [CrossRef]
- Zheng, L.; White, R.H.; Cash, V.L.; Jack, R.F.; Dean, D.R. Cysteine Desulfurase Activity Indicates a Role for NIFS in Metallocluster Biosynthesis. Proc. Natl. Acad. Sci. USA 1993, 90, 2754–2758. [Google Scholar] [CrossRef]
- Land, T.; Rouault, T.A. Targeting of a Human Iron-Sulfur Cluster Assembly Enzyme, Nifs, to Different Subcellular Compartments Is Regulated through Alternative AUG Utilization. Mol. Cell 1998, 2, 807–815. [Google Scholar] [CrossRef]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The Mitochondrial Proteins Atm1p and Nfs1p Are Essential for Biogenesis of Cytosolic Fe/S Proteins. EMBO J 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cowan, J.A. Glutathione-Coordinated [2Fe–2S] Cluster: A Viable Physiological Substrate for Mitochondrial ABCB7 Transport. Chem. Commun. 2015, 51, 2253–2255. [Google Scholar] [CrossRef]
- Srinivasan, V.; Pierik, A.J.; Lill, R. Crystal Structures of Nucleotide-Free and Glutathione-Bound Mitochondrial ABC Transporter Atm1. Science 2014, 343, 1137–1140. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.C.; Bornhövd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 Interacting Protein Isd11 Has an Essential Role in Fe/S Cluster Biogenesis in Mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef]
- Wiedemann, N.; Urzica, E.; Guiard, B.; Müller, H.; Lohaus, C.; Meyer, H.E.; Ryan, M.T.; Meisinger, C.; Mühlenhoff, U.; Lill, R.; et al. Essential Role of Isd11 in Mitochondrial Iron-Sulfur Cluster Synthesis on Isu Scaffold Proteins. EMBO J. 2006, 25, 184–195. [Google Scholar] [CrossRef]
- Boniecki, M.T.; Freibert, S.A.; Mühlenhoff, U.; Lill, R.; Cygler, M. Structure and Functional Dynamics of the Mitochondrial Fe/S Cluster Synthesis Complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Elsasser, H.-P.; Muhlenhoff, U.; Webert, H.; Hobler, A.; Hannemann, F.; Bernhardt, R.; Lill, R. Humans Possess Two Mitochondrial Ferredoxins, Fdx1 and Fdx2, with Distinct Roles in Steroidogenesis, Heme, and Fe/S Cluster Biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 11775–11780. [Google Scholar] [CrossRef] [PubMed]
- Webert, H.; Freibert, S.A.; Gallo, A.; Heidenreich, T.; Linne, U.; Amlacher, S.; Hurt, E.; Mühlenhoff, U.; Banci, L.; Lill, R. Functional Reconstitution of Mitochondrial Fe/S Cluster Synthesis on Isu1 Reveals the Involvement of Ferredoxin. Nat. Commun. 2014, 5, 5013. [Google Scholar] [CrossRef]
- Cai, K.; Tonelli, M.; Frederick, R.O.; Markley, J.L. Human Mitochondrial Ferredoxin 1 (FDX1) and Ferredoxin 2 (FDX2) Both Bind Cysteine Desulfurase and Donate Electrons for Iron-Sulfur Cluster Biosynthesis. Biochemistry 2017, 56, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Barondeau, D.P. Human Frataxin Is an Allosteric Switch That Activates the Fe-S Cluster Biosynthetic Complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian Frataxin Directly Enhances Sulfur Transfer of NFS1 Persulfide to Both ISCU and Free Thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Singh, A.; Uhrigshardt, H.; Saxena, N.; Tong, W.H.; Rouault, T.A. Cochaperone Binding to LYR Motifs Confers Specificity of Iron Sulfur Cluster Delivery. Cell Metab. 2014, 19, 445–457. [Google Scholar] [CrossRef]
- Bych, K.; Kerscher, S.; Netz, D.J.A.; Pierik, A.J.; Zwicker, K.; Huynen, M.A.; Lill, R.; Brandt, U.; Balk, J. The Iron-Sulphur Protein Ind1 Is Required for Effective Complex I Assembly. EMBO J. 2008, 27, 1736–1746. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Netz, D.J.A.; Kerscher, S.; Elsässer, H.-P.; Wittig, I.; Balk, J.; Brandt, U.; Lill, R. Human Ind1, an Iron-Sulfur Cluster Assembly Factor for Respiratory Complex I. Mol. Cell. Biol. 2009, 29, 6059–6073. [Google Scholar] [CrossRef] [PubMed]
- Weiler, B.D.; Brück, M.C.; Kothe, I.; Bill, E.; Lill, R.; Mühlenhoff, U. Mitochondrial [4Fe-4S] Protein Assembly Involves Reductive [2Fe-2S] Cluster Fusion on ISCA1-ISCA2 by Electron Flow from Ferredoxin FDX2. Proc. Natl. Acad. Sci. USA 2020, 117, 20555–20565. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Wilbrecht, C.; Stehling, O.; Niggemeyer, B.; Elsasser, H.P.; Muhlenhoff, U.; Lill, R. The Human Mitochondrial ISCA1, ISCA2, and IBA57 Proteins Are Required for [4Fe-4S] Protein Maturation. Mol. Biol. Cell 2012, 23, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Gourdoupis, S.; Nasta, V.; Calderone, V.; Ciofi-Baffoni, S.; Banci, L. IBA57 Recruits ISCA2 to Form a [2Fe-2S] Cluster-Mediated Complex. J. Am. Chem. Soc. 2018, 140, 14401–14412. [Google Scholar] [CrossRef]
- Brancaccio, D.; Gallo, A.; Mikolajczyk, M.; Zovo, K.; Palumaa, P.; Novellino, E.; Piccioli, M.; Ciofi-Baffoni, S.; Banci, L. Formation of [4Fe-4S] Clusters in the Mitochondrial Iron–Sulfur Cluster Assembly Machinery. J. Am. Chem. Soc. 2014, 136, 16240–16250. [Google Scholar] [CrossRef]
- Brancaccio, D.; Gallo, A.; Piccioli, M.; Novellino, E.; Ciofi-Baffoni, S.; Banci, L. [4Fe-4S] Cluster Assembly in Mitochondria and Its Impairment by Copper. J. Am. Chem. Soc. 2017, 139, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, L.A.; Gómez-Gallardo, M.; Díaz-Pérez, A.L.; Cortés-Rojo, C.; Campos-García, J. Iba57p Participates in Maturation of a [2Fe-2S]-Cluster Rieske Protein and in Formation of Supercomplexes III/IV of Saccharomyces Cerevisiae Electron Transport Chain. Mitochondrion 2019, 44, 75–84. [Google Scholar] [CrossRef]
- Melber, A.; Na, U.; Vashisht, A.; Weiler, B.D.; Lill, R.; Wohlschlegel, J.A.; Winge, D.R. Role of Nfu1 and Bol3 in Iron-Sulfur Cluster Transfer to Mitochondrial Clients. eLife 2016, 5, e15991. [Google Scholar] [CrossRef]
- Uzarska, M.A.; Przybyla-Toscano, J.; Spantgar, F.; Zannini, F.; Lill, R.; Mühlenhoff, U.; Rouhier, N. Conserved Functions of Arabidopsis Mitochondrial Late-Acting Maturation Factors in the Trafficking of Iron-sulfur Clusters. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1250–1259. [Google Scholar] [CrossRef]
- Jain, A.; Singh, A.; Maio, N.; Rouault, T.A. Assembly of the [4Fe-4S] Cluster of NFU1 Requires the Coordinated Donation of Two [2Fe-2S] Clusters from the Scaffold Proteins, ISCU2 and ISCA1. Hum. Mol. Genet. 2020, 29, 3165–3182. [Google Scholar] [CrossRef] [PubMed]
- Nasta, V.; Suraci, D.; Gourdoupis, S.; Ciofi-Baffoni, S.; Banci, L. A Pathway for Assembling [4Fe-4S]2+ Clusters in Mitochondrial Iron–Sulfur Protein Biogenesis. FEBS J. 2019, 287, 2312–2327. [Google Scholar] [CrossRef] [PubMed]
- Suraci, D.; Saudino, G.; Nasta, V.; Ciofi-Baffoni, S.; Banci, L. ISCA1 Orchestrates ISCA2 and NFU1 in the Maturation of Human Mitochondrial [4Fe-4S] Proteins. J. Mol. Biol. 2021, 433, 166924. [Google Scholar] [CrossRef]
- Cameron, J.M.; Janer, A.; Levandovskiy, V.; Mackay, N.; Rouault, T.A.; Tong, W.-H.; Ogilvie, I.; Shoubridge, E.A.; Robinson, B.H. Mutations in Iron-Sulfur Cluster Scaffold Genes NFU1 and BOLA3 Cause a Fatal Deficiency of Multiple Respiratory Chain and 2-Oxoacid Dehydrogenase Enzymes. Am. J. Hum. Genet. 2011, 89, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A Fatal Mitochondrial Disease Is Associated with Defective NFU1 Function in the Maturation of a Subset of Mitochondrial Fe-S Proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef]
- Nizon, M.; Boutron, A.; Boddaert, N.; Slama, A.; Delpech, H.; Sardet, C.; Brassier, A.; Habarou, F.; Delahodde, A.; Correia, I.; et al. Leukoencephalopathy with Cysts and Hyperglycinemia May Result from NFU1 Deficiency. Mitochondrion 2014, 15, 59–64. [Google Scholar] [CrossRef]
- Lebigot, E.; Gaignard, P.; Dorboz, I.; Slama, A.; Rio, M.; de Lonlay, P.; Héron, B.; Sabourdy, F.; Boespflug-Tanguy, O.; Cardoso, A.; et al. Impact of Mutations within the [Fe-S] Cluster or the Lipoic Acid Biosynthesis Pathways on Mitochondrial Protein Expression Profiles in Fibroblasts from Patients. Mol. Genet. Metab. 2017, 122, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, F.; Ardissone, A.; Lamantea, E.; Garavaglia, B.; Zeviani, M.; Farina, L.; Ghezzi, D.; Moroni, I. Cavitating Leukoencephalopathy with Multiple Mitochondrial Dysfunction Syndrome and NFU1 Mutations. Front. Genet. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Tonduti, D.; Dorboz, I.; Imbard, A.; Slama, A.; Boutron, A.; Pichard, S.; Elmaleh, M.; Vallée, L.; Benoist, J.F.; Ogier, H.; et al. New Spastic Paraplegia Phenotype Associated to Mutation of NFU1. Orphanet J. Rare Dis. 2015, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Mayr, J.A.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.L.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, Biochemical, and Genetic Spectrum of Seven Patients with NFU1 Deficiency. Front. Genet. 2015, 6, 123. [Google Scholar] [CrossRef]
- Ferrer-Cortès, X.; Narbona, J.; Bujan, N.; Matalonga, L.; del Toro, M.; Arranz, J.A.; Riudor, E.; Garcia-Cazorla, A.; Jou, C.; O’Callaghan, M.; et al. A Leaky Splicing Mutation in NFU1 Is Associated with a Particular Biochemical Phenotype. Consequences for the Diagnosis. Mitochondrion 2016, 26, 72–80. [Google Scholar] [CrossRef]
- Jin, D.; Yu, T.; Zhang, L.; Wang, T.; Hu, J.; Wang, Y.; Yang, X.A. Novel NFU1 Variants Induced MMDS Behaved as Special Leukodystrophy in Chinese Sufferers. J. Mol. Neurosci. 2017, 62, 255–261. [Google Scholar] [CrossRef]
- De Souza, P.V.S.; Bortholin, T.; Burlin, S.; Naylor, F.G.M.; de Rezende Pinto, W.B.V.; Oliveira, A.S.B. NFU1-Related Disorders as Key Differential Diagnosis of Cavitating Leukoencephalopathy. J. Pediatr. Genet. 2018, 7, 40–42. [Google Scholar] [CrossRef]
- Paquay, S.; Barrea, C.; Sluysmans, T.; Vachiery, J.-L.; Loeckx, I.; Seneca, S.; Vô, C.; Nassogne, M.-C. Idiopathic Pulmonary Arterial Hypertension in Early Infancy: Excluding NFU1 Deficiency. Ann. Pediatr. Cardiol. 2019, 12, 325–328. [Google Scholar] [CrossRef]
- Uzunhan, T.A.; Çakar, N.E.; Seyhan, S.; Aydin, K. A Genetic Mimic of Cerebral Palsy: Homozygous NFU1 Mutation with Marked Intrafamilial Phenotypic Variation. Brain Dev. 2020, 42, 756–761. [Google Scholar] [CrossRef]
- Birjiniuk, A.; Glinton, K.E.; Villafranco, N.; Boyer, S.; Laufman, J.; Mizerik, E.; Scott, D.; Elsea, S.H.; Galambos, C.; Varghese, N.P.; et al. Multiple Mitochondrial Dysfunctions Syndrome 1: An Unusual Cause of Developmental Pulmonary Hypertension. Am. J. Med. Genet. 2020, 182, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.G.; Neville, K.L.; McNamara, N.A.; Keegan, C.E.; Elsea, S.H. Clinical Reasoning: A 12-Month-Old Child with Hypotonia and Developmental Delays. Neurology 2020, 95, 184–187. [Google Scholar] [CrossRef]
- Touraine, B.; Boutin, J.P.; Marion-Poll, A.; Briat, J.F.; Peltier, G.; Lobréaux, S. Nfu2: A Scaffold Protein Required for [4Fe-4S] and Ferredoxin Iron-Sulphur Cluster Assembly in Arabidopsis Chloroplasts. Plant J. 2004, 40, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Py, B.; Gerez, C.; Angelini, S.; Planel, R.; Vinella, D.; Loiseau, L.; Talla, E.; Brochier-Armanet, C.; Garcia Serres, R.; Latour, J.-M.; et al. Molecular Organization, Biochemical Function, Cellular Role and Evolution of NfuA, an Atypical Fe-S Carrier. Mol. Microbiol. 2012, 86, 155–171. [Google Scholar] [CrossRef]
- Wachnowsky, C.; Wesley, N.A.; Fidai, I.; Cowan, J.A. Understanding the Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 1 (MMDS1)—Impact of a Disease-Causing Gly208Cys Substitution on Structure and Activity of NFU1 in the Fe/S Cluster Biosynthetic Pathway. J. Mol. Biol. 2017, 429, 790–807. [Google Scholar] [CrossRef]
- Niihori, M.; Eccles, C.A.; Kurdyukov, S.; Zemskova, M.; Varghese, M.V.; Stepanova, A.A.; Galkin, A.; Rafikov, R.; Rafikova, O. Rats with Human Mutation of NFU1 Develop Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 231–242. [Google Scholar] [CrossRef]
- Haack, T.B.; Rolinski, B.; Haberberger, B.; Zimmermann, F.; Schum, J.; Strecker, V.; Graf, E.; Athing, U.; Hoppen, T.; Wittig, I.; et al. Homozygous Missense Mutation in BOLA3 Causes Multiple Mitochondrial Dysfunctions Syndrome in Two Siblings. J. Inherit. Metab. Dis. 2013, 36, 55–62. [Google Scholar] [CrossRef]
- Baker, P.R.; Friederich, M.W.; Swanson, M.A.; Shaikh, T.; Bhattacharya, K.; Scharer, G.H.; Aicher, J.; Creadon-Swindell, G.; Geiger, E.; MacLean, K.N.; et al. Variant Non Ketotic Hyperglycinemia Is Caused by Mutations in LIAS, BOLA3 and the Novel Gene GLRX5. Brain 2014, 137, 366–379. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in Children with Mitochondrial Disease: Prognosis and Genetic Background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Bindu, P.S.; Sonam, K.; Chiplunkar, S.; Govindaraj, P.; Nagappa, M.; Vekhande, C.C.; Aravinda, H.R.; Ponmalar, J.J.; Mahadevan, A.; Gayathri, N.; et al. Mitochondrial Leukoencephalopathies: A Border Zone between Acquired and Inherited White Matter Disorders in Children? Mult. Scler. Relat. Disord. 2018, 20, 84–92. [Google Scholar] [CrossRef]
- Nikam, R.M.; Gripp, K.W.; Choudhary, A.K.; Kandula, V. Imaging Phenotype of Multiple Mitochondrial Dysfunction Syndrome 2, a Rare BOLA3-Associated Leukodystrophy. Am. J. Med. Genet. 2018, 176, 2787–2790. [Google Scholar] [CrossRef]
- Nishioka, M.; Inaba, Y.; Motobayashi, M.; Hara, Y.; Numata, R.; Amano, Y.; Shingu, K.; Yamamoto, Y.; Murayama, K.; Ohtake, A.; et al. An Infant Case of Diffuse Cerebrospinal Lesions and Cardiomyopathy Caused by a BOLA3 Mutation. Brain Dev. 2018, 40, 484–488. [Google Scholar] [CrossRef]
- Stutterd, C.A.; Lake, N.J.; Peters, H.; Lockhart, P.J.; Taft, R.J.; van der Knaap, M.S.; Vanderver, A.; Thorburn, D.R.; Simons, C.; Leventer, R.J. Severe Leukoencephalopathy with Clinical Recovery Caused by Recessive BOLA3 Mutations. JIMD Rep. 2019, 43, 63–70. [Google Scholar] [CrossRef]
- Meldau, S.; Fratter, C.; Bhengu, L.N.; Sergeant, K.; Khan, K.; Riordan, G.T.; Berman, P.A.M. Pitfalls of Relying on Genetic Testing Only to Diagnose Inherited Metabolic Disorders in Non-Western Populations-5 Cases of Pyruvate Dehydrogenase Deficiency from South Africa. Mol. Genet. Metab. Rep. 2020, 24, 100629. [Google Scholar] [CrossRef]
- Uzarska, M.A.; Nasta, V.; Weiler, B.D.; Spantgar, F.; Ciofi-Baffoni, S.; Saviello, M.R.; Gonnelli, L.; Mühlenhoff, U.; Banci, L.; Lill, R. Mitochondrial Bol1 and Bol3 Function as Assembly Factors for Specific Iron-Sulfur Proteins. eLife 2016, 5, e16673. [Google Scholar] [CrossRef]
- Torraco, A.; Ardissone, A.; Invernizzi, F.; Rizza, T.; Fiermonte, G.; Niceta, M.; Zanetti, N.; Martinelli, D.; Vozza, A.; Verrigni, D.; et al. Novel Mutations in IBA57 Are Associated with Leukodystrophy and Variable Clinical Phenotypes. J. Neurol. 2017, 264, 102–111. [Google Scholar] [CrossRef]
- Saudino, G.; Suraci, D.; Nasta, V.; Ciofi-Baffoni, S.; Banci, L. Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by Cys59tyr Bola3 Mutation. Int. J. Mol. Sci. 2021, 22, 4848. [Google Scholar] [CrossRef]
- Yu, Q.; Tai, Y.; Tang, Y.; Zhao, J.; Negi, V.; Culley, M.K.; Pilli, J.; Sun, W.; Brugger, K.; Saggar, R.; et al. BOLA3 Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2020, 139, 2238–2255. [Google Scholar] [CrossRef]
- Ajit Bolar, N.; Vanlander, A.V.; Wilbrecht, C.; van der Aa, N.; Smet, J.; de Paepe, B.; Vandeweyer, G.; Kooy, F.; Eyskens, F.; de Latter, E.; et al. Mutation of the Iron-Sulfur Cluster Assembly Gene IBA57 Causes Severe Myopathy and Encephalopathy. Hum. Mol. Genet. 2013, 22, 2590–2602. [Google Scholar] [CrossRef]
- Lossos, A.; Stümpfig, C.; Stevanin, G.; Gaussen, M.; Zimmerman, B.-E.; Mundwiller, E.; Asulin, M.; Chamma, L.; Sheffer, R.; Misk, A.; et al. Fe/S Protein Assembly Gene IBA57 Mutation Causes Hereditary Spastic Paraplegia. Neurology 2015, 84, 659–667. [Google Scholar] [CrossRef]
- Debray, F.-G.; Stümpfig, C.; Vanlander, A.V.; Dideberg, V.; Josse, C.; Caberg, J.-H.; Boemer, F.; Bours, V.; Stevens, R.; Seneca, S.; et al. Mutation of the Iron-Sulfur Cluster Assembly Gene IBA57 Causes Fatal Infantile Leukodystrophy. J. Inherit. Metab. Dis. 2015, 38, 1147–1153. [Google Scholar] [CrossRef]
- Ishiyama, A.; Sakai, C.; Matsushima, Y.; Noguchi, S.; Mitsuhashi, S.; Endo, Y.; Hayashi, Y.K.; Saito, Y.; Nakagawa, E.; Komaki, H.; et al. IBA57 Mutations Abrogate Iron-Sulfur Cluster Assembly Leading to Cavitating Leukoencephalopathy. Neurol. Genet. 2017, 3, e184. [Google Scholar] [CrossRef]
- Hamanaka, K.; Miyatake, S.; Zerem, A.; Lev, D.; Blumkin, L.; Yokochi, K.; Fujita, A.; Imagawa, E.; Iwama, K.; Nakashima, M.; et al. Expanding the Phenotype of IBA57 Mutations: Related Leukodystrophy Can Remain Asymptomatic. J. Hum. Genet. 2018, 63, 1223–1229. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, J.; Zhang, Z.; Zhou, L.; Jiang, Y.; Wang, J.; Xiao, J.; Wu, Y. Phenotypic Spectrum of Mutations in IBA57, a Candidate Gene for Cavitating Leukoencephalopathy. Clin. Genet. 2018, 93, 235–241. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, M.; Zhang, Z.; Zhou, L.; Kong, W.; Jiang, Y.; Wang, J.; Xiao, J.; Wu, Y. Genotypic Spectrum and Natural History of Cavitating Leukoencephalopathies in Childhood. Pediatr. Neurol. 2019, 94, 38–47. [Google Scholar] [CrossRef]
- Hu, C.; Li, X.; Zhao, L.; Shi, Y.; Zhou, S.; Wu, B.; Wang, Y. Clinical and Molecular Characterization of Pediatric Mitochondrial Disorders in South of China. Eur. J. Med. Genet. 2020, 63, 103898. [Google Scholar] [CrossRef]
- Al-Hassnan, Z.N.; Al-Dosary, M.; Alfadhel, M.; Faqeih, E.A.; Alsagob, M.; Kenana, R.; Almass, R.; Al-Harazi, O.S.; Al-Hindi, H.; Malibari, O.I.; et al. ISCA2 Mutation Causes Infantile Neurodegenerative Mitochondrial Disorder. J. Med. Genet. 2015, 52, 186–194. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Nashabat, M.; Alrifai, M.T.; Alshaalan, H.; al Mutairi, F.; Al-Shahrani, S.A.; Plecko, B.; Almass, R.; Alsagob, M.; Almutairi, F.B.; et al. Further Delineation of the Phenotypic Spectrum of ISCA2 Defect: A Report of Ten New Cases. Eur. J. Paediatr. Neurol. 2018, 22, 46–55. [Google Scholar] [CrossRef]
- Alaimo, J.T.; Besse, A.; Alston, C.L.; Pang, K.; Appadurai, V.; Samanta, M.; Smpokou, P.; McFarland, R.; Taylor, R.W.; Bonnen, P.E. Loss-of-Function Mutations in ISCA2 Disrupt 4Fe-4S Cluster Machinery and Cause a Fatal Leukodystrophy with Hyperglycinemia and MtDNA Depletion. Hum. Mutat. 2018, 39, 537–549. [Google Scholar] [CrossRef]
- Alfadhel, M. Multiple Mitochondrial Dysfunctions Syndrome 4 Due to ISCA2 Gene Defects: A Review. Child Neurol. Open 2019, 6, 2329048X1984737. [Google Scholar] [CrossRef]
- Toldo, I.; Nosadini, M.; Boscardin, C.; Talenti, G.; Manara, R.; Lamantea, E.; Legati, A.; Ghezzi, D.; Perilongo, G.; Sartori, S. Neonatal Mitochondrial Leukoencephalopathy with Brain and Spinal Involvement and High Lactate: Expanding the Phenotype of ISCA2 Gene Mutations. Metab. Brain Dis. 2018, 33, 805–812. [Google Scholar] [CrossRef]
- Eidi, M.; Garshasbi, M. A Novel ISCA2 Variant Responsible for an Early-Onset Neurodegenerative Mitochondrial Disorder: A Case Report of Multiple Mitochondrial Dysfunctions Syndrome 4. BMC Neurol. 2019, 19, 153. [Google Scholar] [CrossRef]
- Hartman, T.G.; Yosovich, K.; Michaeli, H.G.; Blumkin, L.; Ben-Sira, L.; Lev, D.; Lerman-Sagie, T.; Zerem, A. Expanding the Genotype-Phenotype Spectrum of ISCA2-Related Multiple Mitochondrial Dysfunction Syndrome-Cavitating Leukoencephalopathy and Prolonged Survival. Neurogenetics 2020, 21, 243–249. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe-S Cluster Biogenesis and Its Implication in Disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Shukla, A.; Hebbar, M.; Srivastava, A.; Kadavigere, R.; Upadhyai, P.; Kanthi, A.; Brandau, O.; Bielas, S.; Girisha, K.M. Homozygous p.(Glu87Lys) Variant in ISCA1 Is Associated with a Multiple Mitochondrial Dysfunctions Syndrome. J. Hum. Genet. 2017, 62, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Kaur, P.; Girisha, K. Report of the Third Family with Multiple Mitochondrial Dysfunctions Syndrome 5 Caused by the Founder Variant p.(Glu87Lys) in ISCA1. J. Pediatr. Genet. 2018, 7, 130–133. [Google Scholar] [CrossRef]
- Torraco, A.; Stehling, O.; Stümpfig, C.; Rösser, R.; de Rasmo, D.; Fiermonte, G.; Verrigni, D.; Rizza, T.; Vozza, A.; di Nottia, M.; et al. ISCA1 Mutation in a Patient with Infantile-Onset Leukodystrophy Causes Defects in Mitochondrial [4Fe-4S] Proteins. Hum. Mol. Genet. 2018, 27, 2739–2754. [Google Scholar] [CrossRef] [PubMed]
- Lebigot, E.; Hully, M.; Amazit, L.; Gaignard, P.; Michel, T.; Rio, M.; Lombès, M.; Thérond, P.; Boutron, A.; Golinelli-Cohen, M.P. Expanding the Phenotype of Mitochondrial Disease: Novel Pathogenic Variant in ISCA1 Leading to Instability of the Iron-Sulfur Cluster in the Protein. Mitochondrion 2020, 52, 75–82. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S.; Daruich, A. The Eye on Mitochondrial Disorders. J. Child Neurol. 2016, 31, 652–662. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Nb of Patients (Nb and Origin of Families) | Age at Onset/Age at Death | Metabolic Findings | Impaired Mitochondrial Proteins | Pulmonary Hypertension | Clinical Feature | Brain MRI/ Spectroscopy | Genotype (NFU1:NM_001002755.4) |
|---|---|---|---|---|---|---|---|---|
| [47] | 3 (1F, Mexico) | Neonatal/1st m | High lactate and glycine levels (bl) | Complexes I + III, II, IV, PDH, KGDH, lipoylated proteins | No | FD, hypotonia, respiratory failure | NA | c.[545G > A]; [545G > A]; p.[(Arg182Gln)]; [(Arg182Gln)] |
| [48] | 10 (9F incl. 4F of basque origin) | 1–9 m/2–15 m | High lactate and glycine (bl and CSF) and amino-adipate, α-KG, 2 ketoadipate, tyglyglycine (ur) levels | PDH, lipoylated proteins | Yes (7/10) | FD (6/10), dysmorphia (1/10), skills regression (6/10) | LD and semioval necrosis (n = 1), semioval center and cerebellum lesions (n = 1), cerebral atrophy (n = 1) | c.[622G > T]; [622G > T], p.[(Gly208Cys)]; [(Gly208Cys)] (9/10) c.[622G > T]; [545 + 5G > A], p.[(Gly208Cys)]; [Pro183Glnfs*10] (1/10) |
| [49,50] | 1 (France) | 5 m/3 y | High lactate (bl), glycine (bl and CSF) and α-KG (ur) levels | Complexes I, II, PDH, KGDH, lipoylated proteins | In acute episodes | Severe DR, transient MD, myoclonies, hypotonia | Progressive LE, kysts in PV WM and in CC | c.[565G > A]; [622G > T], p.[(Gly189Arg)]; [(Gly208Cys)] |
| [51] | 1 (Italia) | 7 m/18 m | High lactate, pyruvate, glycine (bl), succinate, fumarate, glutarate (ur) levels | Complexes I, II, IV, PDH | No | FD, spasticity, severe DR | Diffuse LE (deep WM), cavitations/peak of lactate | c.[565G > A]; [629G > T], p.[(Gly189Arg)]; [(Cys210Phe)] |
| [52] | 1 (unknown) | 18 m/alive at 30 y | High lactate, pyruvate (bl), glycine (bl and CSF) levels | Complexes II, IV, PDH, KGDH, | No | Spastic tetraparesis, scoliosis, DR after fever, MSN | PV WM and CC abnormal signal, CC atrophy/reduced NAA peak, mild lactate, and high choline peaks | c.[146del]; [565G > A], p.[(Pro49Leufs*8)]; [(Gly189Arg)] |
| [53] | 7 (5F, Germany (G), Romania/Serbia (RS) Pakistan (P), Belgium (B)) | Birth–5 m/3 m–30 m | High lactate (6/7), glycine (7/7) levels (bl) | Complexes I (2/5), II (3/3), IV (1/4), PDH (5/5) | Yes (4/7) | Muscular hypotonia (6/7), apnea (4/7), FD (4/7), tubulopathy (1/7), CMP (2/7) | LE (3/4) affecting PV (2/4), capsula interna (3/4), or upper cervical cord (1/4); hypomyelination (2/4) | g.[69400462C > A]; [69592691_69648327del], p.[(Gly208Cys)]; [?] (G) c.[565G > A]; [568G > A], p.[(Gly189Arg)]; [(Gly190Arg)] (G) c.[544C > T]; [?], p.[(Arg182Trp)]; [?]) (RS) c.[302 + 3A > G]; [302 + 3A > G], p.[(Val56Glyfs∗9)]; [(Val56Glyfs∗9)] (3/7, P) c.[62G > C]; [622G > T], p.[(Arg21Pro)]; [(Gly208Cys)] (B) |
| [54] | 2 (2F, unknown) | 2–15 m/14–19 m | High lactate (CSF) and glycine (ur, bl and CSF), 2-amino and 2-hydroxyadipate, tyglylglycine, isovalerylglycine (ur) levels | GCS (1/1), lipoylated proteins (2/2), CII + CIII (1/1) | Yes (1/2) | Severe spasticity, axial hypotonia, apnea, DR | cysts in temporal lobes with glycine peak (1/2), WM spongiosis, PV WM and CC LD (1/2) | c.[622G> T]; [545 + 5G > A], p.[(Gly208Cys)]; [Pro183Glnfs*10] |
| [55] | 2 (2F, China) | 3–4 m/3–4 m | High lactate level (bl, ur, 1/2), normal glycine levels (2/2) | NA | Yes (1/2) | DR (2/2), seizures, muscular hypertonia, and ascitis (1/2) | LD in subcortical WM (internal—external capsule, CC, medulla) or midbrain, thalamus, lateral ventricle, and centrum ovale | c.[545 + 5G > A]; [545 + 5G > A], p.[(Pro183Glnfs*10)]; [(Pro183Glnfs*10)] c.[721G > T]; [303_369del], p.[(Val241Phe)]; [?] |
| [50] | 3 (3F, unknown) | 3–3, 5–7 m/4 m to 3 y | High lactate (bl 3/3) and glycine (bl:2/2, ur:1/2 and CSF:1/1), fumarate (1/1), 2-amino-adipate (pl) or 2 keto-adipate (ur) levels | Complexes I (3/3), II (2/3), IV (1/3), PDH (3/3), KGDH (2/3), lipoylated proteins (2/3) | Yes (1/3) | Seizures (2/3), spasticity (1/3), apnea (1/3), PMD (2/3) | Normal (1/3), WM abnormalities (1/3), necrotizing LD (1/3) /lactate peak (1/2) | c.[146del], [303–1988_c.369 + 1021del], p.[(Pro49Leufs*8)]; [?] c.[622G > T]; [629G > T], p.[(Gly208Cys)]; [(Cys210Phe)] c.[179T > G]; [179T > G], p.[(Phe60Cys)]; [(Phe60Cys)] |
| [56] | 1 (Brazil) | 4 m/8 m | High lactate (bl, ur) and glutaric acid (ur) levels | NA | No (normal ECG) | Severe hypotonia, spastic tetraparesis, DR, lethargy | Cavitating cystic LE (PV, deep, subcortical areas) | c.[545G > A]; [622G > T], p.[(Arg182Gln)]; [(Gly208Cys)] |
| [57] | 1 (Portugal) | 1 m/10 m | Elevated glycine level (bl) | NA | Severe (onset) | FD, DR, seizures, spasticity, and axial hypotonia | WM necrotic areas, PV WM abnormal signals | c.[622G > T]; [622G > T], p.[(Gly208Cys)]; [(Gly208Cys)] |
| [58] | 1 (Turkey) | 23 m/alive at 41 m | High lactate and glycine (bl) levels | NA | No (normal ECG) | Episodes of DR and legs spasticity after fever | Frontal PV cysts and frontal WM and CC hyperintensity, | c.[565G > A]; [565G > A], p.[(Gly189Arg)]; [(Gly189Arg)] |
| [59] | 2 (1F, Guatemala) | 2–3 m/5–7 m | High lactate (bl) and α-KG (ur) levels | E3 and PDH | Yes (with cardiomegaly or dilated CMP) | FD, emesis, developmental delay, spasticity | NA | c.[544C > T]; [622G > T], p.[(Arg182Trp)]; [(Gly208Cys)] |
| [60] | 1 (unknown) | 7 m/12 m | High glycine (bl, CSF) and lactate (CSF) levels | NA | Yes (mild) | PMD, bradypnea, hypotonia | Bilateral basal ganglia, thalami, frontoparietal WM, cerebral peduncles, spinal cord abnormal signals/lactate peak (MRS) | c.[299C > G]; [398T > C], p.[(Ala100Gly)]; [(Leu133Pro)] |
| Ref. | Nb of Patients (Nb and Origin of Families) | Age at Onset/Age at Death | Metabolic Findings | Impaired Mitochondrial Proteins | Cardiomyo- pathy | Clinical Feature | Brain MRI/Spectroscopy | Genotype (BOLA3:NM_212552.2) |
|---|---|---|---|---|---|---|---|---|
| [47] | 1 (East Indian) | 4 m/11 m | High glycine level (bl, ur) | Complexes I and II, PDH, lipoylated protein | Dilated | FD, lethargy, respiratory distress, HMG, DD | NA | c.[123dupA]; [123dupA], p.[(Glu42Argfs*13)]; [(Glu42Argfs*13)] |
| [65] | 2 (1F, unknown) | <1 m/3 m | High pyruvate (bl, ur), lactate (bl, ur), and glycine levels (bl) | Complexes I, II, IV, PDH, lipoylated protein | Hyper-trophic | FD, SE, hypotonia, progressive encephalopathy | Delayed myelination, supra-, PV, and bifrontal and biparietal hyperintense lesions (T2) | c.[200T > A]; [200T > A], p.[(Ile67Asn)]; [(Ile67Asn)] |
| [66] | 3 (3F: Australia, India, African- American) | 6–8 m/7 m, 22 m, 11 y | High lactate (bl), glycine (CSF) levels | Complex II (n = 1/3), PDH (n = 1/2), lipoylated protein (n = 2/2), GCS (n = 3/3) | Hyper-trophic (n = 2/2) | FD, SE (n = 1/3), Myoclonus (2/3), SP and severe PMDD (1/3), skills regression (2/3), optic atrophy (n = 2/3) | PV WM and spinal cord lesions (n = 1/3), subcortical fibers lesions (n = 1/3) LD (n = 2/3), cerebral and cerebellar atrophy (n = 1/3) | c.[136C > T]; [136C > T], p.[(Arg46*)]; [(Arg46*)] |
| [67,68] | 4 (4F, Japan) | <1, 2, 6 m/3–11 m | Metabolic acidosis | Combined complex deficiencies (n = 4/4) | Hyper-trophic (n = 3/4) | Renal, respiratory, and/or liver failures, SE, FCMD (n = 1) | NA | c.[287A > G]; [287A > G], p.[(His96Arg)]; [(His96Arg)] (3/4) c.[225_229del]; [287A > G], p.[(Lys75Asnfs*9)]; [(His96Arg)] |
| [50] | 2 (1F, India) | 5–7 m/8 m | High pyruvate (bl), lactate (bl, CSF), glycine (bl, CSF, ur) levels | Complexes I, II, IV, PDH, lipoylated protein | No | FD, tetraparesis, hypotonia, PMDD | Diffuse demyelination/lactate peak | c.[136C > T]; [136C > T], p.[(Arg46*)]; [(Arg46*)] |
| [69] | 2 (unknown) | 18 m/alive at 12 y (n = 1) | NA | NA | NA | SPT (n = 1/2), skills regression, gait difficulty | PV and deep WM or fronto-parietal lesions/lactate peak (n = 1/1) | c.[295C > T]; [295C > T], p.[(Arg99Trp)]; [(Arg99Trp)] |
| [70] | 1 (unknown) | 8 m/15 m | High lactate (bl), tiglylglycine (ur) levels | PDH, lipoylated protein | NA | FD, hypertonicity then hypotonia, nystagmus | Symmetric ovoid areas and CC, cervical cord regions abnormal signal. PV, deep, subcortical WM lesions/lactate peak | c.[200T > A]; [220_222del], p.[(Ile67Asn)]; [(Glu74del)] |
| [71] | 1 (Japan) | 6 m/10 m | High lactate (bl, CSF), glycine (bl) levels | Complexes I, II, IV | Hypertrophic | FD, SE, eyelid movements, hypotonia, skill regression | Deep WM to spinal cord lesions | c.[287A > G]; [287A > G], p.[(His96Arg); [(His96Arg)] |
| [72] | 1 (England) | 18 m/alive at 8 y | Normal lactate (bl), high glycine (bl, ur) levels | PDH, normal RCC activity but low quantity of protein in complexes I and II | No | Ataxia, acute hemiparesis, cognitive regression, congenital hypothyroidism | subcortical WM, basal ganglia, brainstem and cerebellum abnormal signal and dysmyelination | c.[136C > T]; [176G > A], p.[(Arg46*)]; [(Cys59Tyr)] |
| [73] | 1 (South Africa) | 7 m/17 m | High lactate (bl), glycine (ur) levels | PDH | No | Sudden skill regression, floppiness, encephalopathy | Demyelination | c.[159dupT]; [159dupT], p.[(Asp54*)]; [(Asp54*)] |
| Ref. | Nb of Patients (Nb and Origin of Families) | Age at Onset/Age at Death | Metabolic Findings | Impaired Mitochondrial Proteins | Ophthalmological Findings | Clinical Feature | Brain MRI/Spectroscopy | Genotype (IBA57: NM_001010867) |
|---|---|---|---|---|---|---|---|---|
| [78] | 2 (1F, Morocco) | Antenatal/1 d, 15 d | Increased lactate (bl) and glycine (bl, CSF) levels | Complexes I, II, and IV, lipoylated proteins, IBA57 protein | NA | Polyhydramnios, microcephaly, severe hypotonia | Cerebral atrophy, polymicrogyria, CC hypoplasia | c.[941A > C]; [941A > C], p.[(Gln314Pro)]; [(Gln314Pro)] |
| [79] | 12 (1F, Palestine) | After 3 y/alive (12–65 y) | Normal lactate level | Complexes I, II, lipoylated proteins | Optic atrophy | Peripheral neuropathy, progressive spastic paraplegia | Hyperintensity in WM, cystic cavitation. Cerebellar, CC, CSC atrophy | c.[678A > G]; [678A > G], p.[Gly227Valfs]; [Gly227Valfs] |
| [80] | 1 (Morocco) | 6 m/17 m | Increased lactate and glycine levels (bl, CSF) | Complexes I, II, and IV, lipoylated proteins, | NA | FD with vomiting, opistothonos crises, progressive hypotonia, skills regression | Central and PV LE involving CC IC, USC, and cerebellar WM/lactate peak | c.[436C > T]; [436C > T], p.[(Arg146Trp)]; [(Arg146Trp)] |
| [50] | 2 (2F, unknown) | 3 w–8 m/2–11 m | High lactate level (bl, CSF) | Complexes I, II, lipoylated proteins, PDHc | NA | Hypotonia, with recurrent myoclonus, pyramidal syndrome, apnea | PV LD, myelination delay | c.[335T > G]; [437G > C], p.[(Leu112Trp)]; [(Arg146Pro)] c.[316A > G]; [738C > G]; p.[(Thr106Ala)]; [(Asn246Lys)] |
| [81] | 3 (2F, unknown) | 11 m–2 y/alive at 6–7 y | Intermittent high lactate level (bl, CSF) | Complexes I, II, lipoylated proteins, IBA57 protein level | NA | Spasticity, axial hypotonia, PMR | PV and CC LE with cysts or cavitation | c.[323A > C]; [940C > T], p.[(Tyr108Ser)]; [(Gln314*)] c.[323A > C]; [150C > A], p.[(Tyr108Ser)]; [(Cys50*)] |
| [75] | 4 (4F, Tunisia, unknown) | 4–18 m/2 y (n = 2) or alive at 12–16 y | High lactate and pyruvate levels (bl or CSF, 3/4) | Complex II (fib: 3/4, muscle 2/3)), complex I fib (1/4) | Nystagmus (2/4), abnormal VEP (3/3) | Severe PMR (4/4), FD (3/4), spastic tetraparesis (3/4), severe hypotonia (1/4), epilepsy (1/4) | Cavitating LE or LD (3/4), cerebral and cerebellar WM involvement (3/4)/lactate peak (3/4) | c.[586T > G]; [c.686C > T], p.[(Trp196Gly]; [Pro229Leu] c.[87insGCCCAAGGTGC]; [313C > T], p.[(Arg30Alafs)]; [(Arg105Trp)] c.[706C > T]; [706C > T], p.[(Pro236Ser)]; [(Pro236Ser)] c.[316A > G]; [757G > C]; p.[(Thr106Ala]; [(Val253Leu)] |
| [69] | 2 (2F, unknown)) | 18–20 m/alive at 19–31 m | NA | NA | NA | Milestones loss, difficulty in standing, progressive quadriparesis | Large hyperintense lesions in FP WM (2/2), in CC, and multiple WM cysts (1/2) | c.[738C > G]; [802C > T], p.[(Asn246Lys)]; [(Arg268Cys)] c.[656A > G]; [706C > T], p.[(Tyr219Cys)]; [(Pro236Ser)] |
| [82] | 3 (2F, Israel, Japan) | 7–20 m/alive at 7–19–29 y | Normal lactate level (1/1) | Normal RCC activity (1/1) | Reduced vision with pale discs (2/3), pendular nystagmus (1/3); no (1/3) | No neurological signs (1/3), spasticity with learning disabilities (1/3), PMR, spasticity, seizures, severe hypotonia and PMD (1/3) | PV WM hypersignal, thin optic nerves, PV rarefaction with thin CC (2/3), progressive WM atrophy, and cystic degeneration (1/3) | c.[335T > C]; [c.588dup], p.[(Leu112Ser)]; [(Arg197Alafs)] c.[386A > T]; [731A > C], p.[(Asp129Val)]; [(Glu244Ala)] |
| [83] | 11 (9F, China) | 5–15 m/Alive at 11 m to 10 y | High blood lactate level (2/11) | NA | Nystagmus (3/11), visual impairment 3/11 | Spasticity and motor regression (11/11), seizures (2/11) | Cavitating lesions in the frontal, parieto-occipital WM, CC (7/11), temporal (4/11), cerebellar, or IC (1/11) | c.[286T > C]; [316A > G], p.[(Tyr96His)]; [(Thr106Ala)] (n = 2) c.[701A > G]; [782T > C], p.[(Asp234Gly)]; [(Ile261Thr)] c.[286T > C]; [697C > T], p.[(Tyr96His)]; [(Arg223*)] (n = 3) c.[188G > A]; [286T > C], p.[(Gly63Asp)]; [(Tyr96His)] c.[286T > C]; [307C > T], p.[(Tyr96His)]; [(Gln103*)] (n = 2) c.[286T > C]; [754G > T], p.[(Tyr96His)]; [(Gly252Cys)] c.[22C > T]; [286T > C], p.[(Arg8*)]; [(Tyr96His)] |
| [84] | 17 (unknown, China) | NA | NA | NA | Visual impairment (8/17) | NA | No progressive cavitating LE: involving diffuse (12/17 or deep WM (5/17), | c.[22C > T]; [286T > C], p.[(Arg8*)]; [(Tyr96His)] c.[188G > A]; [286T > C], p.[(Gly63Asp)]; [(Tyr96His)] (n = 2) c.[236C > T]; [307C > T], p.[(Pro79Leu)]; [(Gln103*)] c.[286T > C]; [697C > T], p.[(Tyr96His)]; [(Arg223*)] (n = 4) c.[286T > C]; [307C > T], p.[(Tyr96His)]; [(Gln103*)] (n = 2) c.[286T > C]; [316A > G], p.[(Tyr96His)]; [(Thr106Ala)] (n = 2) c.[286T > C]; [522_523del], p.[(Tyr96His)]; [(Leu175Alafs)] c.[286T > C]; [589_590del], p.[(Tyr96His)]; [(Arg197Alafs)] c.[286T > C]; [754G > T], p.[(Tyr96His)]; [(Gly252Cys)] c.[286T > C]; [1053G > A], p.[(Tyr96His)]; [(Trp351*)] c.[701A > G]; [782T > C], p.[(Asp234Gly)]; [(Ile261Thr)] |
| [85] | 2 (2F, China) | 5–15 m/unknown | Normal lactate level | NA | NA | PMR | LE | c.[286T > C]; [697C > T], p.[(Tyr96His)]; [(Arg223*)] c.[286T > C]; [589_590del], p.[(Tyr96His)]; [(Arg197Alafs)] |
| Ref. | Nb of Patients (Nb and Origin of Families) | Age at Onset/Age at Death | Metabolic Findings | Impaired Mitochondrial Proteins | Ophthalmological Findings | Clinical Feature | Brain MRI/Spectroscopy | Genotype (ISCA2: NM_194279) |
|---|---|---|---|---|---|---|---|---|
| [86] | 6 (5F, Saudi Arabia) | 3–7 m/11 m to 5 y (n = 4), alive (13–16 m, n = 2) | Negative | Complex I | Optic atrophy (6/6) | Spasticity, severe PMR | Lesions in cerebral (PV region to U fibers) and cerebellar WM, CC, internal capsule | c.[229G > A]; [229G > A], p.[(Gly77Ser)]; [(Gly77Ser)] |
| [87] | 1 (Saudi Arabia) | NA | NA | NA | NA | Neurodegeneration | Lesions in WM/Lactate peak | c.[229G > A]; [229G > A], p.[(Gly77Ser)]; [(Gly77Ser)] |
| [50] | 2 (2F, unknown) | 2 d, NA/12 d (n = 1), alive at 12 y | Elevated lactate (bl, CSF 1/2) | Complexes I, II, lipoylated protein | No | FD, spasticity (1/2), severe hypotonia | Leukodystrophy (1/2) WM atrophy (1/2) | c.[154C > T]; [154C > T], p.[(Leu52Phe)]; [(Leu52Phe)] c.[313A > G]; [313A > G], p.[(Arg105Gly)]; [(Arg105Gly)] |
| [88] | 10 (9F, Saudi Arabia) | 3–7 m/11–28 m (n = 5), alive between 10 to 34 m (n = 5) | Elevated lactate and glycine levels (bl, 2/2) | NA | Optic atrophy and nystagmus | Seizures 2/10, axial hypotonia with peripheral spasticity | Severe PMR/lactate peak (1/2), glycine peak (2/2) | c.[229G > A]; [229G > A], p.[(Gly77Ser)]; [(Gly77Ser)] |
| [89] | 2 (2F, Saudi Arabia) | 3–6 m/2 y, alive at 3 y | Elevated lactate (CSF), glycine, and glutamate (CSF, ur) levels | Complexes II, IV, lipoylated proteins, mtDNA depletion | Optic atrophy and nystagmus | PMR (2/2), spasticity, macrocephaly and syndactyly (1/2) | Lesions in cerebral (PV region to U fibers 1/2) and cerebellar WM (2/2), CC, internal capsule, spinal cord (1/2)/lactate peak (2/2) | c.[229G > A]; [229G > A], p.[(Gly77Ser)]; [(Gly77Ser)] |
| [91] | 1 (Italy) | 2 m/3 m | Initially normal, elevated lactate level (CSF, later) | Complexes II and IV (muscle) | Nystagmus, normal fundus oculi | Progressive severe hypotonia | Lesions in cerebral grey and WM (U fibers, CC, spinal cord) | c.[295delT]; [334A > G], p.[(Phe99Leufs*18)]; [(Ser112Gly)] |
| [92] | 1 (Iran) | 7 m/NA | NA | NA | NA | Hypotonia, irritability, malaise, muscle stiffness | Lesions in PV WM; centrum semiovale, cerebellar peduncles/peak of lactate, increase choline | c.[355G > A]; [355G > A], p.[(Ala119Thr)]; [(Ala119Thr)] |
| [93] | 1 (Yemen/Tunisia) | 11 m/alive 11 y | Mildly elevated lactate (bl, CSF) | All respiratory chain complexes | Nystagmus | Severe hypotonia, PMR, spasticity later, | Progressive lesions in PV region, genu, splenium (edema), thick CC, no spinal cord involvement/lactate peak | c.[5C > A]; [413C > G], p.[(Ala2Asp)]; [(Pro138Arg)] |
| Ref. | Nb of Patients (Nb and Origin of Families) | Age at Onset/Age at Death | Metabolic Findings | Impaired Mitochondrial Proteins | Ophthalmic Findings | Clinical Feature | Brain MRI Spectroscopy | Genotype (ISCA1:NM_030940.3) |
|---|---|---|---|---|---|---|---|---|
| [95] | 4 (2F, India) | Neonate (2/4) 2–3 m (2/4)/11 m to 5 y | Elevated CPK (1/2) | NA | Nystagmus (1/4) fundus pigmentation (1/2) | Major PMDD, SP, SE, FD | Pachygyria (2/4), VMG, cerebral and cerebellar WM LD/lactate peak | c.[259G > A]; [259G > A], p.[(Gln87Lys)]; [(Gln87Lys)] |
| [97] | 1 (Italia) | 3 m/11 y | High lactate (bl) and glycine (ur) levels | Complexes II, III, and IV, ATP synthesis, lipoylated proteins | Nystagmus | Mild PMDD, SP, FD | Vacuolating LD, TCC | c.[29T > G]; [29T > G], p.[(Val10Gly)]; [(Val10Gly)] |
| [96] | 1 (India) | 3 m/NA | NA | NA | No | Major PMDD, SP, SE | Pachygyria (n = 2/4), VMG, cerebral and cerebellar WM LD/lactate peak | c.[259G > A]; [259G > A], p.[(Gln87Lys)]; [(Gln87Lys)] |
| [98] | 1 (Egypt) | 20 m/alive at 7 y | High lactate (bl) level | Complexes I, II and IV, lipoylated proteins | No | Mild PMDD, SP, hemiparesis | Vacuolating LD, TCC, VMG/lactate peak | c.[302A > G]; [302A > G], p.[(Tyr101Cys)]; [(Tyr101Cys)] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebigot, E.; Schiff, M.; Golinelli-Cohen, M.-P. A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation. Biomedicines 2021, 9, 989. https://doi.org/10.3390/biomedicines9080989
Lebigot E, Schiff M, Golinelli-Cohen M-P. A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation. Biomedicines. 2021; 9(8):989. https://doi.org/10.3390/biomedicines9080989
Chicago/Turabian StyleLebigot, Elise, Manuel Schiff, and Marie-Pierre Golinelli-Cohen. 2021. "A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation" Biomedicines 9, no. 8: 989. https://doi.org/10.3390/biomedicines9080989
APA StyleLebigot, E., Schiff, M., & Golinelli-Cohen, M.-P. (2021). A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation. Biomedicines, 9(8), 989. https://doi.org/10.3390/biomedicines9080989