
Acute Respiratory Distress Syndrome: Focus on Viral Origin and Role of Pulmonary Lymphatics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
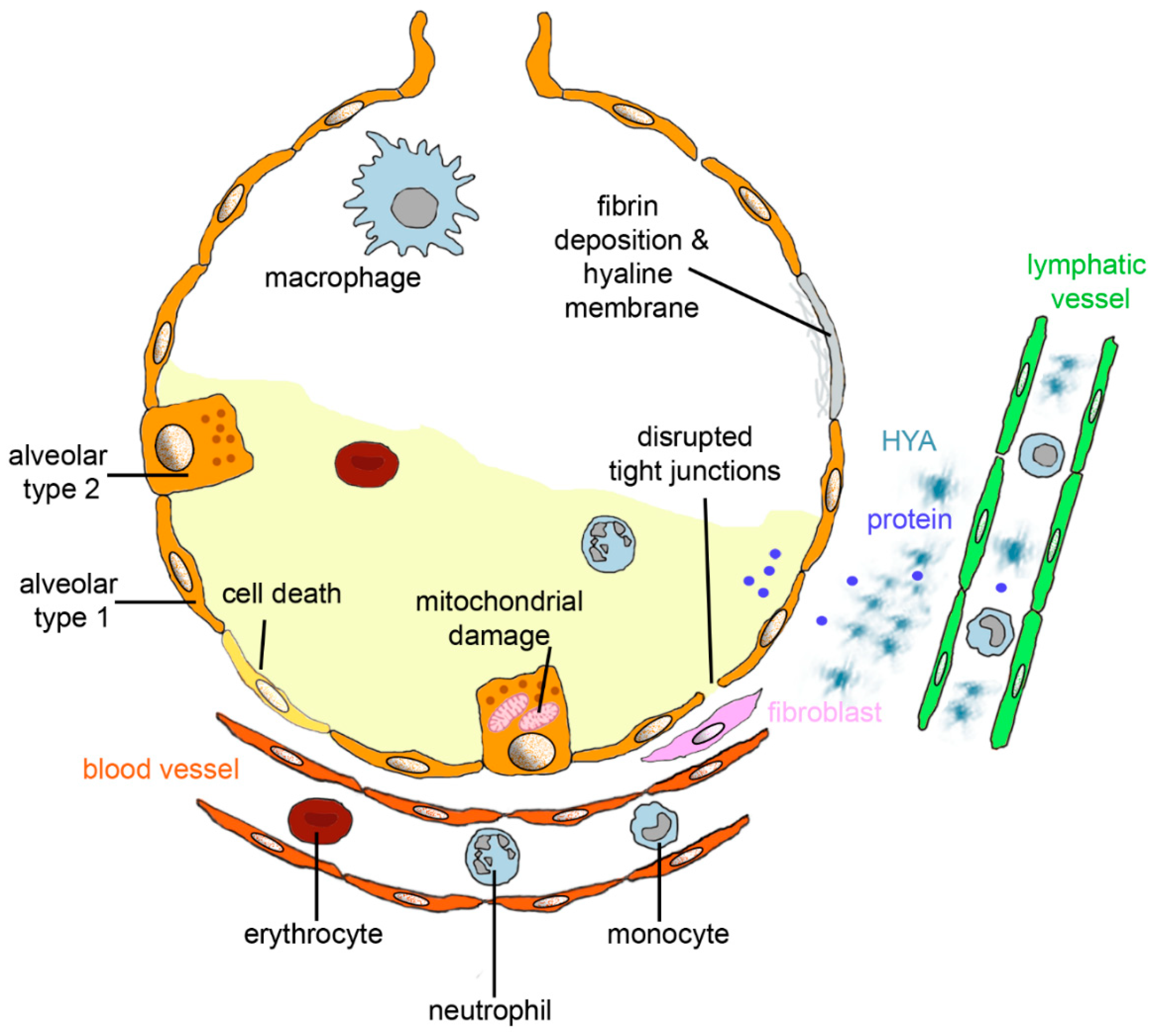
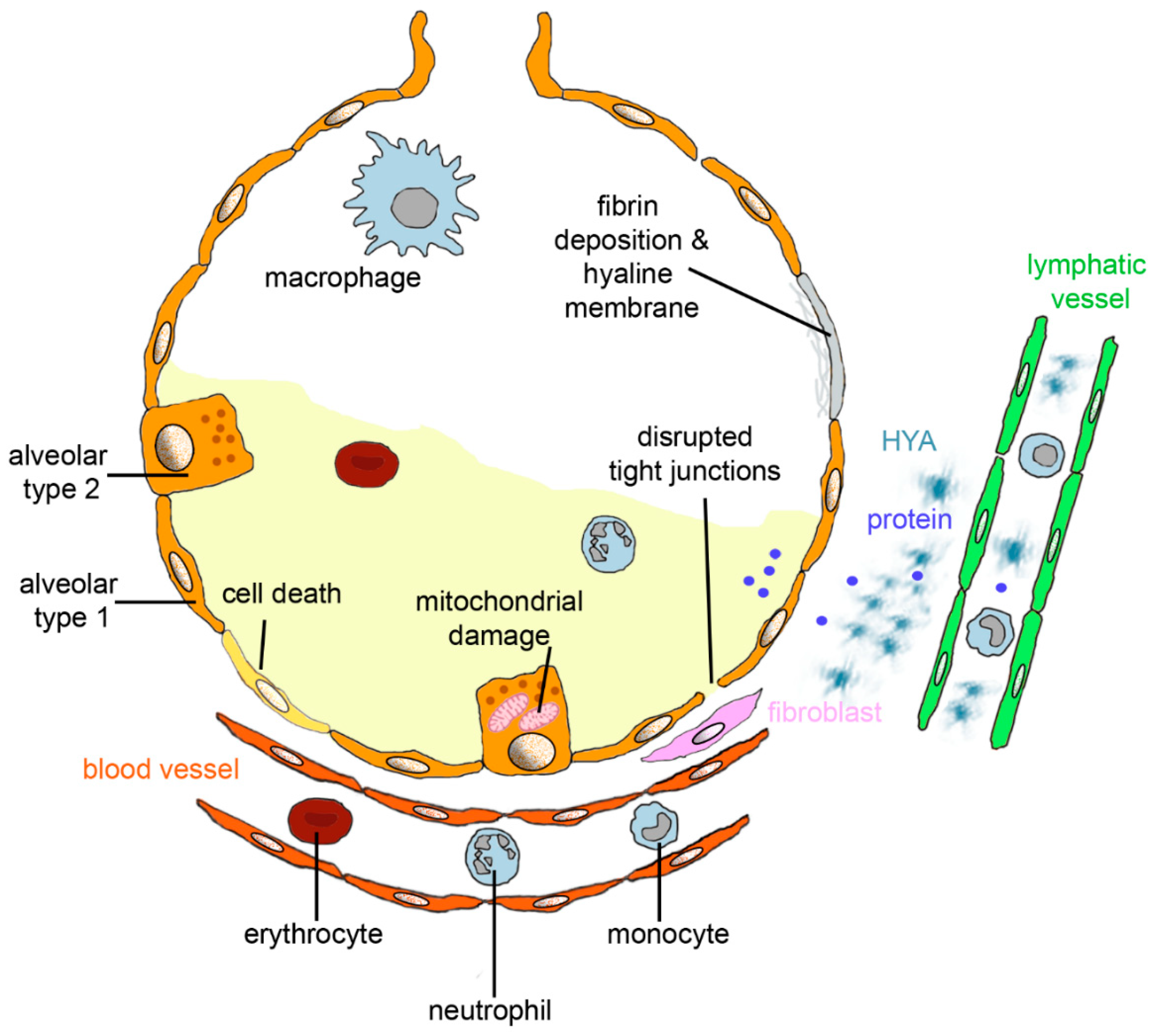
2. Lung Changes in ARDS
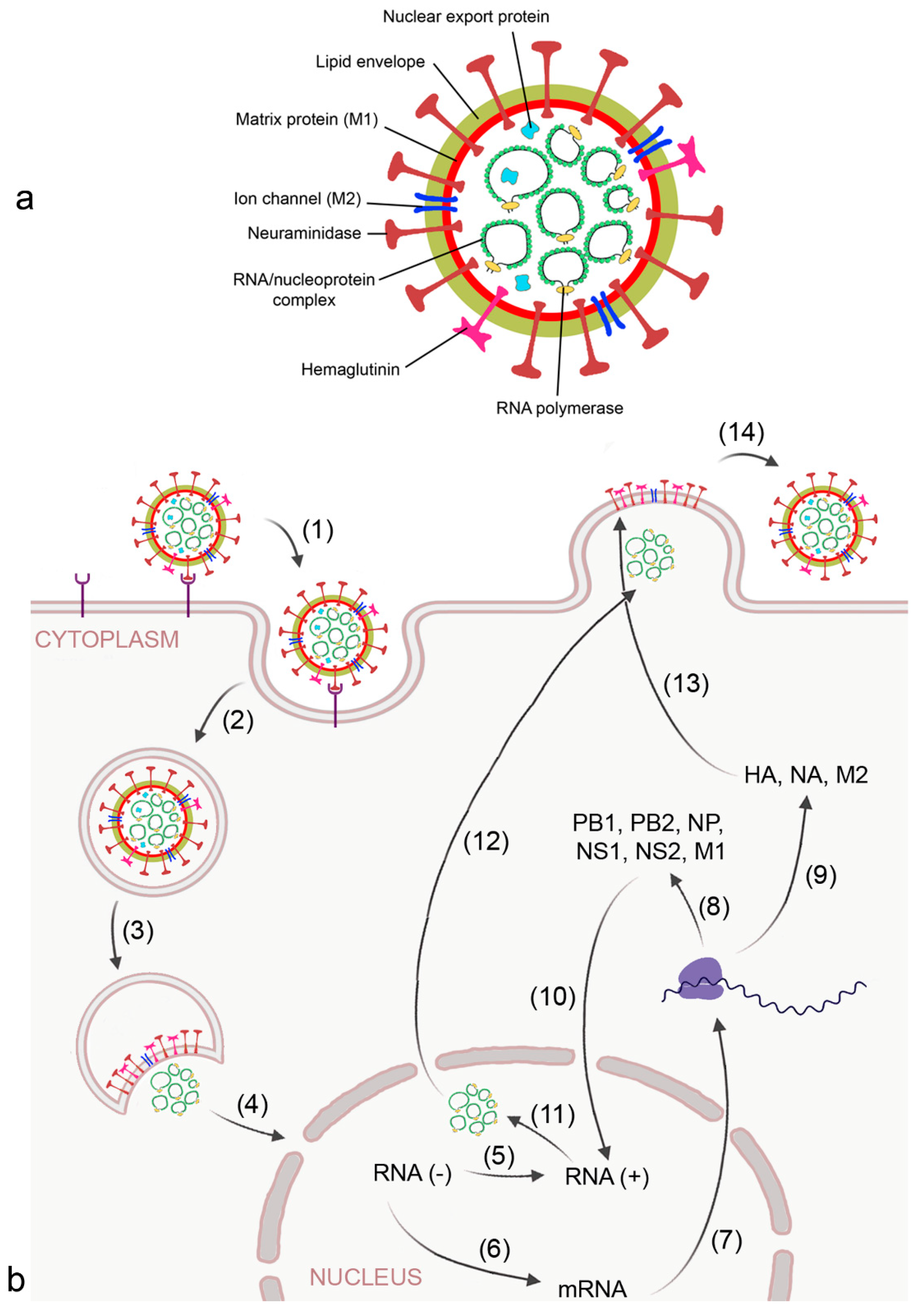
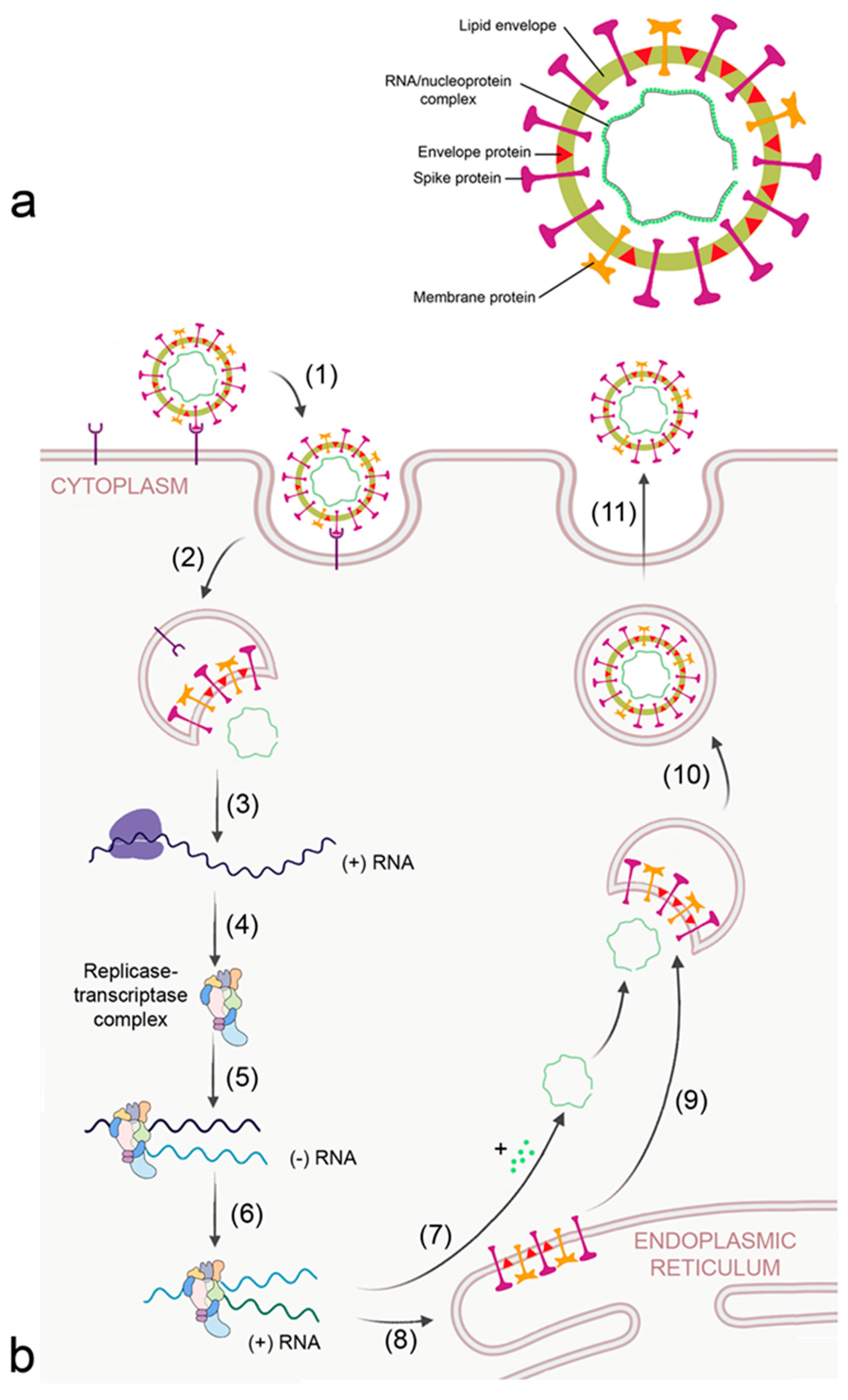
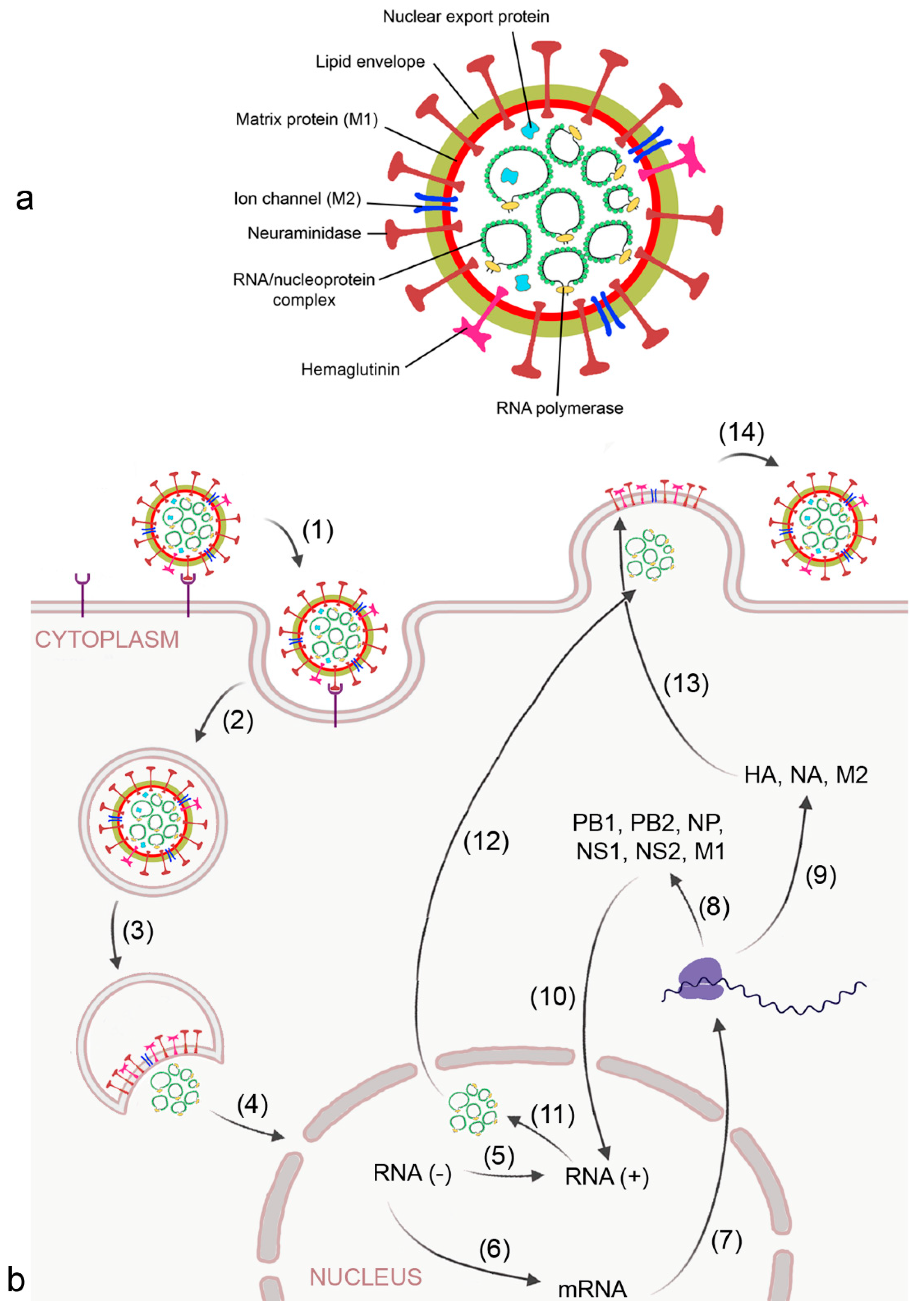
2.1. Virus-Induced ARDS
2.2. Treatment Options for ARDS
2.3. Pulmonary Lymphatic System
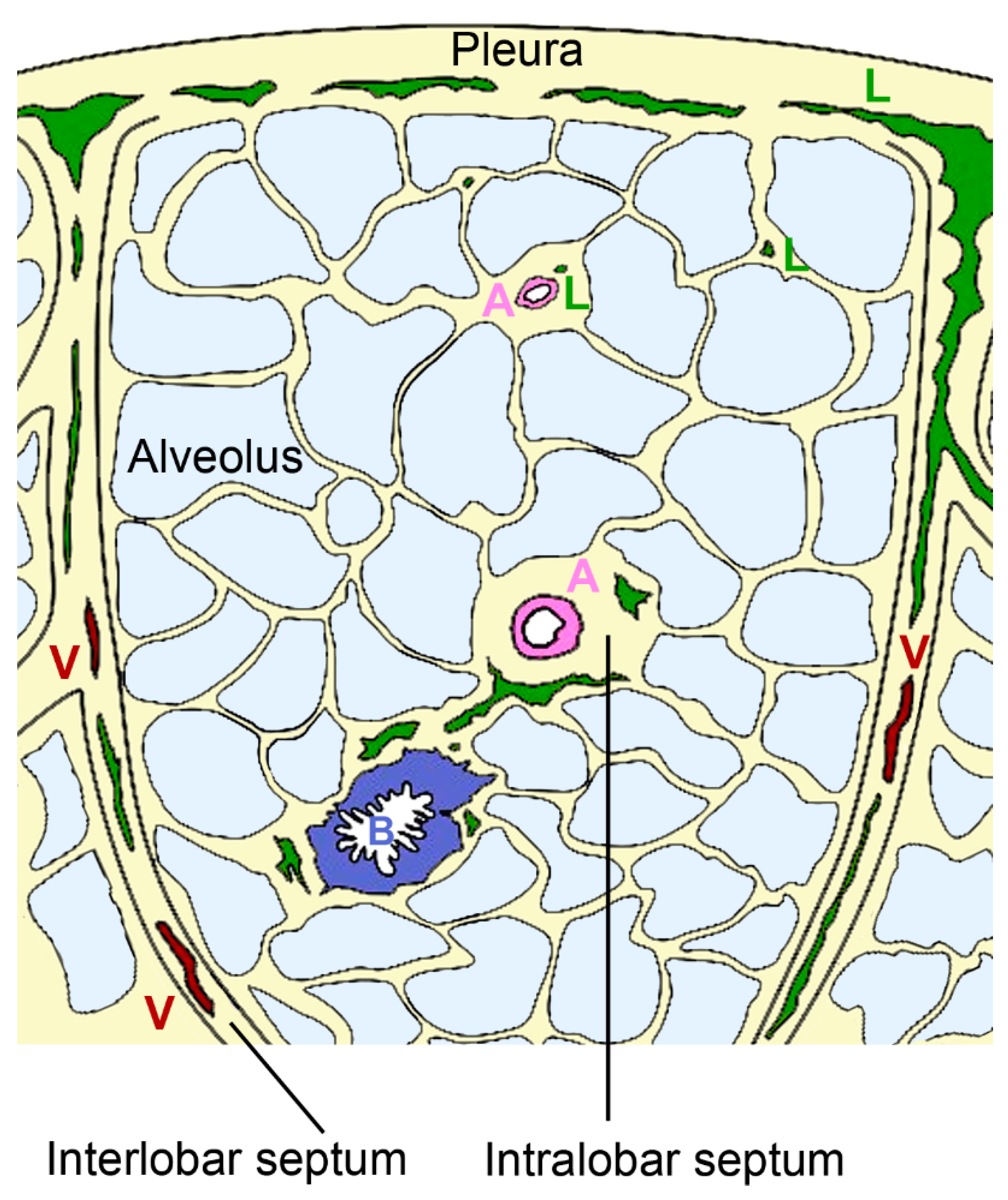
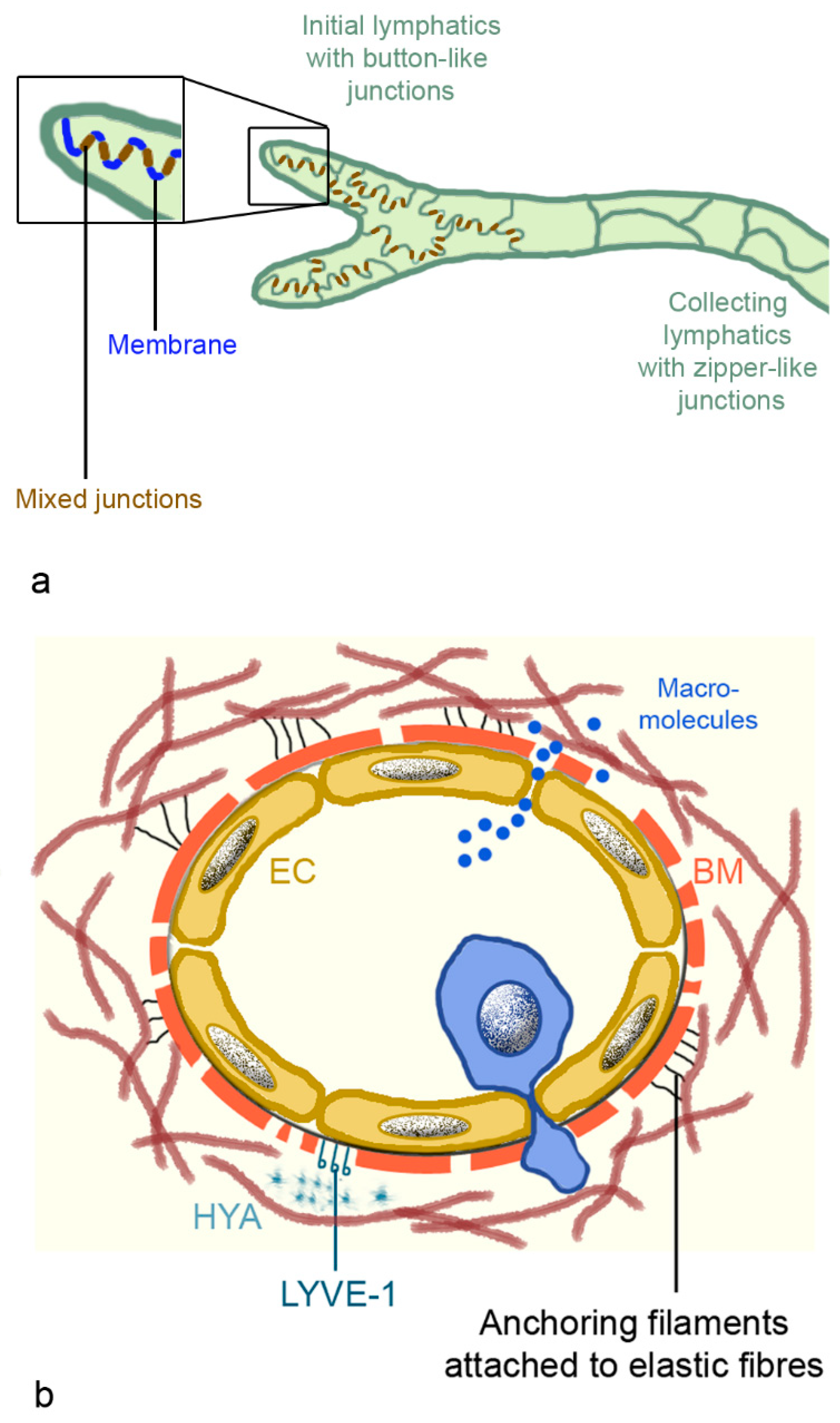
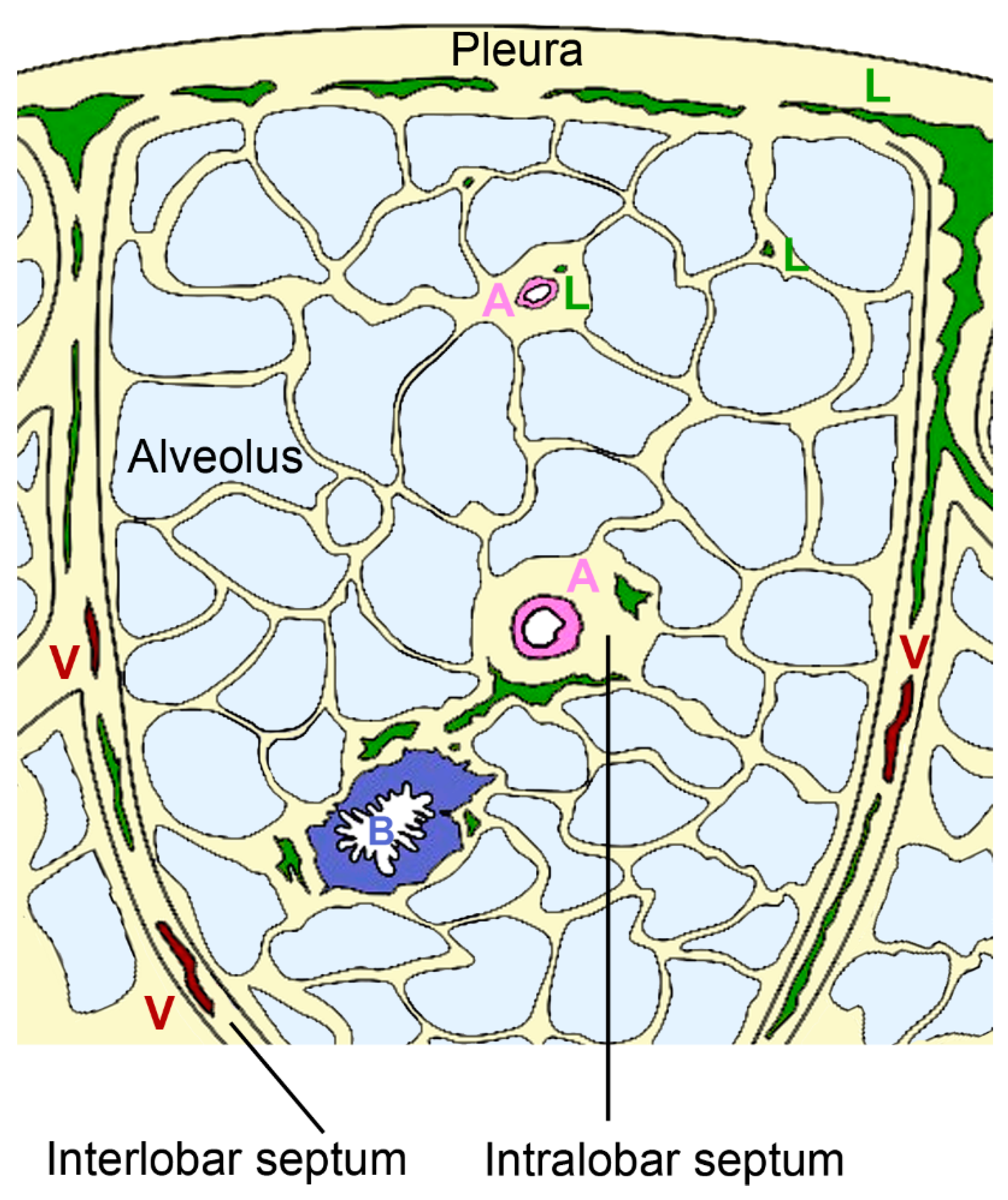
2.3.1. Architecture of the Pulmonary Lymphatic System
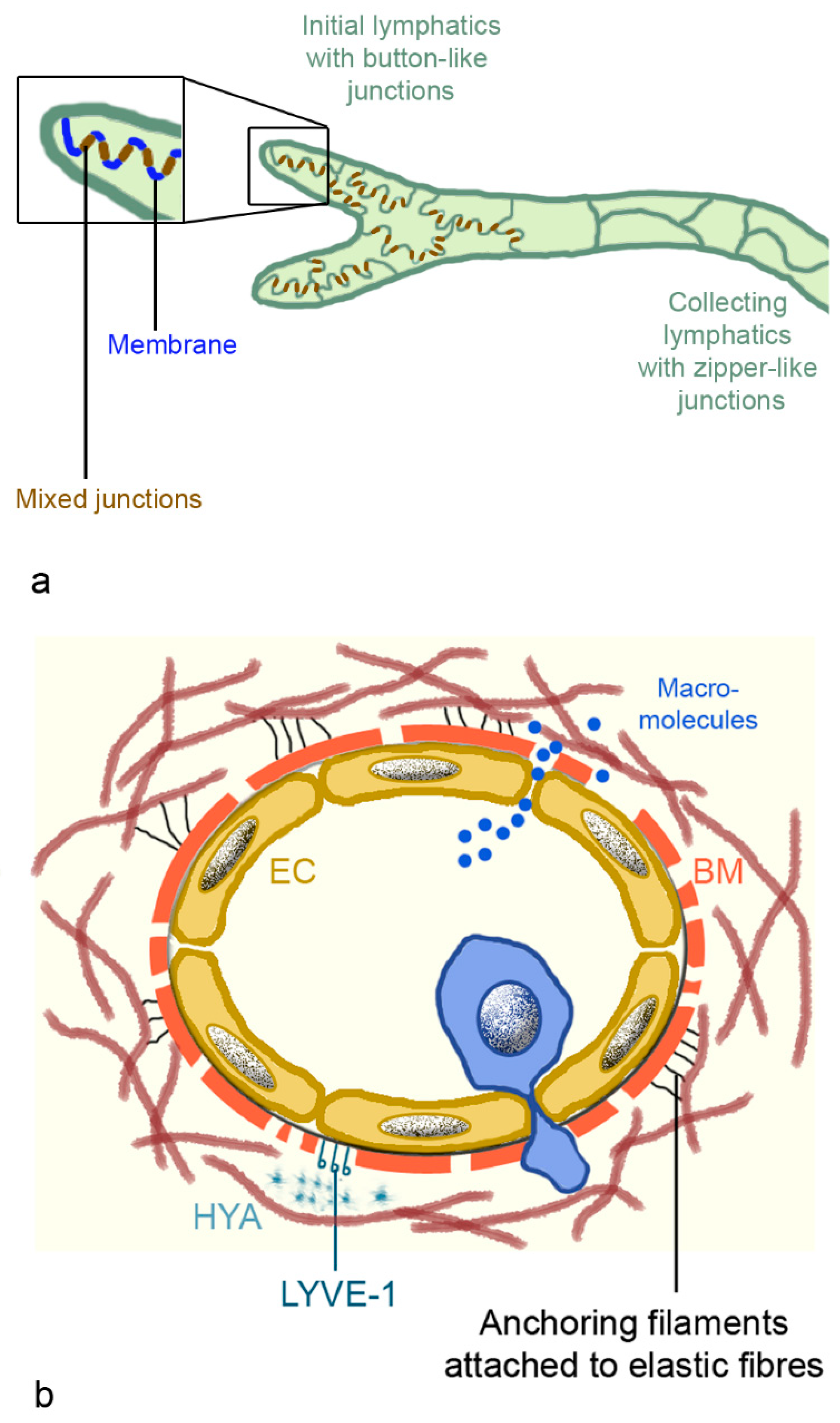
2.3.2. Morphology of Lymphatic Vessels
2.4. Lymphatic Vessels in Pulmonary Diseases
2.4.1. Role in Acute Lung Injury/Pulmonary Edema
2.4.2. Role in Chronic Pulmonary Diseases
3. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- De Luca, D.; Cogo, P.; Kneyber, M.C.; Biban, P.; Semple, M.G.; Perez-Gil, J.; Conti, G.; Tissieres, P.; Rimensberger, P.C. Surfactant Therapies for Pediatric and Neonatal ARDS: ESPNIC Expert Consensus Opinion for Future Research Steps. Crit. Care 2021, 25, 75. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, A. Respiratory Distress Syndrome Treatment & Management. In Respiratory Distress Syndrome; Rosenkrantz, T., Ed.; Medscape, 2020; Available online: https://emedicine.medscape.com/article/976034-treatment (accessed on 20 August 2021).
- Reilly, J.P.; Calfee, C.S.; Christie, J. Acute Respiratory Distress Syndrome Phenotypes. Semin. Respir. Crit. Care Med. 2019, 40, 19–30. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; Van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Siegel, M. Acute Respiratory Distress Syndrome: Epidemiology, Pathophysiology, Pathology, and Etiology in Adults; Parsons, P., Ed.; Medscape, 2020. [Google Scholar]
- Brown, R.; McKelvey, M.C.; Ryan, S.; Creane, S.; Linden, D.; Kidney, J.C.; McAuley, D.F.; Taggart, C.C.; Weldon, S. The Impact of Aging in Acute Respiratory Distress Syndrome: A Clinical and Mechanistic Overview. Front. Med. 2020, 7, 589553. [Google Scholar] [CrossRef]
- Gibson, P.G.; Qin, L.; Puah, S.H. COVID-19 Acute Respiratory Distress Syndrome (ARDS): Clinical Features and Differences From Typical Pre-COVID-19 ARDS. Med. J. Aust. 2020, 213, 54–56e1. [Google Scholar] [CrossRef]
- Absolute Reports. Acute Respiratory Distress Syndrome (ARDS) Market Insight, Epidemiology and Market Forecast -2030. DelveInsight 2020. Available online: https://www.delveinsight.com/report-store/acute-respiratory-distress-syndrome-ards-market (accessed on 20 August 2021).
- Zambon, M.; Vincent, J.-L. Mortality Rates for Patients with Acute Lung Injury/ARDS Have Decreased over Time. Chest 2008, 133, 1120–1127. [Google Scholar] [CrossRef]
- Griffiths, M.J.D.; McAuley, D.F.; Perkins, G.D.; Barrett, N.; Blackwood, B.; Boyle, A.; Chee, N.; Connolly, B.; Dark, P.; Finney, S.; et al. Guidelines on the Management of Acute Respiratory Distress Syndrome. BMJ Open Respir. Res. 2019, 6, e000420. [Google Scholar] [CrossRef] [PubMed]
- Gehr, P.; Bachofen, M.; Weibel, E.R. The Normal Human Lung: Ultrastructure and Morphometric Estimation of Diffusion Capacity. Respir. Physiol. 1978, 32, 121–140. [Google Scholar] [CrossRef]
- Rehfeld, A.; Nylander, M.; Karnov, K. The Respiratory System. In Compendium of Histology; Rehfeld, A., Nylander, M., Karnov, K., Eds.; Springer: Cham, Switzerland, 2017; pp. 351–377. [Google Scholar] [CrossRef]
- MacLaren, R.; Stringer, K.A. Emerging Role of Anticoagulants and Fibrinolytics in the Treatment of Acute Respiratory Distress Syndrome. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2007, 27, 860–873. [Google Scholar] [CrossRef]
- Bongard, F.S.; Matthay, M.; Mackersie, R.C.; Lewis, F.R. Morphologic and Physiologic Correlates of Increased Extravascular Lung Water. Surgery 1984, 96, 395–403. [Google Scholar] [PubMed]
- Jacobson, J.; Garcia, J. Pulmonary Circulation and Regulation of Fluid Balance. In Murray and Nadel’s Textbook of Respiratory Medicine; Mason, R., Broaddus, V., Martin, T., King, T., Schraufnagel, D., Murray, J., Nadel, J., Eds.; Saunders: Philadelphia, PA, USA, 2010; pp. 108–133. [Google Scholar]
- Aberle, D.R.; Wiener-Kronish, J.P.; Webb, W.R.; Matthay, A.M. Hydrostatic versus Increased Permeability Pulmonary Edema: Diagnosis Based on Radiographic Criteria in Critically Ill Patients. Radiology 1988, 168, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Graziano, P.; Smith, M. Acute Lung Injury. In Practical Pulmonary Pathology: A Diagnostic Approach; Leslie, K., Wick, M., Eds.; Elsevier, 2018; pp. 125–146. [Google Scholar] [CrossRef]
- Revercomb, L.; Hanmandlu, A.; Wareing, N.; Akkanti, B.; Karmouty-Quintana, H. Mechanisms of Pulmonary Hypertension in Acute Respiratory Distress Syndrome (ARDS). Front. Mol. Biosci. 2021, 7, 624093. [Google Scholar] [CrossRef]
- Beiderlinden, M.; Kuehl, H.; Boes, T.; Peters, J. Prevalence of Pulmonary Hypertension Associated with Severe Acute Respiratory Distress Syndrome: Predictive Value of Computed Tomography. Intensiv. Care Med. 2006, 32, 852–857. [Google Scholar] [CrossRef]
- Ñamendys-Silva, S.; Santos-Martínez, L.; Pulido, T.; Rivero-Sigarroa, E.; Baltazar-Torres, J.A.; Dominguez-Cherit, G.; Sandoval, J. Pulmonary Hypertension Due to Acute Respiratory Distress Syndrome. Braz. J. Med Biol. Res. 2014, 47, 904–910. [Google Scholar] [CrossRef]
- Sadigov, A.; Akhundov, S.S.; Agayeva, A. Post-Acute Respiratory Distress Syndrome Pulmonary Fibrosis and Pulmonary Artery Hypertension in Patients Affected by Severe COVID19. Am. J. Respir. Crit. Care Med. 2021, 203. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute Respiratory Distress Syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Perez, R.; Gorbett, D.; Mueller, J.; Daniels, C.J. Pulmonary Hypertension Idiopathic Pulmonary Fibrosis: A Dastardly Duo. Am. J. Med Sci. 2013, 346, 221–225. [Google Scholar] [CrossRef]
- Thille, A.W.; Esteban, A.; Fernández-Segoviano, P.; Rodriguez, J.-M.; Aramburu, J.-A.; Vargas-Errázuriz, P.; Martín-Pellicer, A.; Lorente, A.J.; Frutos-Vivar, F. Chronology of Histological Lesions in Acute Respiratory Distress Syndrome with Diffuse Alveolar Damage: A Prospective Cohort Study of Clinical Autopsies. Lancet Respir. Med. 2013, 1, 395–401. [Google Scholar] [CrossRef]
- Del Sorbo, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome and Multiple Organ Failure. Curr. Opin. Crit. Care 2011, 17, 1–6. [Google Scholar] [CrossRef]
- Chang, J.C. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin. Appl. Thromb. 2019, 25, 1076029619887437. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.D.; Lazrak, A.; Trombley, J.E.; Shei, R.-J.; Adewale, A.T.; Tipper, J.L.; Yu, Z.; Ashtekar, A.R.; Rowe, S.M.; Matalon, S.; et al. Influenza-Mediated Reduction of Lung Epithelial Ion Channel Activity Leads to Dysregulated Pulmonary Fluid Homeostasis. JCI Insight 2018, 3, e123467. [Google Scholar] [CrossRef] [PubMed]
- Luyt, C.; Combes, A.; Trouillet, J.-L.; Nieszkowska, A.; Chastre, J. Virus-Induced Acute Respiratory Distress Syndrome: Epidemiology. Manag. Outcome 2011, 40, e561–e568. [Google Scholar] [CrossRef]
- Clementi, N.; Ghosh, S.; De Santis, M.; Castelli, M.; Criscuolo, E.; Zanoni, I.; Clementi, M.; Mancini, N. Viral Respiratory Pathogens and Lung Injury. Clin. Microbiol. Rev. 2021, 34. [Google Scholar] [CrossRef]
- Archer, S.L.; Sharp, W.W.; Weir, E.K. Differentiating COVID-19 Pneumonia from Acute Respiratory Distress Syndrome and High Altitude Pulmonary: EdemaTherapeutic Implications. Circulation 2020, 142, 101–104. [Google Scholar] [CrossRef]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.S. Influenza Virus-Induced Lung Injury: Pathogenesis and Implications for Treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [PubMed]
- Flerlage, T.; Boyd, D.F.; Meliopoulos, V.; Thomas, P.G.; Schultz-Cherry, S. Influenza Virus and SARS-CoV-2: Pathogenesis and Host Responses in the Respiratory Tract. Nat. Rev. Genet. 2021, 19, 425–441. [Google Scholar] [CrossRef]
- Fenizia, C.; Galbiati, S.; Vanetti, C.; Vago, R.; Clerici, M.; Tacchetti, C.; Daniele, T. SARS-CoV-2 Entry: At the Crossroads of CD147 and ACE2. Cells 2021, 10, 1434. [Google Scholar] [CrossRef]
- Gadanec, L.; McSweeney, K.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Pronier, C.; Gacouin, A.; Lagathu, G.; Le Tulzo, Y.; Tadié, J.-M.; Thibault, V. Respiratory Influenza Viral Load as a Marker of Poor Prognosis in Patients with Severe Symptoms. J. Clin. Virol. 2021, 136, 104761. [Google Scholar] [CrossRef]
- Blot, M.; Jacquier, M.; Manoha, C.; Piroth, L.; Charles, P.-E.; Glele, L.-S.A.; Beltramo, G.; Nguyen, M.; Bonniaud, P.; Prin, S.; et al. Alveolar SARS-CoV-2 Viral Load Is Tightly Correlated with Severity in COVID-19 ARDS. Clin. Infect. Dis. 2020, 72, e446–e447. [Google Scholar] [CrossRef] [PubMed]
- Brosnahan, S.B.; Jonkman, A.H.; Kugler, M.C.; Munger, J.; Kaufman, D.A. COVID-19 and Respiratory System Disorders. Arter. Thromb. Vasc. Biol. 2020, 40, 2586–2597. [Google Scholar] [CrossRef]
- Quan, C.; Li, C.; Ma, H.; Li, Y.; Zhang, H. Immunopathogenesis of Coronavirus-Induced Acute Respiratory Distress Syndrome (ARDS): Potential Infection-Associated Hemophagocytic Lymphohistiocytosis. Clin. Microbiol. Rev. 2020, 34. [Google Scholar] [CrossRef]
- Piroth, L.; Cottenet, J.; Mariet, A.-S.; Bonniaud, P.; Blot, M.; Tubert-Bitter, P.; Quantin, C. Comparison of the Characteristics, Morbidity, and Mortality of COVID-19 and Seasonal Influenza: A Nationwide, Population-Based Retrospective Cohort Study. Lancet Respir. Med. 2021, 9, 251–259. [Google Scholar] [CrossRef]
- Donnelly, A.C.; Ghani, A.; Leung, G.; Hedley, A.J.; Fraser, C.; Riley, S.; Abu-Raddad, L.; Ho, L.-M.; Thach, T.Q.; Chau, P.; et al. Epidemiological Determinants of Spread of Causal Agent of Severe Acute Respiratory Syndrome in Hong Kong. Lancet 2003, 361, 1761–1766. [Google Scholar] [CrossRef]
- World Health Organization. Consensus Document on the Epidemiology of Severe Acute Respiratory Syndrome (SARS); Department of Communicable Disease Surveillance and Response: Geneva, Switzerland, 2003. [Google Scholar]
- Lemaitre, M.; Carrat, F. Comparative Age Distribution of Influenza Morbidity and Mortality During Seasonal Influenza Epidemics and the 2009 H1N1 Pandemic. BMC Infect. Dis. 2010, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Mineo, G.; Ciccarese, F.; Modolon, C.; Landini, M.P.; Valentino, M.; Zompatori, M. Post-ARDS Pulmonary Fibrosis in Patients with H1N1 Pneumonia: Role of Follow-Up CT. La Radiol. Med. 2011, 117, 185–200. [Google Scholar] [CrossRef]
- Das, K.M.; Lee, E.Y.; Singh, R.; A. Enani, M.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-Up Chest Radiographic Findings in Patients with MERS-CoV after Recovery. Indian J. Radiol. Imaging 2017, 27, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; To, K.-W.; Tong, M.; Hui, D.S. The Long-Term Impact of Severe Acute Respiratory Syndrome on Pulmonary Function, Exercise Capacity and Health Status. Respirology 2010, 15, 543–550. [Google Scholar] [CrossRef]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary Fibrosis in the Aftermath of the Covid-19 Era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Zhou, Y.; Yu, H.; Li, R.; Zhan, Q.; Ni, F.; Fang, S.; Lu, Y.; Ding, X.; et al. 3-Month, 6-Month, 9-Month, and 12-Month Respiratory Outcomes in Patients Following COVID-19-Related Hospitalisation: A Prospective Study. Lancet Respir. Med. 2021, 9, 747–754. [Google Scholar] [CrossRef]
- Yang, S.-C.; Tsai, Y.-F.; Pan, Y.-L.; Hwang, T.-L. Understanding the Role of Neutrophils in Acute Respiratory Distress Syndrome. Biomed. J. 2020, 44, 439–446. [Google Scholar] [CrossRef]
- Sinha, P.; Calfee, C.S.; Cherian, S.; Brealey, D.; Cutler, S.; King, C.; Killick, C.; Richards, O.; Cheema, Y.; Bailey, C.; et al. Prevalence of Phenotypes of Acute Respiratory Distress Syndrome in Critically Ill Patients with COVID-19: A Prospective Observational Study. Lancet Respir. Med. 2020, 8, 1209–1218. [Google Scholar] [CrossRef]
- Baker, S.A.; Kwok, S.; Berry, G.J.; Montine, T.J. Angiotensin-Converting Enzyme 2 (ACE2) Expression Increases with Age in Patients Requiring Mechanical Ventilation. PLoS ONE 2021, 16, e0247060. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Boyle, A.J.; Mac Sweeney, R.; McAuley, D.F. Pharmacological Treatments in ARDS; a State-of-the-Art Update. BMC Med. 2013, 11, 166. [Google Scholar] [CrossRef]
- Harman, E.; Riley, L. Acute Respiratory Distress Syndrome (ARDS) Medication; Crit. Care Medscape, 2020. [Google Scholar]
- Horie, S.; McNicholas, B.; Rezoagli, E.; Pham, T.; Curley, G.; McAuley, D.; O’Kane, C.; Nichol, A.; Dos Santos, C.; Rocco, P.R.M.; et al. Emerging Pharmacological Therapies for ARDS: COVID-19 and Beyond. Intensiv. Care Med. 2020, 46, 2265–2283. [Google Scholar] [CrossRef] [PubMed]
- Kassirian, S.; Taneja, R.; Mehta, S. Diagnosis and Management of Acute Respiratory Distress Syndrome in a Time of COVID-19. Diagnostics 2020, 10, 1053. [Google Scholar] [CrossRef]
- Pum, A.; Ennemoser, M.; Adage, T.; Kungl, A.J. Cytokines and Chemokines in SARS-CoV-2 Infections—Therapeutic Strategies Targeting Cytokine Storm. Biomolecules 2021, 11, 91. [Google Scholar] [CrossRef]
- Cornet, A.; Oudemans-van Straaten, H.; Schultz, M.; Juffermans, N.; Tuinman, P. Anticoagulants for ARDS: Facts and Future. Neth. J. Crit. Care 2014, 18, 3–8. [Google Scholar]
- Feng, Y. Efficacy of Statin Therapy in Patients with Acute Respiratory Distress Syndrome/Acute Lung Injury: A Systematic Review and Meta-Analysis. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 3190–3198. [Google Scholar]
- Monsalve-Naharro, J.; Domingo-Chiva, E.; García Castillo, S.; Cuesta-Montero, P.; Jiménez-Vizuete, J. Inhaled Nitric Oxide in Adult Patients with Acute Respiratory Distress Syndrome. Farm. Hosp. 2017, 41, 292–312. [Google Scholar] [PubMed]
- Bo, L.; Jin, F.; Ma, Z.; Li, C. Redox Signaling and Antioxidant Therapies in Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Expert Rev. Respir. Med. 2021, 15, 1355–1365. [Google Scholar] [CrossRef]
- Fröhlich, E. Therapeutic Potential of Mesenchymal Stem Cells and Their Products in Lung Diseases—Intravenous Administration versus Inhalation. Pharmaceutics 2021, 13, 232. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Duarte, J.D.; Hanson, R.L.; Machado, R.F. Pharmacologic Treatments for Pulmonary Hypertension: Exploring Pharmacogenomics. Futur. Cardiol. 2013, 9, 335–349. [Google Scholar] [CrossRef]
- Li, J.; Gao, J.; Xu, Y.-P.; Zhou, T.-L.; Jin, Y.-Y.; Lou, J.-N. Expression of Severe Acute Respiratory Syndrome Coronavirus Receptors, ACE2 and CD209L in Different Organ Derived Microvascular Endothelial Cells. Zhonghua yi xue za zhi 2007, 87, 833–837. [Google Scholar] [PubMed]
- Wu, C.; Li, H.; Zhang, P.; Tian, C.; Luo, J.; Zhang, W.; Bhandari, S.; Jin, S.; Hao, Y. Lymphatic Flow: A Potential Target in Sepsis-Associated Acute Lung Injury. J. Inflamm. Res. 2020, 13, 961–968. [Google Scholar] [CrossRef]
- Margaris, K.; Black, R.A. Modelling the Lymphatic System: Challenges and Opportunities. J. R. Soc. Interface 2012, 9, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Sozio, F.; Borghini, A.; Sestini, P.; Renzoni, E. Pulmonary Lymphatic Vessel Morphology: A Review. Ann. Anat. Anat. Anz. 2018, 218, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Sozio, F.; Rossi, A.; Weber, E.; Abraham, D.J.; Nicholson, A.G.; Wells, A.U.; Renzoni, E.A.; Sestini, P. Morphometric Analysis of Intralobular, Interlobular and Pleural Lymphatics in Normal Human Lung. J. Anat. 2012, 220, 396–404. [Google Scholar] [CrossRef]
- Kambouchner, M.; Bernaudin, J.-F. Intralobular Pulmonary Lymphatic Distribution in Normal Human Lung Using D2-40 Antipodoplanin Immunostaining. J. Histochem. Cytochem. 2009, 57, 643–648. [Google Scholar] [CrossRef]
- Partanen, T.A.; Arola, J.; Saaristo, A.; Jussila, L.; Ora, A.; Miettinen, M.; Stacker, S.A.; Achen, M.G.; Alitalo, K. VEGF-C and VEGF-D Expression in Neuroendocrine Cells and their Receptor, VEGFR-3, in Fenestrated Blood Vessels in Human Tissues. FASEB J. 2000, 14, 2087–2096. [Google Scholar] [CrossRef]
- Stump, B.; Cui, Y.; Kidambi, P.; LaMattina, A.M.; El-Chemaly, S. Lymphatic Changes in Respiratory Diseases: More than Just Remodeling of the Lung? Am. J. Respir. Cell Mol. Biol. 2017, 57, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Schraufnagel, D.E. Lung Lymphatic Anatomy and Correlates. Pathophysiology 2010, 17, 337–343. [Google Scholar] [CrossRef]
- Barile, M. Pulmonary Edema: A Pictorial Review of Imaging Manifestations and Current Understanding of Mechanisms of Disease. Eur. J. Radiol. Open 2020, 7, 100274. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature—Mechanisms and Consequences. Front. Immunol. 2019, 10, 471. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, K.; LaMattina, A.M.; Visner, G.; El-Chemaly, S. Lymphatic Vessels: The Next Frontier in Lung Transplant. Ann. Am. Thorac. Soc. 2017, 14, S226–S232. [Google Scholar] [CrossRef]
- Zhang, P.; Han, J.; Cao, F.; Liu, Y.; Tian, C.; Wu, C.; Smith, F.G.; Hao, Y.; Jin, S. PCTR1 improves Pulmonary Edema Fluid Clearance Through Activating the Sodium Channel and Lymphatic Drainage in Lipopolysaccharide-Induced ARDS. J. Cell. Physiol. 2020, 235. [Google Scholar] [CrossRef]
- Laurent, T.C.; Fraser, J.R. Hyaluronan. Faseb J. 1992, 6, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Tengblad, A.; Laurent, U.B.G.; Lilja, K.; Cahill, R.N.P.; Engström-Laurent, A.; Fraser, J.R.R.; E Hansson, H.; Laurent, T.C. Concentration and Relative Molecular Mass of Hyaluronate in Lymph and Blood. Biochem. J. 1986, 236, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.C.; Lall, R.; Srivastava, A.; Sinha, A. Hyaluronic Acid: Molecular Mechanisms and Therapeutic Trajectory. Front. Veter- Sci. 2019, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Lauer, M.E.; Dweik, R.A.; Garantziotis, S.; Aronica, M.A. The Rise and Fall of Hyaluronan in Respiratory Diseases. Int. J. Cell Biol. 2015, 2015, 712507. [Google Scholar] [CrossRef]
- Modig, J.; Hällgren, R. Increased Hyaluronic Acid Production in Lung—A Possible Important Factor in Interstitial and Alveolar Edema during General Anesthesia and in Adult Respiratory Distress Syndrome. Resuscitation 1989, 17, 223–231. [Google Scholar] [CrossRef]
- Hellman, U.; Karlsson, M.G.; Engström-Laurent, A.; Cajander, S.; Dorofte, L.; Ahlm, C.; Laurent, C.; Blomberg, A. Presence of Hyaluronan in Lung Alveoli in Severe Covid-19: An Opening for New Treatment Options? J. Biol. Chem. 2020, 295, 15418–15422. [Google Scholar] [CrossRef]
- Lee-Sayer, S.S.M.; Dong, Y.; Arif, A.A.; Olsson, M.; Brown, K.L.; Johnson, P. The Where, When, How, and Why of Hyaluronan Binding by Immune Cells. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Fries, E.; Kaczmarczyk, A. Inter-Alpha-Inhibitor, Hyaluronan and Inflammation. Acta Biochim. Pol. 2003, 50, 735–742. [Google Scholar] [CrossRef]
- Johnsson, H.; Eriksson, L.; Sedin, G. Antenatal Betamethasone Administration Decreases the Lung Hyaluronan Concentration in Preterm Rabbit Pups. Pediatr. Res. 2001, 49, 566–571. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Pacheco-Rodriguez, G.; Ikeda, Y.; Malide, D.; Moss, J. Lymphatics in Idiopathic Pulmonary Fibrosis: New Insights into an Old Disease. Lymphat. Res. Biol. 2009, 7, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Ebina, M.; Shibata, N.; Ohta, H.; Hisata, S.; Tamada, T.; Ono, M.; Okaya, K.; Kondo, T.; Nukiwa, T. The Disappearance of Subpleural and Interlobular Lymphatics in Idiopathic Pulmonary Fibrosis. Lymphat. Res. Biol. 2010, 8, 199–207. [Google Scholar] [CrossRef]
- Baluk, P.; Naikawadi, R.P.; Kim, S.; Rodriguez, F.; Choi, D.; Hong, Y.-K.; Wolters, P.J.; McDonald, D.M. Lymphatic Proliferation Ameliorates Pulmonary Fibrosis after Lung Injury. Am. J. Pathol. 2020, 190, 2355–2375. [Google Scholar] [CrossRef]
- Mori, M.; Andersson, C.K.; Graham, G.J.; Löfdahl, C.-G.; Erjefält, J.S. Increased Number and Altered Phenotype of Lymphatic Vessels in Peripheral Lung Compartments of Patients with COPD. Respir. Res. 2013, 14, 65. [Google Scholar] [CrossRef]
- Meinecke, A.-K.; Nagy, N.; Lago, G.D.; Kirmse, S.; Klose, R.; Schrödter, K.; Zimmermann, A.; Helfrich, I.; Rundqvist, H.; Theegarten, D.; et al. Aberrant Mural Cell Recruitment to Lymphatic Vessels and Impaired Lymphatic Drainage in a Murine Model of Pulmonary Fibrosis. Blood 2012, 119, 5931–5942. [Google Scholar] [CrossRef] [PubMed]
- Forte, A.J.; Boczar, D.; Huayllani, M.T.; Lu, X.; A. McLaughlin, S. Pharmacotherapy Agents in Lymphedema Treatment: A Systematic Review. Cureus 2019, 11, e6300. [Google Scholar] [CrossRef]
- Sheikhi-Mobarakeh, Z.; Yarmohammadi, H.; Mokhatri-Hesari, P.; Fahimi, S.; Montazeri, A.; Heydarirad, G. Herbs as Old Potential Treatments for Lymphedema Management: A Systematic Review. Complement. Ther. Med. 2020, 55, 102615. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fröhlich, E. Acute Respiratory Distress Syndrome: Focus on Viral Origin and Role of Pulmonary Lymphatics. Biomedicines 2021, 9, 1732. https://doi.org/10.3390/biomedicines9111732
Fröhlich E. Acute Respiratory Distress Syndrome: Focus on Viral Origin and Role of Pulmonary Lymphatics. Biomedicines. 2021; 9(11):1732. https://doi.org/10.3390/biomedicines9111732
Chicago/Turabian StyleFröhlich, Eleonore. 2021. "Acute Respiratory Distress Syndrome: Focus on Viral Origin and Role of Pulmonary Lymphatics" Biomedicines 9, no. 11: 1732. https://doi.org/10.3390/biomedicines9111732
APA StyleFröhlich, E. (2021). Acute Respiratory Distress Syndrome: Focus on Viral Origin and Role of Pulmonary Lymphatics. Biomedicines, 9(11), 1732. https://doi.org/10.3390/biomedicines9111732