
Patient-Specific Modeling of Diffuse Large B-Cell Lymphoma


Abstract
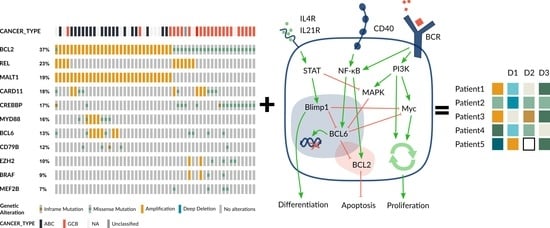
:
1. Introduction
2. Materials and Methods
2.1. Logical Modeling
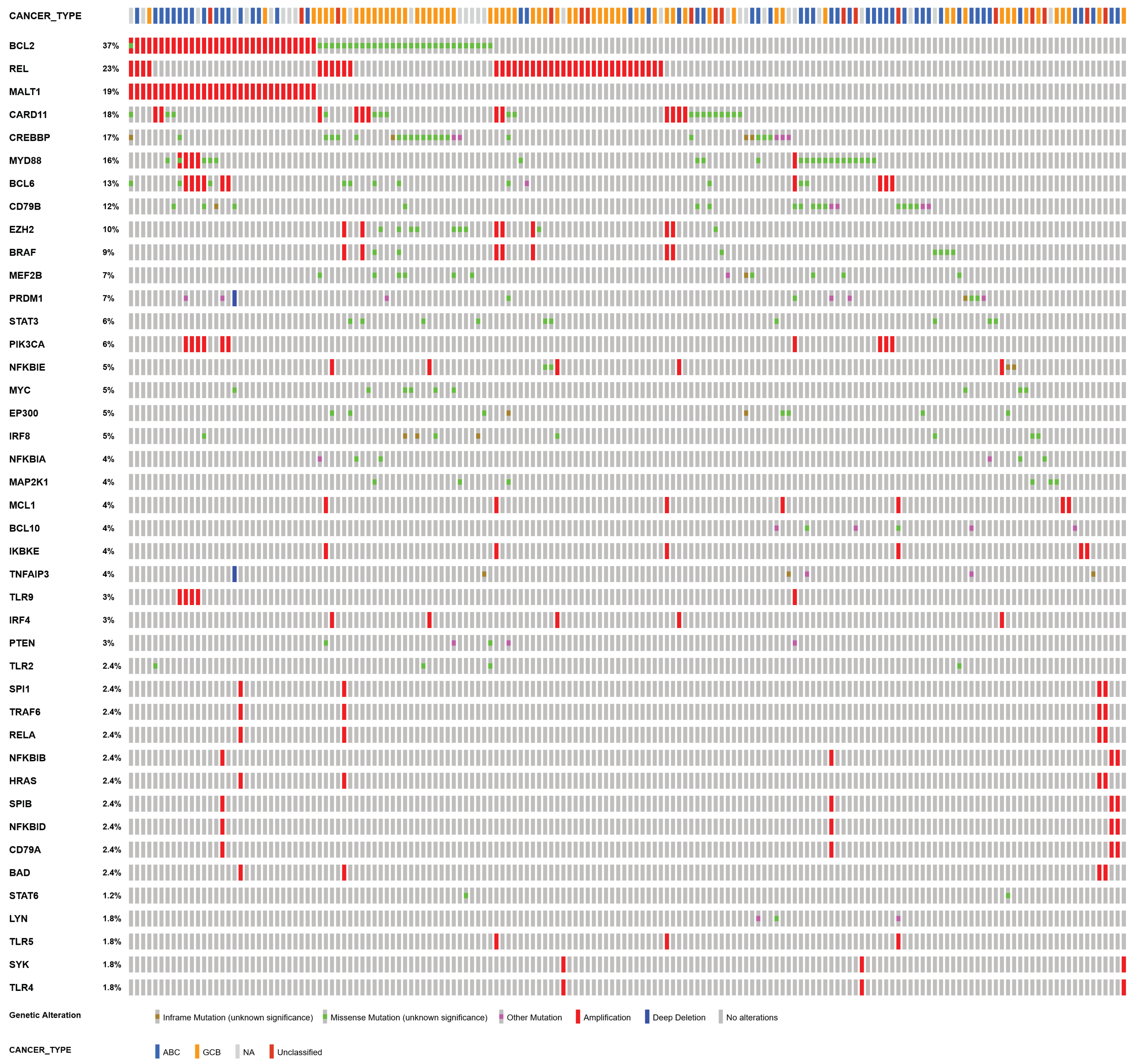
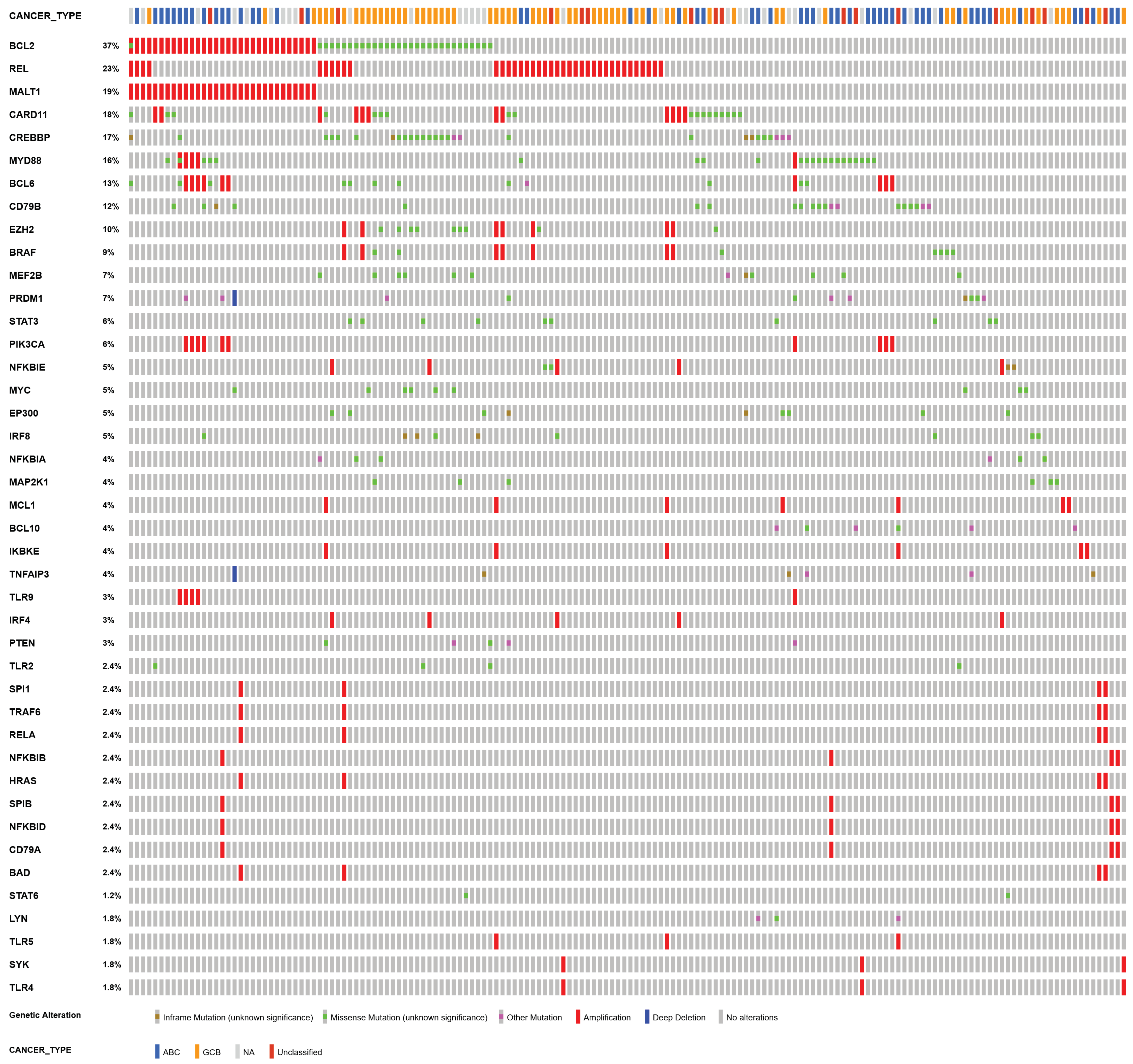
2.2. Patient-Specific Models Derived from Clinical Data
3. Results
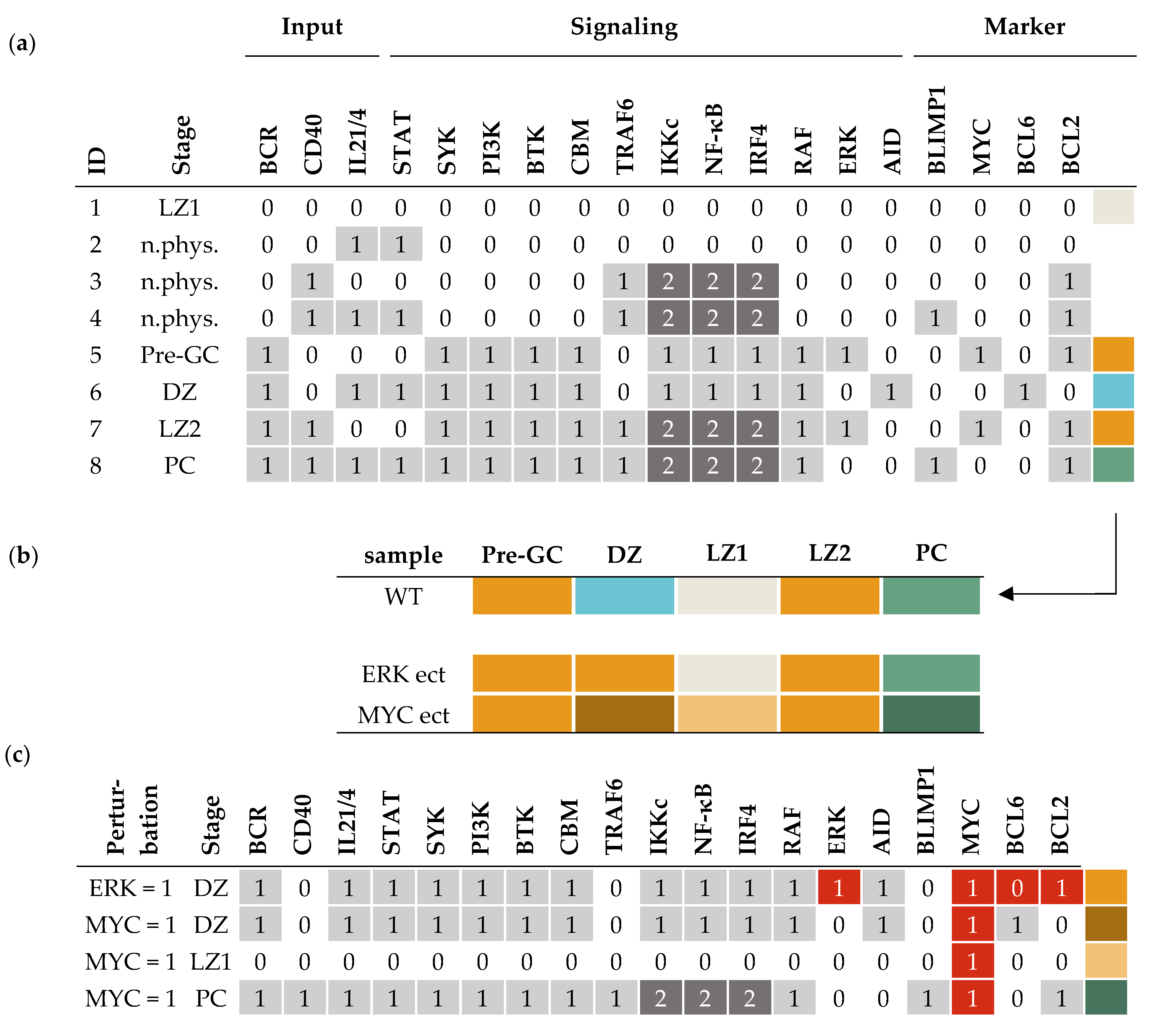
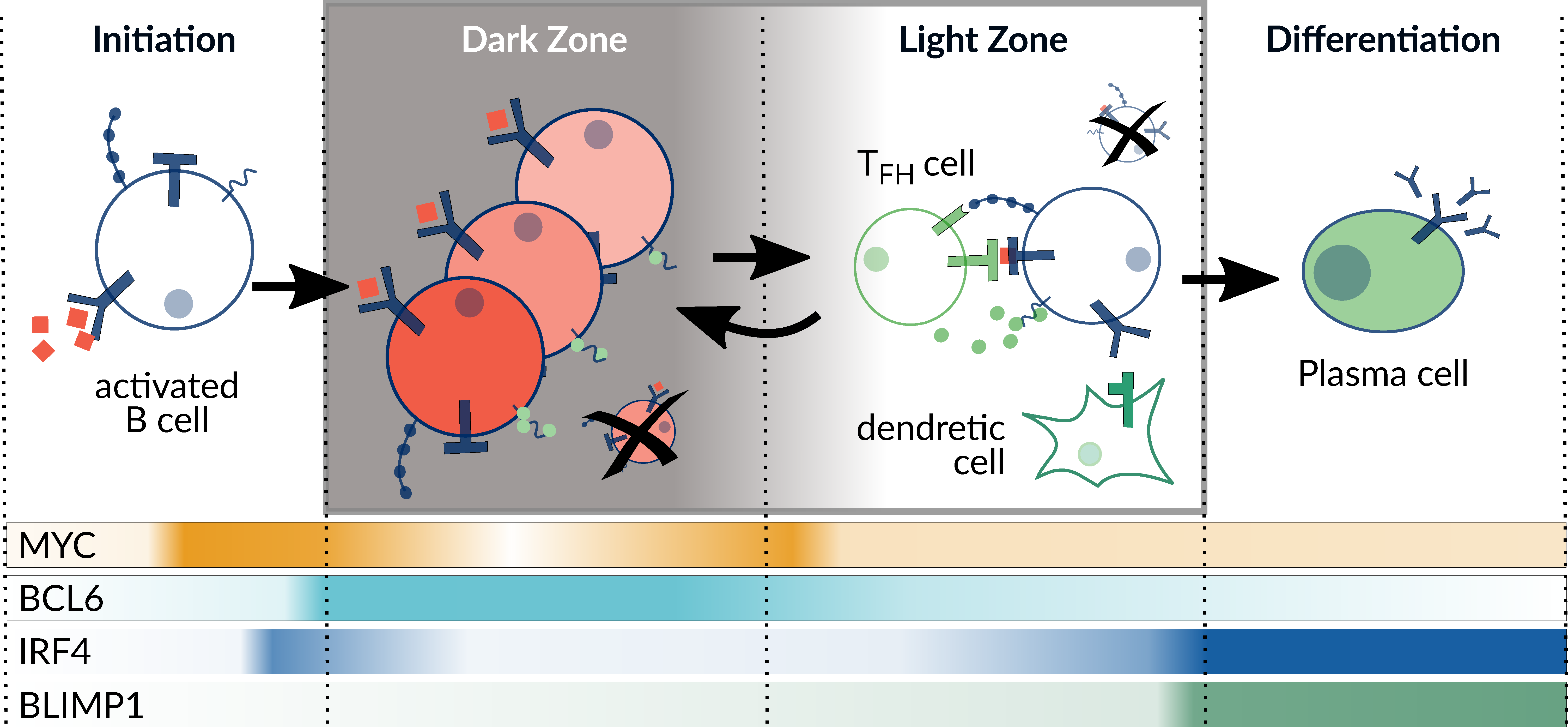
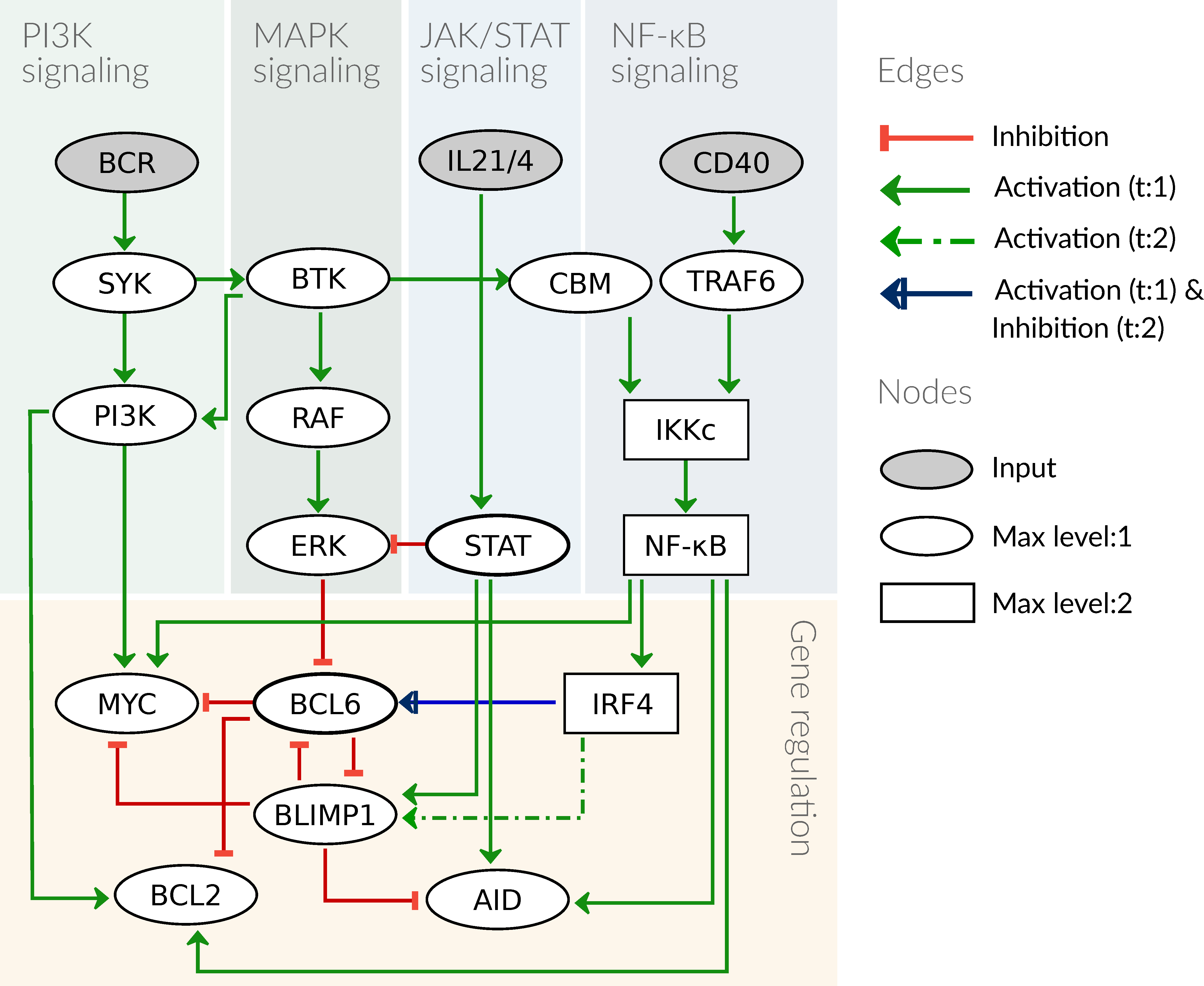
3.1. Computational Model of the Germinal Center Reaction
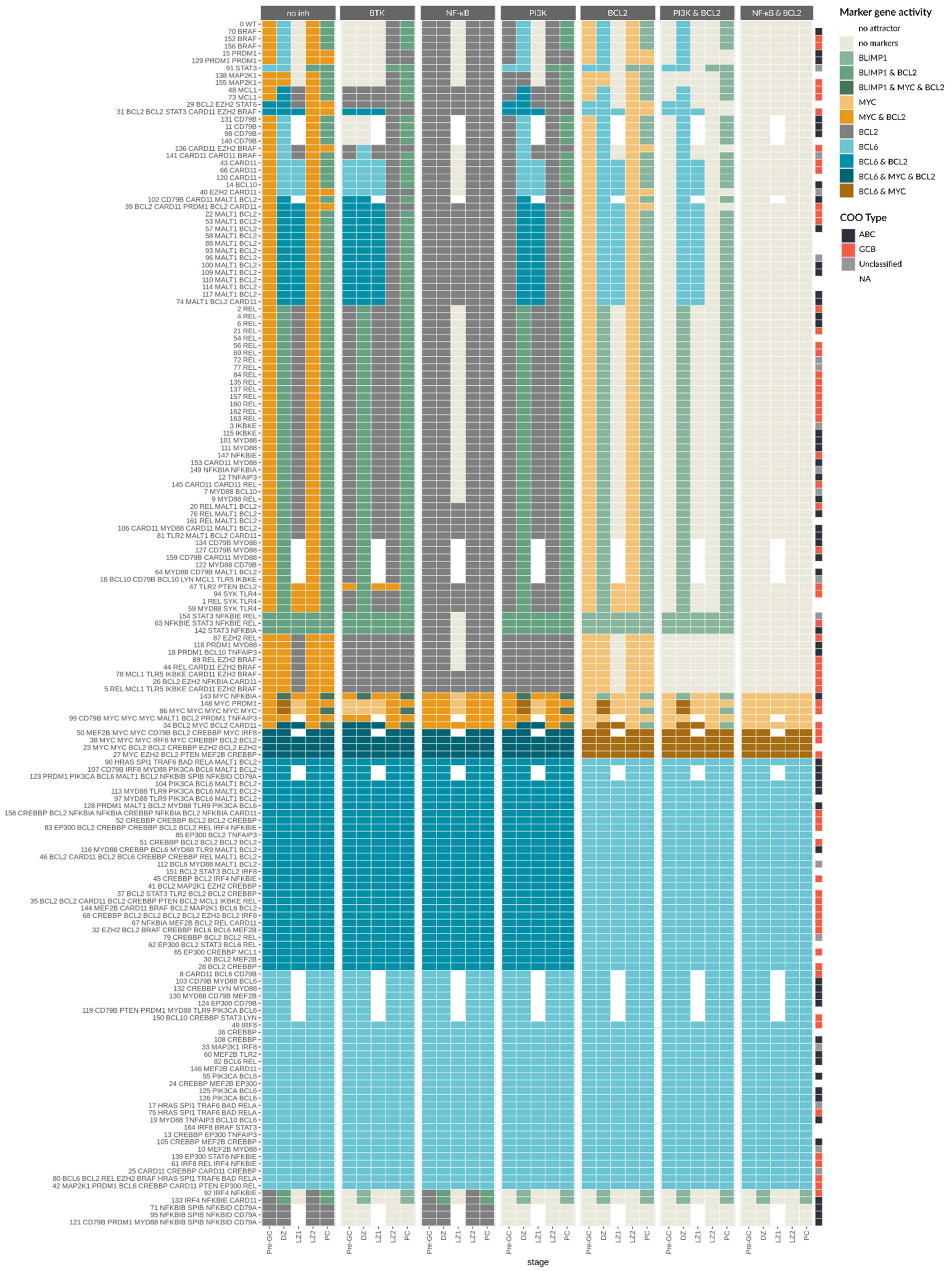
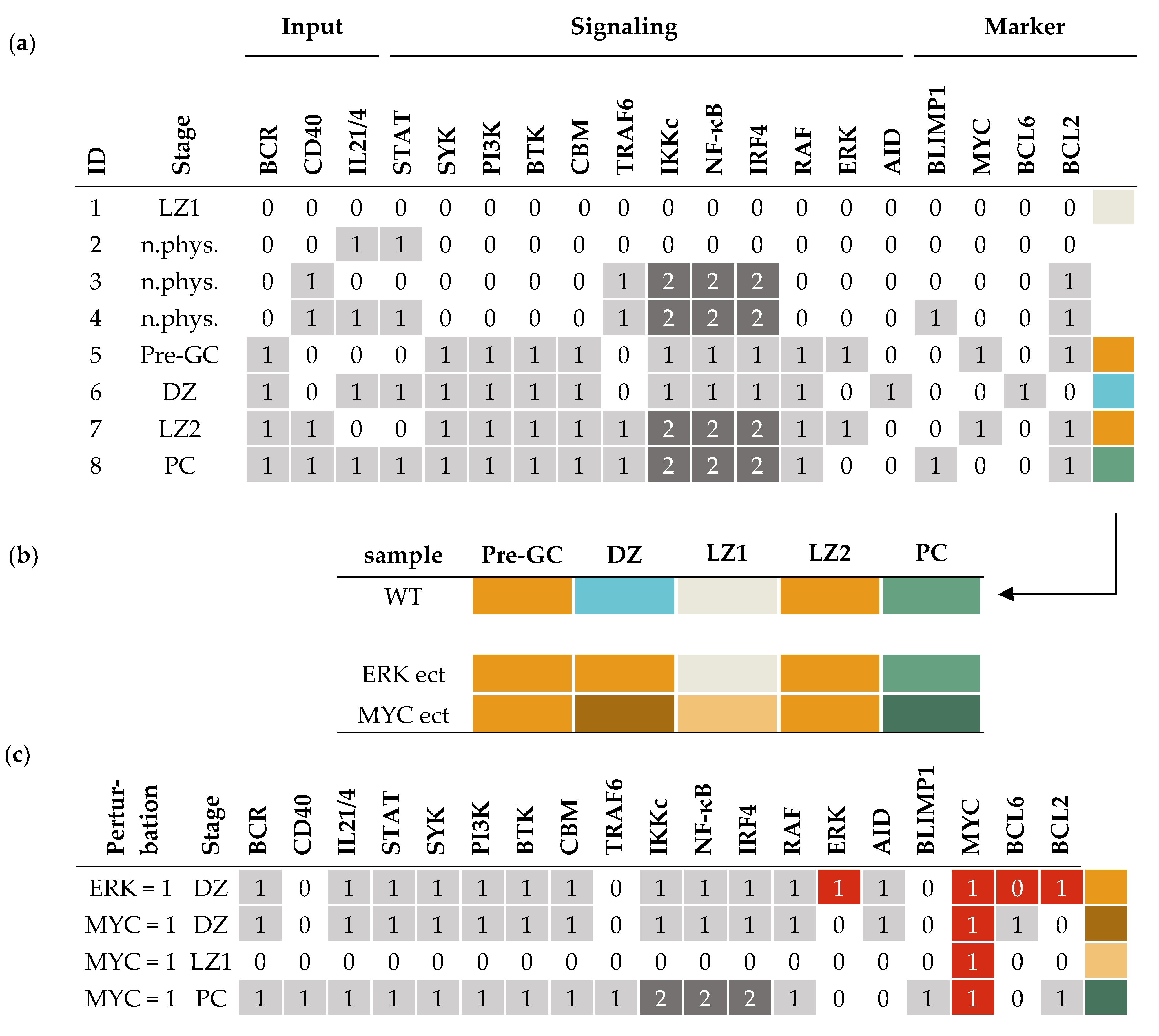
3.2. Attractors of the Model Reflect Germinal Center Stages
3.3. Simulating the Effect of Genetic Lesions and Drug Treatment
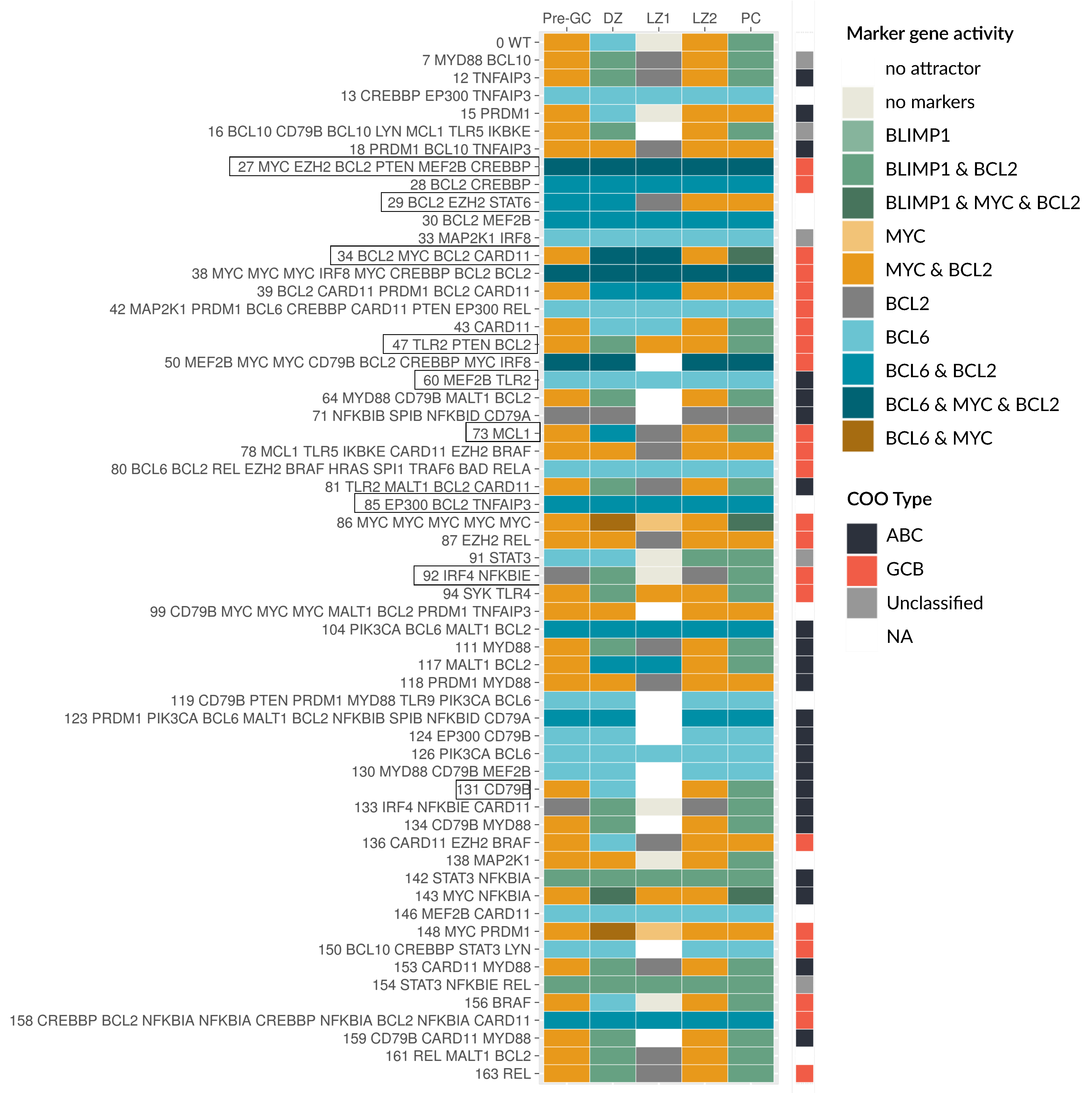
3.4. Patient-Specific Models Often Present a Shift in Attractors

3.5. Oncogenic Attractors Are Quite Heterogenous
3.6. Drug Simulation Shows Resistance for BCL6 Perturbed Samples
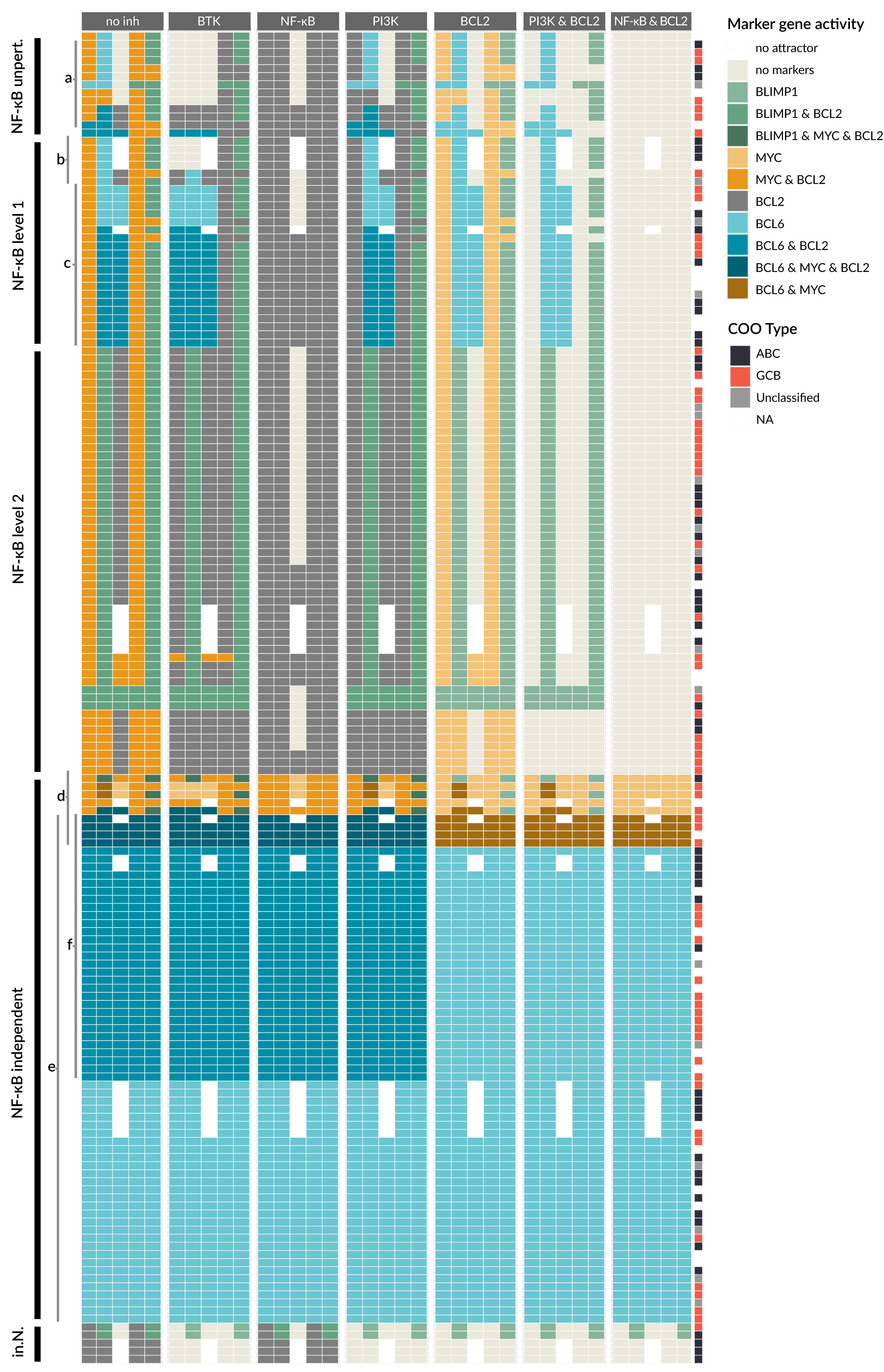
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Function | Reference |
|---|---|---|
| BCR | Input | |
| IL21/4 | Input | |
| CD40 | Input | |
| SYK | BCR | [29] |
| BTK | SYK | [29] |
| RAF | BTK | [44] |
| CBM | BTK | [44] |
| TRAF6 | CD40 | [36] |
| STAT | IL21/4 | [38] |
| PI3K | SYK ∧ BTK | [29] |
| MYC | PI3K ∧ NF-κB ∧ ¬ BLIMP1 ∧ ¬ BCL6 | [30,31,32] |
| ERK | RAF ∧ ¬ STAT | [33,34] |
| IKKc:1 | CBM | [35] |
| IKKc:2 | TRAF6 | [36] |
| NF-κB | IKKc | [71] |
| IRF4 | NF-κB | [47] |
| BCL6 | IRF4:1 ∧ ¬ ERK ∧ ¬ BLIMP1 ∧ ¬ IRF4:2 | [3,34,37] |
| BLIMP1 | IRF4:2 ∧ STAT ∧ ¬ BCL6 | [37,38] |
| AID | NF-κB ∧ STAT ∧ ¬ BLIMP1 | [39,40] |
| BCL2 | (PI3K ∨ NF-κB) ∧ ¬ BCL6 | [41,42,43] |
References
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef]
- MacLennan, I.C.M. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef]
- Hatzi, K.; Melnick, A. Breaking bad in the germinal center: How deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 2014, 20, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Basso, K.; Saito, M.; Sumazin, P.; Margolin, A.A.; Wang, K.; Lim, W.K.; Kitagawa, Y.; Schneider, C.; Alvarez, M.J.; Califano, A.; et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood 2010, 115, 975–984. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.M.; Khalil, A.; Uduman, M.; Hershberg, U.; Louzoun, Y.; Haberman, A.M.; Kleinstein, S.H.; Shlomchik, M.J. Taking Advantage: High-Affinity B Cells in the Germinal Center Have Lower Death Rates, but Similar Rates of Division, Compared to Low-Affinity Cells. J. Immunol. 2009, 183, 7314–7325. [Google Scholar] [CrossRef]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Finkin, S.; Hartweger, H.; Oliveira, T.Y.; Kara, E.E.; Nussenzweig, M.C. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity 2019, 51, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M.J.; Luo, W.; Weisel, F. Linking signaling and selection in the germinal center. Immunol. Rev. 2019, 288, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; de Silva, N.S.; Silva, K.; Carette, A.; Simonetti, G.; Pasparakis, M.; Klein, U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J. Exp. Med. 2014, 211, 2103–2118. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Elsen, M.B.; Davis, R.E.; Ma, C.L.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Minderman, M.; Pals, S.T. Towards genomic-based prognostication and precision therapy for diffuse large B-cell lymphoma. Haematologica 2020, 105, 2194. [Google Scholar] [CrossRef]
- Fontan Gabas, L.; Goldstein, R.L.; Casalena, G.; Durant, M.; Teater, M.R.; Wilson, J.; Phillip, J.M.; Xia, M.; Shah, S.; Us, I.; et al. Identification of MALT1 Feedback Mechanisms Enables Rational Design of Potent Anti-Lymphoma Regimens for ABC-DLBCL. Blood 2020, 137, 788–800. [Google Scholar] [CrossRef]
- Tognetti, M.; Gabor, A.; Yang, M.; Cappelletti, V.; Windhager, J.; Rueda, O.M.; Charmpi, K.; Esmaeilishirazifard, E.; Bruna, A.; de Souza, N.; et al. Deciphering the signaling network of breast cancer improves drug sensitivity prediction. Cell Syst. 2021, 12, 401–418.e12. [Google Scholar] [CrossRef]
- Eduati, F.; Jaaks, P.; Wappler, J.; Cramer, T.; Merten, C.A.; Garnett, M.J.; Saez-Rodriguez, J. Patient-specific logic models of signaling pathways from screenings on cancer biopsies to prioritize personalized combination therapies. Mol. Syst. Biol. 2020, 16, e8664. [Google Scholar] [CrossRef]
- Béal, J.; Pantolini, L.; Noël, V.; Barillot, E.; Calzone, L. Personalized logical models to investigate cancer response to BRAF treatments in melanomas and colorectal cancers. PLoS Comput. Biol. 2021, 17, e1007900. [Google Scholar] [CrossRef]
- Saez-Rodriguez, J.; Blüthgen, N. Personalized signaling models for personalized treatments. Mol. Syst. Biol. 2020, 16, e9042. [Google Scholar] [CrossRef]
- Palma, A.; Iannuccelli, M.; Rozzo, I.; Licata, L.; Perfetto, L.; Massacci, G.; Castagnoli, L.; Cesareni, G.; Sacco, F. Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia. J. Pers. Med. 2021, 11, 117. [Google Scholar] [CrossRef]
- Cacace, E.; Collombet, S.; Thieffry, D. Logical modeling of cell fate specification—Application to T cell commitment. Curr. Top. Dev. Biol. 2020, 139, 205–238. [Google Scholar] [CrossRef]
- Flobak, Å.; Baudot, A.; Remy, E.; Thommesen, L.; Thieffry, D.; Kuiper, M.; Lægreid, A. Discovery of Drug Synergies in Gastric Cancer Cells Predicted by Logical Modeling. PLoS Comput. Biol. 2015, 11, e1004426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Chen, Q.; Wang, R. Logical modeling of thymus and natural killer lymphocyte differentiation. J. Biol. Phys. 2021, 47, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Remy, E.; Rebouissou, S.; Chaouiya, C.; Zinovyev, A.; Radvanyi, F.; Calzone, L. A modeling approach to explain mutually exclusive and co-occurring genetic alterations in bladder tumorigenesis. Cancer Res. 2015, 75, 4042–4052. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, S.A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 1969, 22, 437–467. [Google Scholar] [CrossRef]
- Naldi, A.; Hernandez, C.; Abou-Jaoudé, W.; Monteiro, P.T.; Chaouiya, C.; Thieffry, D. Logical modelling and analysis of cellular regulatory networks with GINsim 3.0. Front. Physiol. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Naldi, A.; Hernandez, C.; Levy, N.; Stoll, G.; Monteiro, P.T.; Chaouiya, C.; Helikar, T.; Zinovyev, A.; Calzone, L.; Cohen-Boulakia, S.; et al. The CoLoMoTo interactive notebook: Accessible and reproducible computational analyses for qualitative biological networks. Front. Physiol. 2018, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Naldi, A.; Remy, E.; Thieffry, D.; Chaouiya, C. A reduction of logical regulatory graphs preserving essential dynamical properties. In Lecture Notes in Computer Science (including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), Proceedings of the International Conference on Computational Methods in Systems Biology, Williamsburg, VA, USA, 5–10 July 2009; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5688 LNBI, pp. 266–280. [Google Scholar] [CrossRef]
- Craxton, A.; Jiang, A.; Kurosaki, T.; Clark, E.A. Syk and Bruton’s tyrosine kinase are required for B cell antigen receptor-mediated activation of the kinase Akt. J. Biol. Chem. 1999, 274, 30644–30650. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wong, K.K.; Calame, K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 1997, 276, 596–599. [Google Scholar] [CrossRef]
- Calado, D.P.; Sasaki, Y.; Godinho, S.A.; Pellerin, A.; Köchert, K.; Sleckman, B.P.; De Alborán, I.M.; Janz, M.; Rodig, S.; Rajewsky, K. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 2012, 13, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Weisel, F.; Shlomchik, M.J. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 2018, 48, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevrier, S.; Kratina, T.; Emslie, D.; Tarlinton, D.M.; Corcoran, L.M. IL4 and IL21 cooperate to induce the high Bcl6 protein level required for germinal center formation. Immunol. Cell Biol. 2017, 95, 925–932. [Google Scholar] [CrossRef]
- Niu, H.; Ye, B.H.; Dalla-Favera, R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev. 1998, 12, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamason, R.; McCully, R.; Lew, S.; Pomerantz, J. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry 2010, 49, 8240–8250. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, C.L.; Manning, E.M.; Erickson, L.D.; O’Connor, B.P.; Lind, E.F.; Pullen, S.S.; Kehry, M.R.; Noelle, R.J. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol. 2002, 3, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, K.; Muto, A.; Tanaka, H.; Takahashi, S.; Igarashi, K. Regulation of the plasma cell transcription factor Blimp-1 gene by Bach2 and Bcl6. Int. Immunol. 2008, 20, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, H.P.; Oh, J.; Tunyaplin, C.; Carotta, S.; Donovan, C.E.; Goldman, M.L.; Tailor, P.; et al. Analysis of Interleukin-21-Induced Prdm1 Gene Regulation Reveals Functional Cooperation of STAT3 and IRF4 Transcription Factors. Immunity 2009, 31, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zan, H.; Casali, P. Regulation of Aicda expression and AID activity. Autoimmunity 2013, 46, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Lin, K.I.; Kuo, T.C.; Yu, X.; Hurt, E.M.; Rosenwald, A.; Giltnane, J.M.; Yang, L.; Zhao, H.; Calame, K.; et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002, 17, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, L.; Sasaki, Y.; Calado, D.; Zhang, B.; Paik, J.; DePinho, R.; Kutok, J.; Kearney, J.; Otipoby, K.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Medeiros, L.J.; Xu-Monette, Z.Y.; Li, J.; Young, K.H. Dysregulation of cell survival in diffuse large B cell lymphoma: Mechanisms and therapeutic targets. Front. Oncol. 2019, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.; O’Reilly, L.A.; Gugasyan, R.; Strasser, A.; Adams, J.M.; Gerondakis, S. The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J. 2000, 19, 6351–6360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Dixit, V.M. Signaling to NF-kappaB: Regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2010, 2, a003350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciammas, R.; Li, Y.; Warmflash, A.; Song, Y.; Dinner, A.R.; Singh, H. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol. Syst. Biol. 2011, 7, 495. [Google Scholar] [CrossRef] [PubMed]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.R.; Corradin, A.; Klein, U.; Álvarez, M.J.; Toffolo, G.M.; Di Camillo, B.; Califano, A.; Stolovitzky, G.A. Quantitative modeling of the terminal differentiation of B cells and mechanisms of lymphomagenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 2672–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Mitchell, S.; Liu, Y.; Ohta, S.; Lin, Y.S.; Metzig, M.O.; Nutt, S.L.; Hoffmann, A. A Regulatory Circuit Controlling the Dynamics of NFκB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity 2019, 50, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Méndez, A.; Mendoza, L. A Network Model to Describe the Terminal Differentiation of B Cells. PLoS Comput. Biol. 2016, 12, e1004696. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, K.; Maienschein-Cline, M.; Simonetti, G.; Chen, J.; Rosenthal, R.; Brink, R.; Chong, A.S.; Klein, U.; Dinner, A.R.; Singh, H.; et al. Transcriptional Regulation of Germinal Center B and Plasma Cell Fates by Dynamical Control of IRF4. Immunity 2013, 38, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Schwickert, T.A.; Victora, G.D.; Fooksman, D.R.; Kamphorst, A.O.; Mugnier, M.R.; Gitlin, A.D.; Dustin, M.L.; Nussenzweig, M.C. A dynamic T cell–limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 2011, 208, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Ise, W.; Kurosaki, T. Plasma cell differentiation during the germinal center reaction. Immunol. Rev. 2019, 288, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Thobe, K.; Kuznia, C.; Sers, C.; Siebert, H. Evaluating Uncertainty in Signaling Networks Using Logical Modeling. Front. Physiol. 2018, 9, 1335. [Google Scholar] [CrossRef] [Green Version]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-B in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, H.; Ziepert, M.; Becher, C.; Barth, T.F.E.; Bernd, H.-W.; Feller, A.C.; Klapper, W.; Hummel, M.; Stein, H.; Hansmann, M.-L.; et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013, 121, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Krull, J.E.; Wenzl, K.; Hartert, K.T.; Manske, M.K.; Sarangi, V.; Maurer, M.J.; Larson, M.C.; Nowakowski, G.S.; Ansell, S.M.; McPhail, E.; et al. Somatic copy number gains in MYC, BCL2, and BCL6 identifies a subset of aggressive alternative-DH/TH DLBCL patients. Blood Cancer J. 2020, 10, 117. [Google Scholar] [CrossRef]
- Thys, A.; Douanne, T.; Bidère, N. Post-translational Modifications of the CARMA1-BCL10-MALT1 Complex in Lymphocytes and Activated B-Cell Like Subtype of Diffuse Large B-Cell Lymphoma. Front. Oncol. 2018, 8, 498. [Google Scholar] [CrossRef]
- Poltz, R.; Naumann, M. Dynamics of p53 and NF-κB regulation in response to DNA damage and identification of target proteins suitable for therapeutic intervention. BMC Syst. Biol. 2012, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Konrath, F.; Mittermeier, A.; Cristiano, E.; Wolf, J.; Loewer, A. A systematic approach to decipher crosstalk in the p53 signaling pathway using single cell dynamics. PLOS Comput. Biol. 2020, 16, e1007901. [Google Scholar] [CrossRef] [PubMed]
- Mothes, J.; Ipenberg, I.; Arslan, S.Ç.; Benary, U.; Scheidereit, C.; Wolf, J. A Quantitative Modular Modeling Approach Reveals the Effects of Different A20 Feedback Implementations for the NF-kB Signaling Dynamics. Front. Physiol. 2020, 11, 896. [Google Scholar] [CrossRef]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative Dissection and Modeling of the NF-κB p100-p105 Module Reveals Interdependent Precursor Proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pone, E.J.; Zhang, J.; Mai, T.; White, C.A.; Li, G.; Sakakura, J.K.; Patel, P.J.; Al-Qahtani, A.; Zan, H.; Xu, Z.; et al. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-κB pathway. Nat. Commun. 2012, 3, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Carrio, R.; Yu, A.; Malek, T. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J. Immunol. 2004, 173, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Sáez, A.I.; Sáez, A.J.; Artiga, M.J.; Pérez-Rosado, A.; Camacho, F.I.; Díez, A.; García, J.F.; Fraga, M.; Bosch, R.; Rodríguez-Pinilla, S.M.; et al. Building an Outcome Predictor Model for Diffuse Large B-Cell Lymphoma. Am. J. Pathol. 2004, 164, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Çağlayan, Ç.; Goldstein, J.S.; Ayer, T.; Rai, A.; Flowers, C.R. A population-based multistate model for diffuse large B-cell lymphoma–specific mortality in older patients. Cancer 2019, 125, 1837–1847. [Google Scholar] [CrossRef]
- Du, W.; Goldstein, R.; Jiang, Y.; Aly, O.; Cerchietti, L.; Melnick, A.; Elemento, O. Effective combination therapies for B-cell lymphoma predicted by a virtual disease model. Cancer Res. 2017, 77, 1818–1830. [Google Scholar] [CrossRef] [Green Version]
- Béal, J.; Montagud, A.; Traynard, P.; Barillot, E.; Calzone, L. Personalization of Logical Models With Multi-Omics Data Allows Clinical Stratification of Patients. Front. Physiol. 2019, 9, 1965. [Google Scholar] [CrossRef]
- Karin, M. The Beginning of the End: IκB Kinase (IKK) and NF-κB Activation*. J. Biol. Chem. 1999, 274, 27339–27342. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Component | Function | Reference |
|---|---|---|
| PI3K | SYK ∧ BTK | [29] |
| MYC | PI3K ∧ NF-κB ∧ ¬ BLIMP1 ∧ ¬ BCL6 | [30,31,32] |
| ERK | RAF ∧ ¬ STAT | [33,34] |
| IKKc:1 | CBM | [35] |
| IKKc:2 | TRAF6 | [36] |
| BCL6 | IRF4:1 ∧ ¬ IRF4:2 ∧ ¬ (ERK ∨ BLIMP1) | [3,34,37] |
| BLIMP1 | IRF4:2 ∧ STAT ∧ ¬ BCL6 | [37,38] |
| AID | NF-κB ∧ STAT ∧ ¬ BLIMP1 | [39,40] |
| BCL2 | (PI3K ∨ NF-κB) ∧ ¬ BCL6 | [41,42,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thobe, K.; Konrath, F.; Chapuy, B.; Wolf, J. Patient-Specific Modeling of Diffuse Large B-Cell Lymphoma. Biomedicines 2021, 9, 1655. https://doi.org/10.3390/biomedicines9111655
Thobe K, Konrath F, Chapuy B, Wolf J. Patient-Specific Modeling of Diffuse Large B-Cell Lymphoma. Biomedicines. 2021; 9(11):1655. https://doi.org/10.3390/biomedicines9111655
Chicago/Turabian StyleThobe, Kirsten, Fabian Konrath, Björn Chapuy, and Jana Wolf. 2021. "Patient-Specific Modeling of Diffuse Large B-Cell Lymphoma" Biomedicines 9, no. 11: 1655. https://doi.org/10.3390/biomedicines9111655
APA StyleThobe, K., Konrath, F., Chapuy, B., & Wolf, J. (2021). Patient-Specific Modeling of Diffuse Large B-Cell Lymphoma. Biomedicines, 9(11), 1655. https://doi.org/10.3390/biomedicines9111655