
Mechanical Stretching-Induced Traumatic Brain Injury Is Mediated by the Formation of GSK-3β-Tau Complex to Impair Insulin Signaling Transduction

 , and
, and 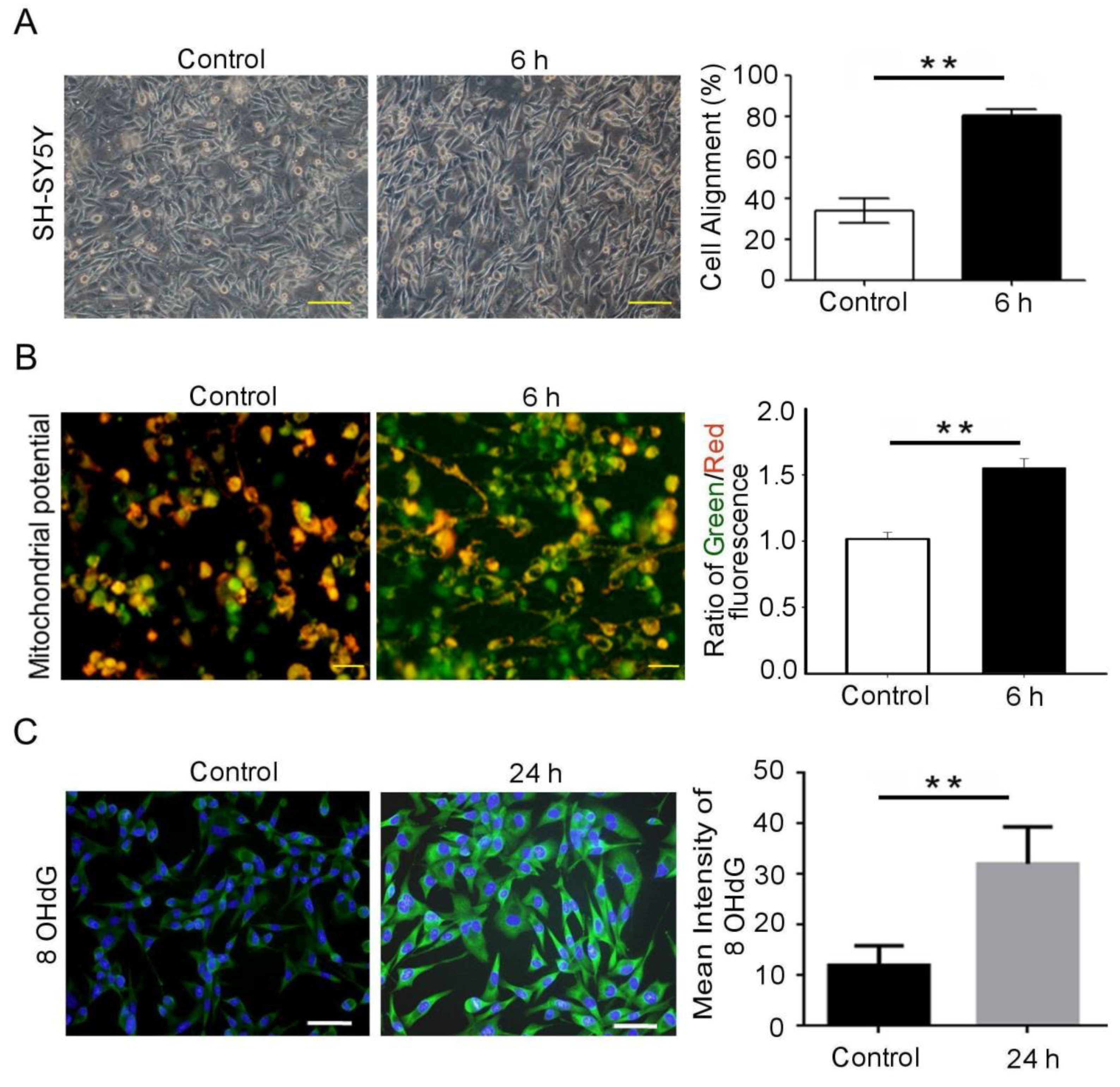{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Stretch Device
2.3. Stretched-Injury Model
2.4. Cell Alignment Measurement
2.5. Immunoblotting Assay
2.6. Measuring BDNF Levels
2.7. Immunofluorescence Assay
2.8. Mitochondrial Membrane Potential
2.9. Co-Immunoprecipitation
2.10. Statistical Analyses
3. Results
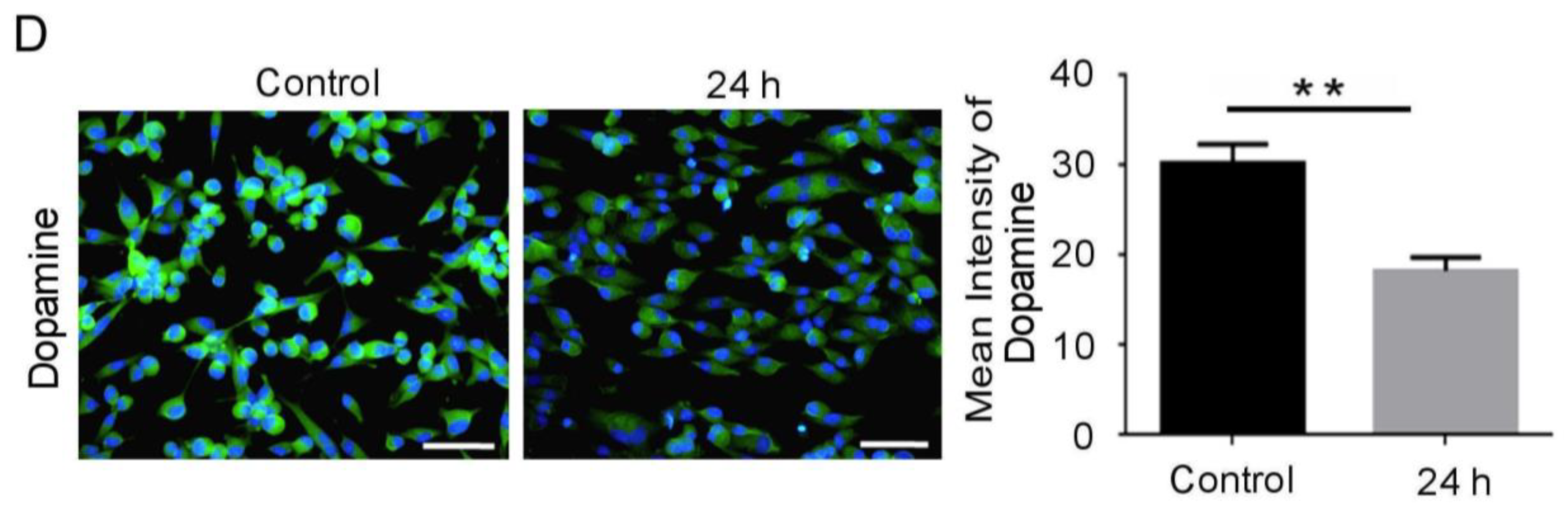
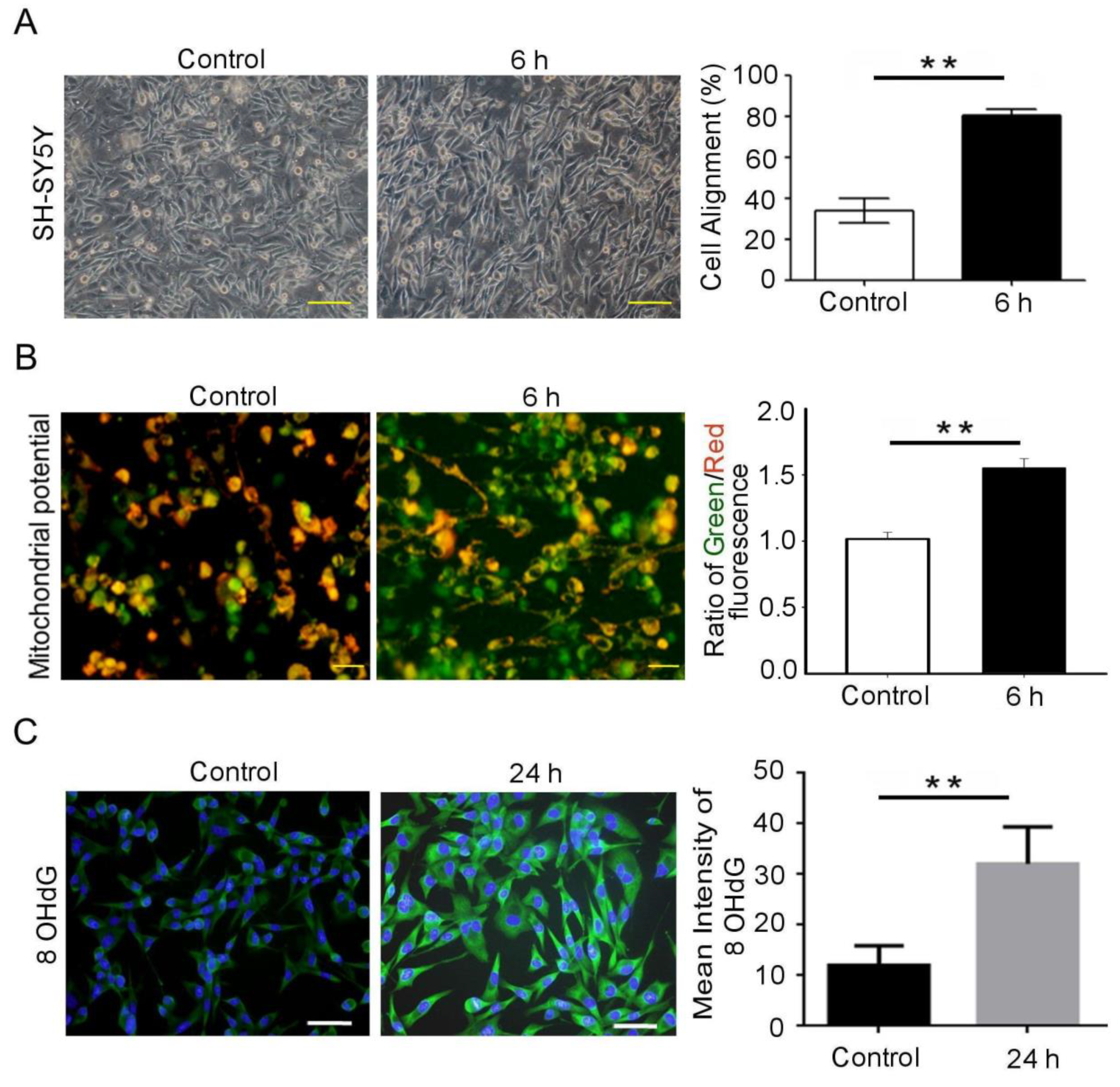
3.1. Mechanical Injury Induces Neuron Injury through Stretching, thus Altering the Mitochondrial Membrane Potential and Inducing Oxidative DNA Damage
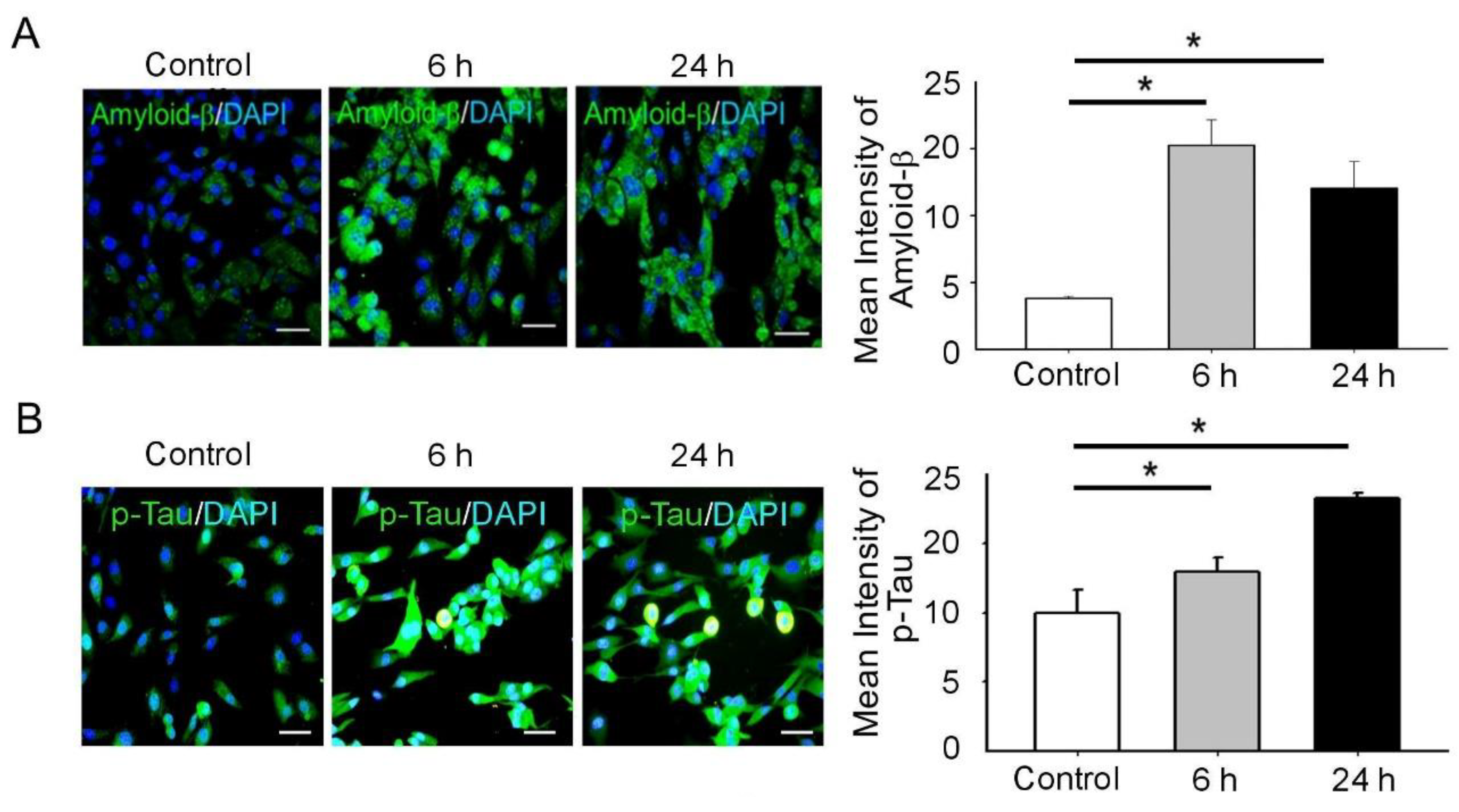
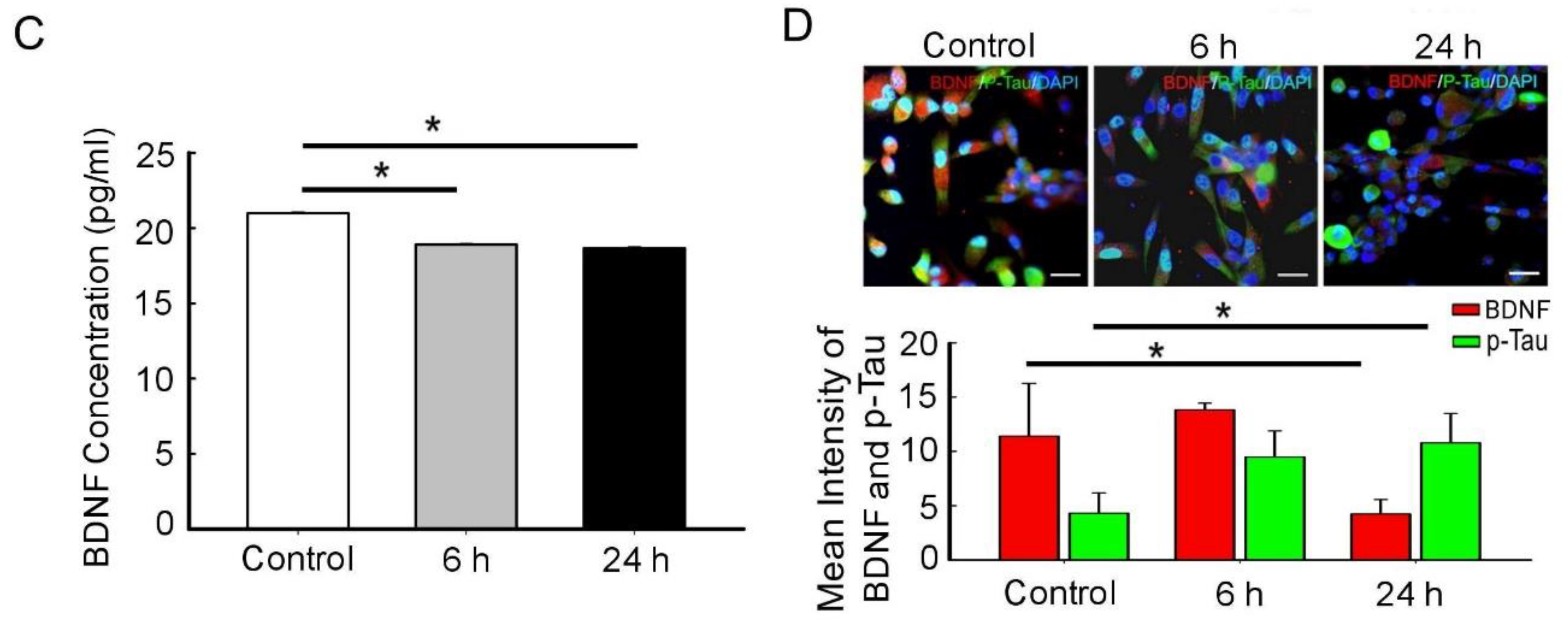
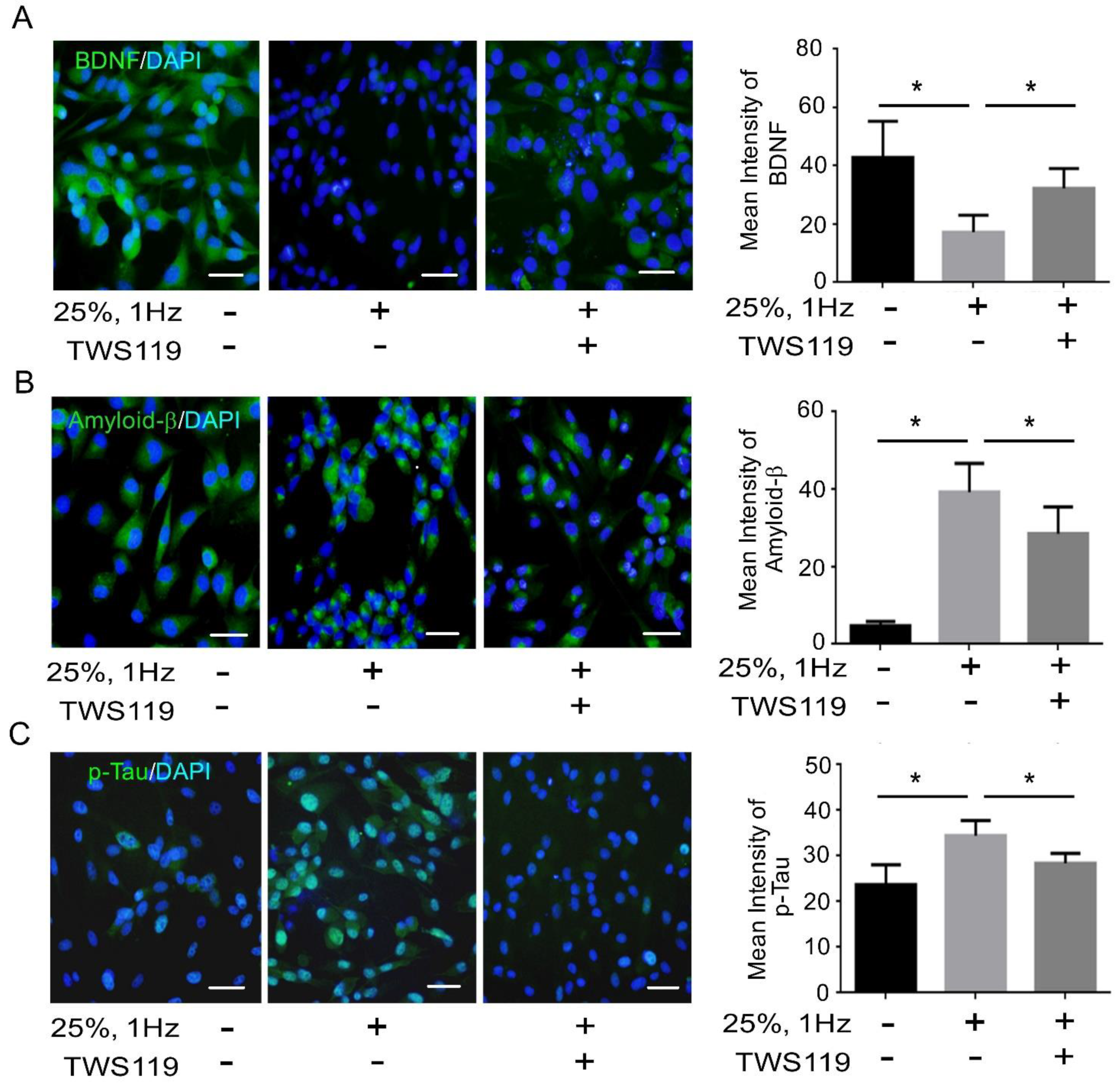
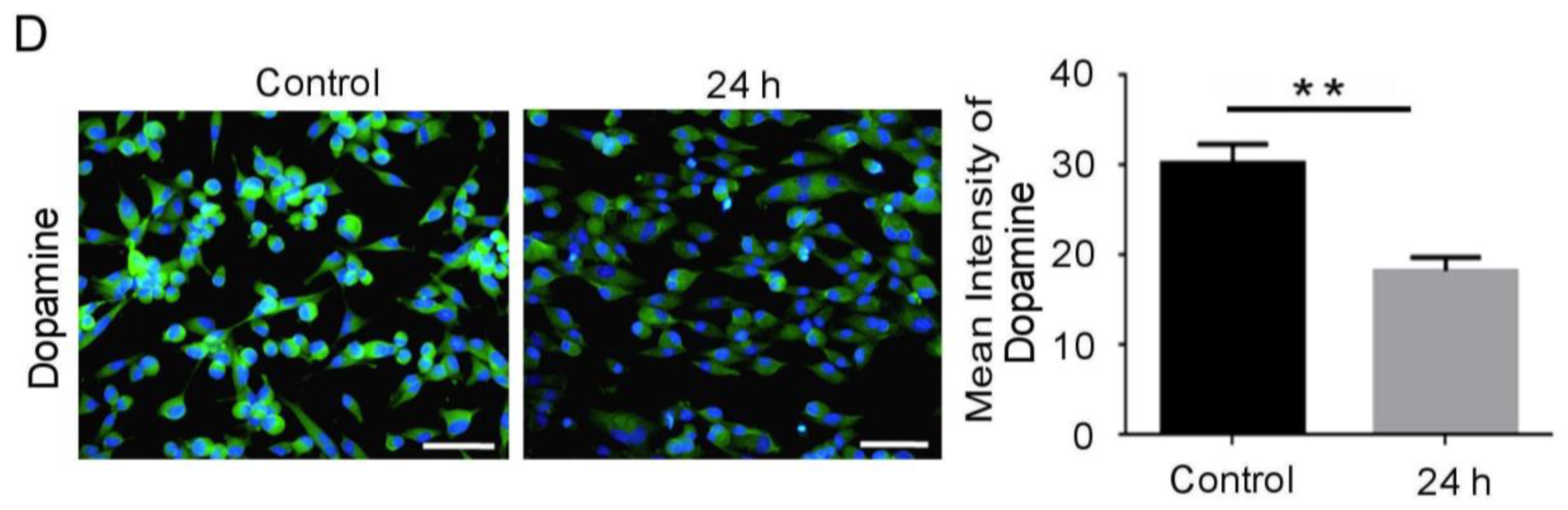
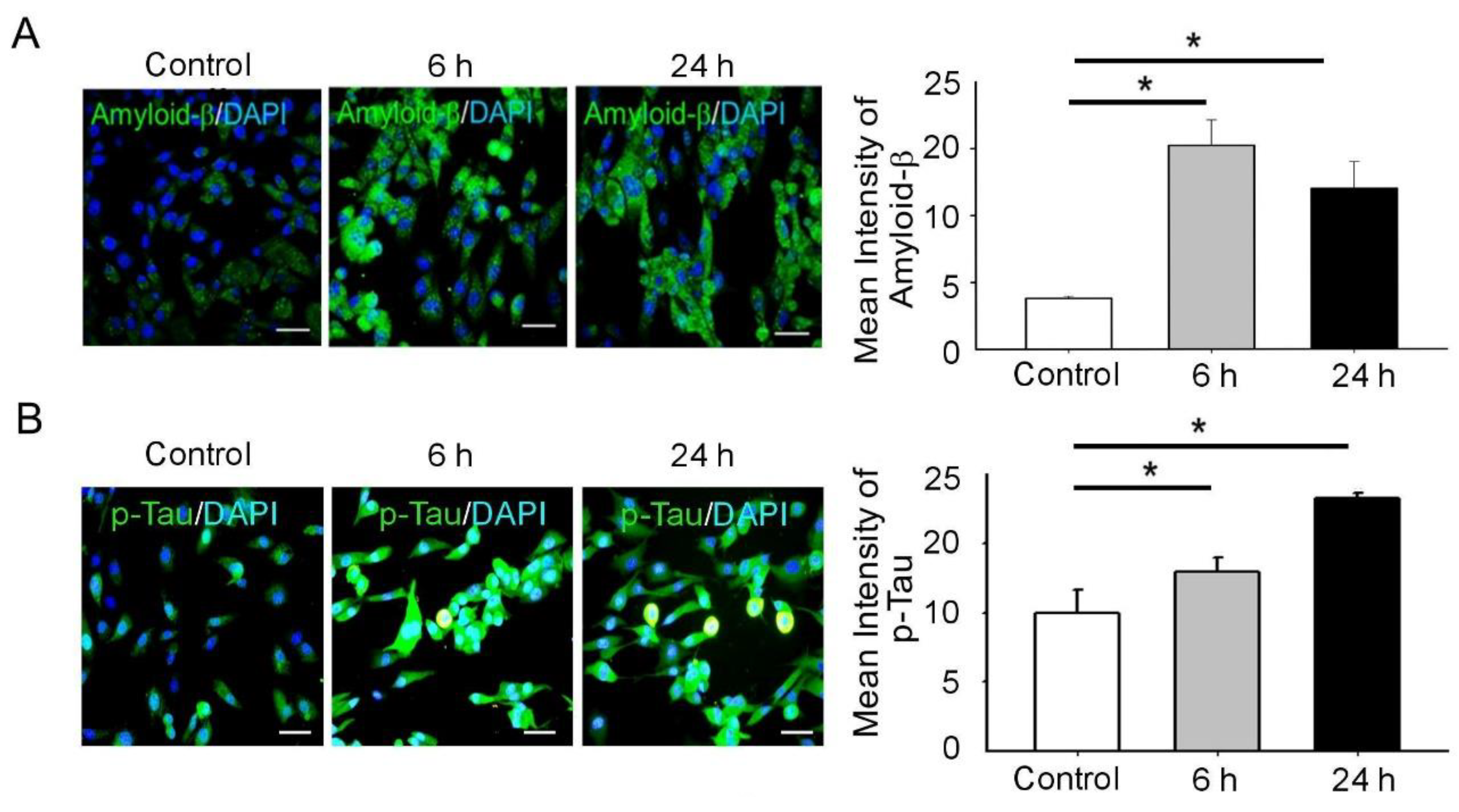
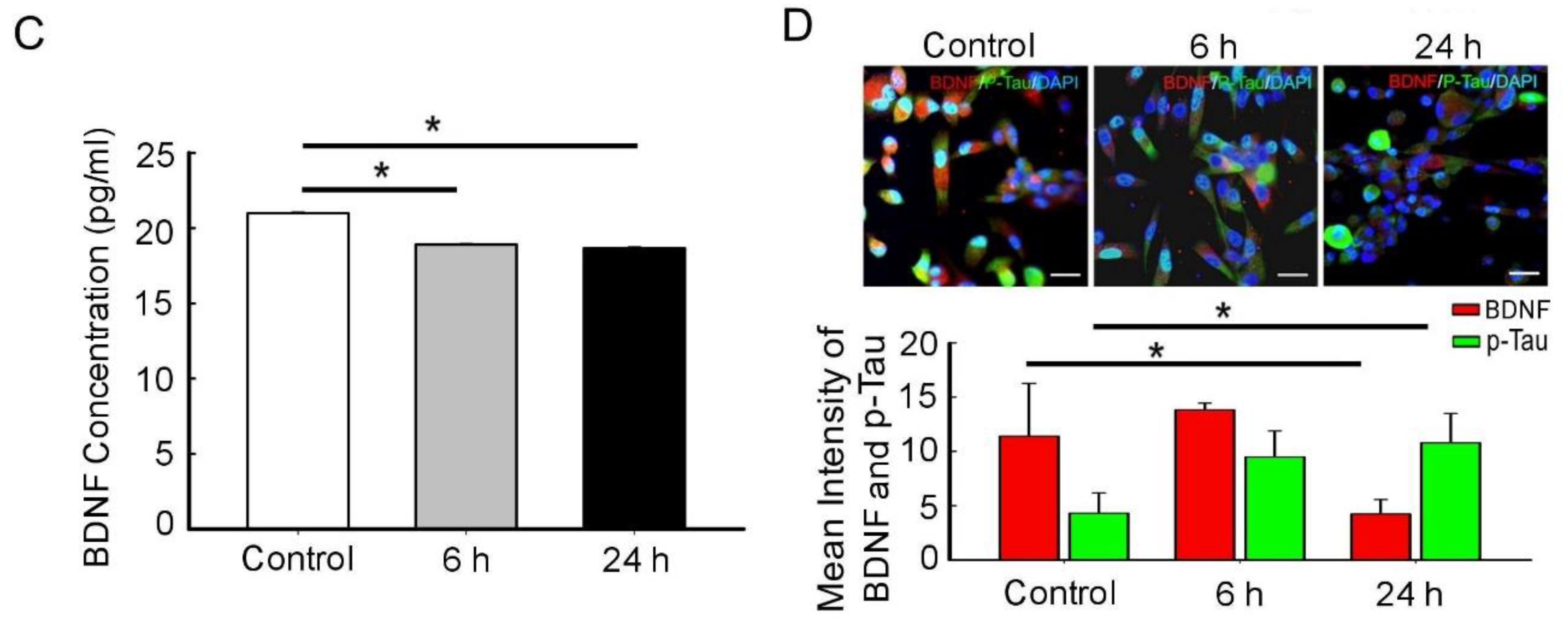
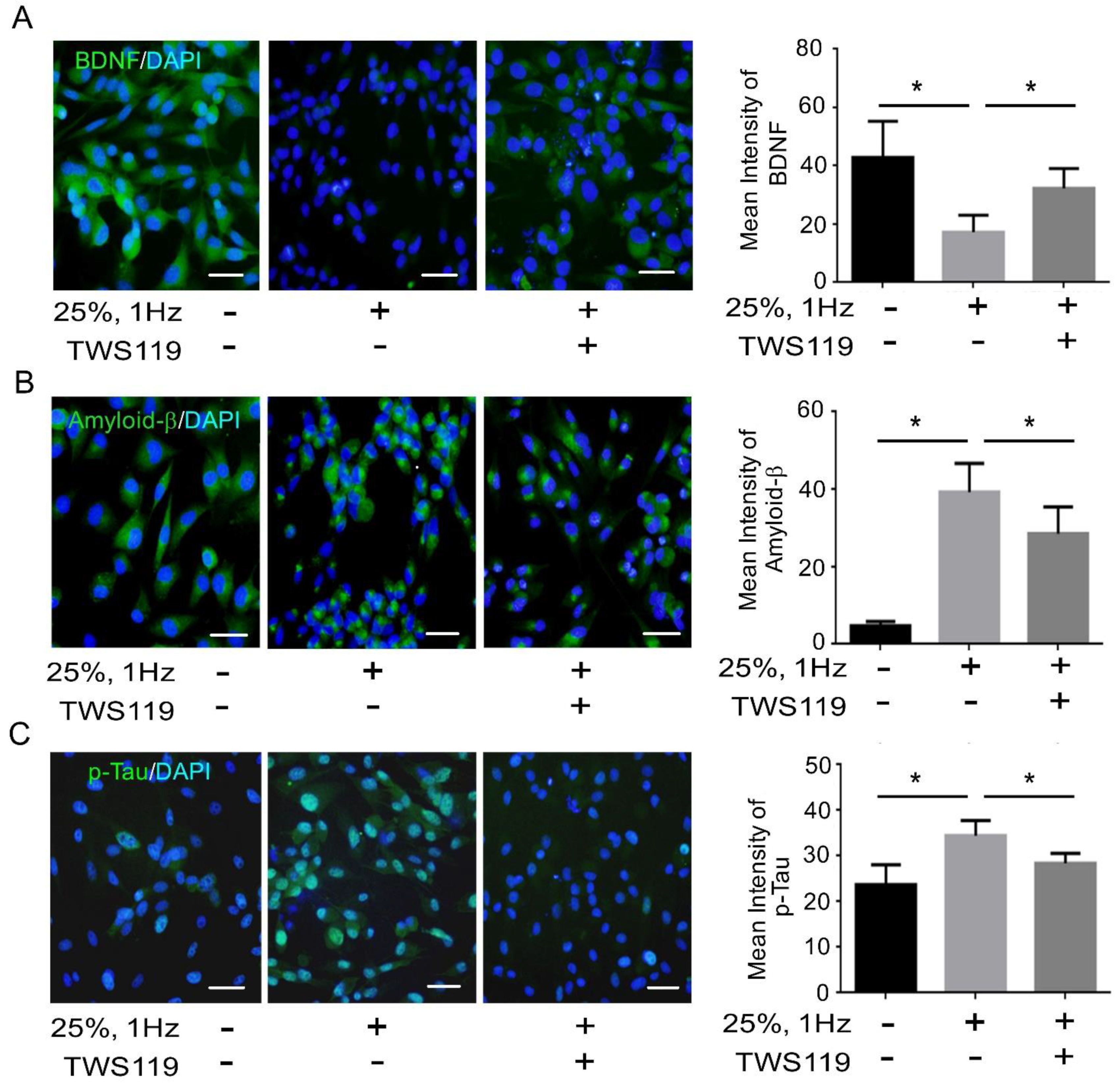
3.2. BDNF Reduction Is Associated with Increased Amyloid-β/p-Tau
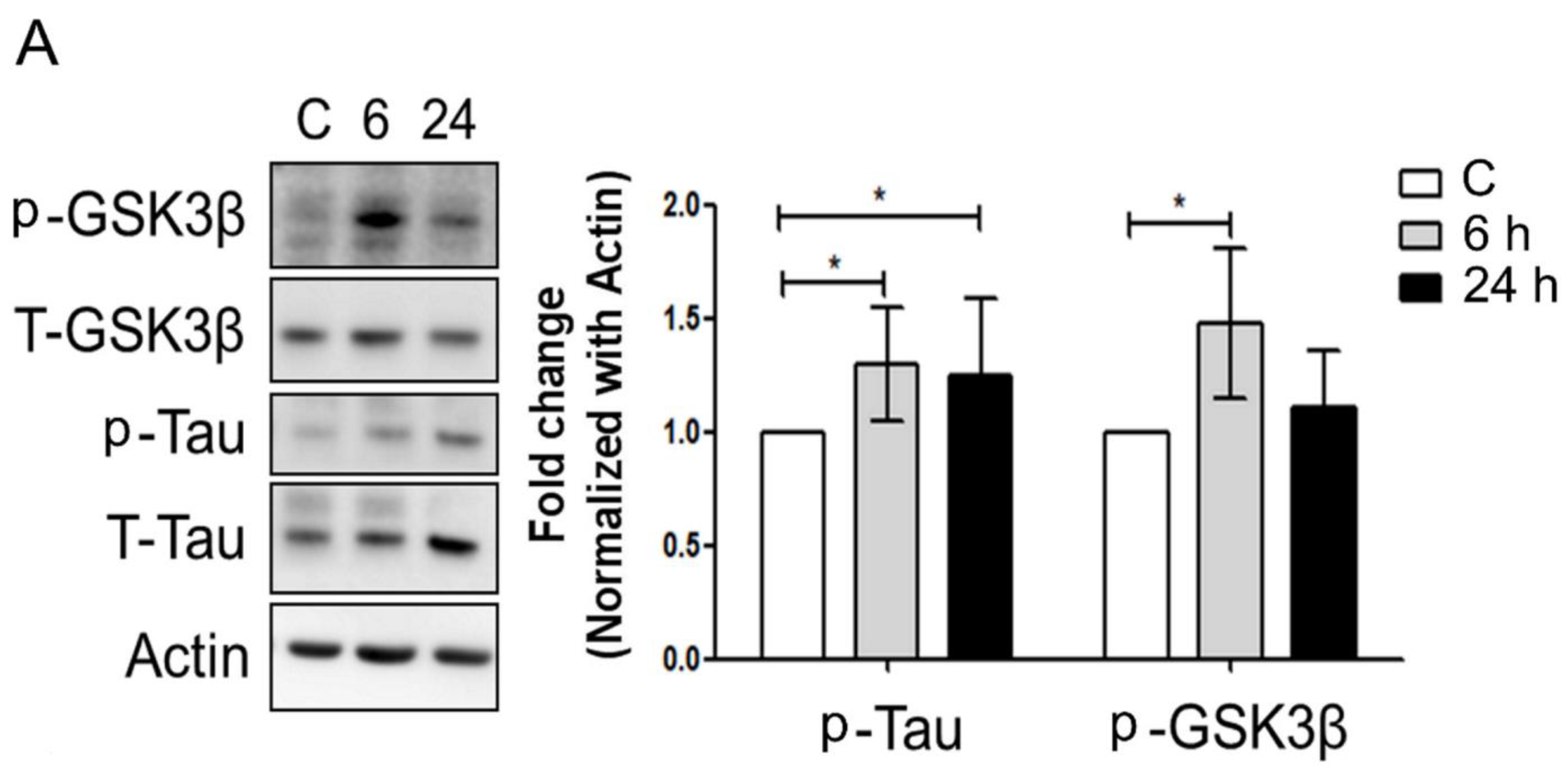
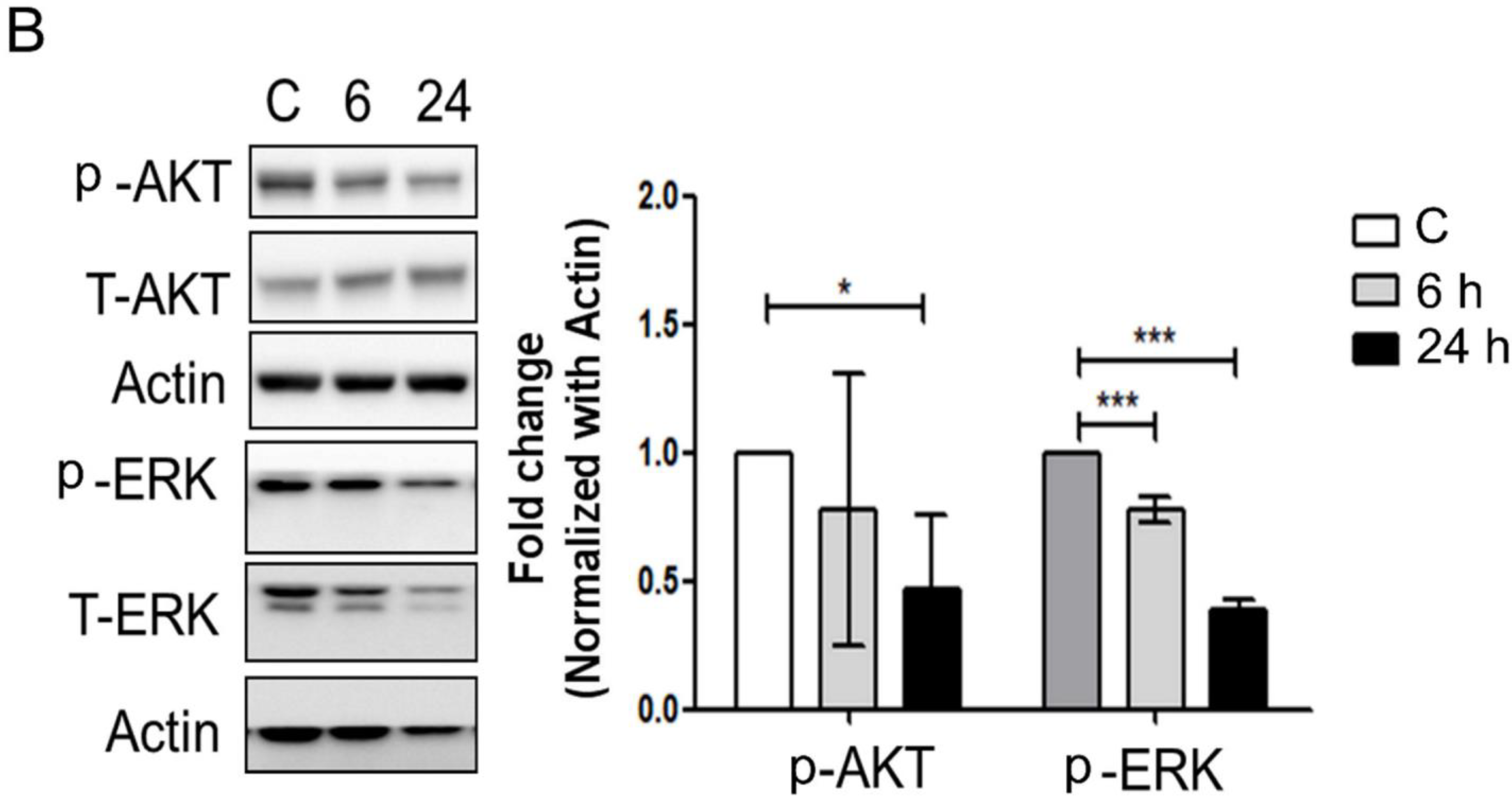
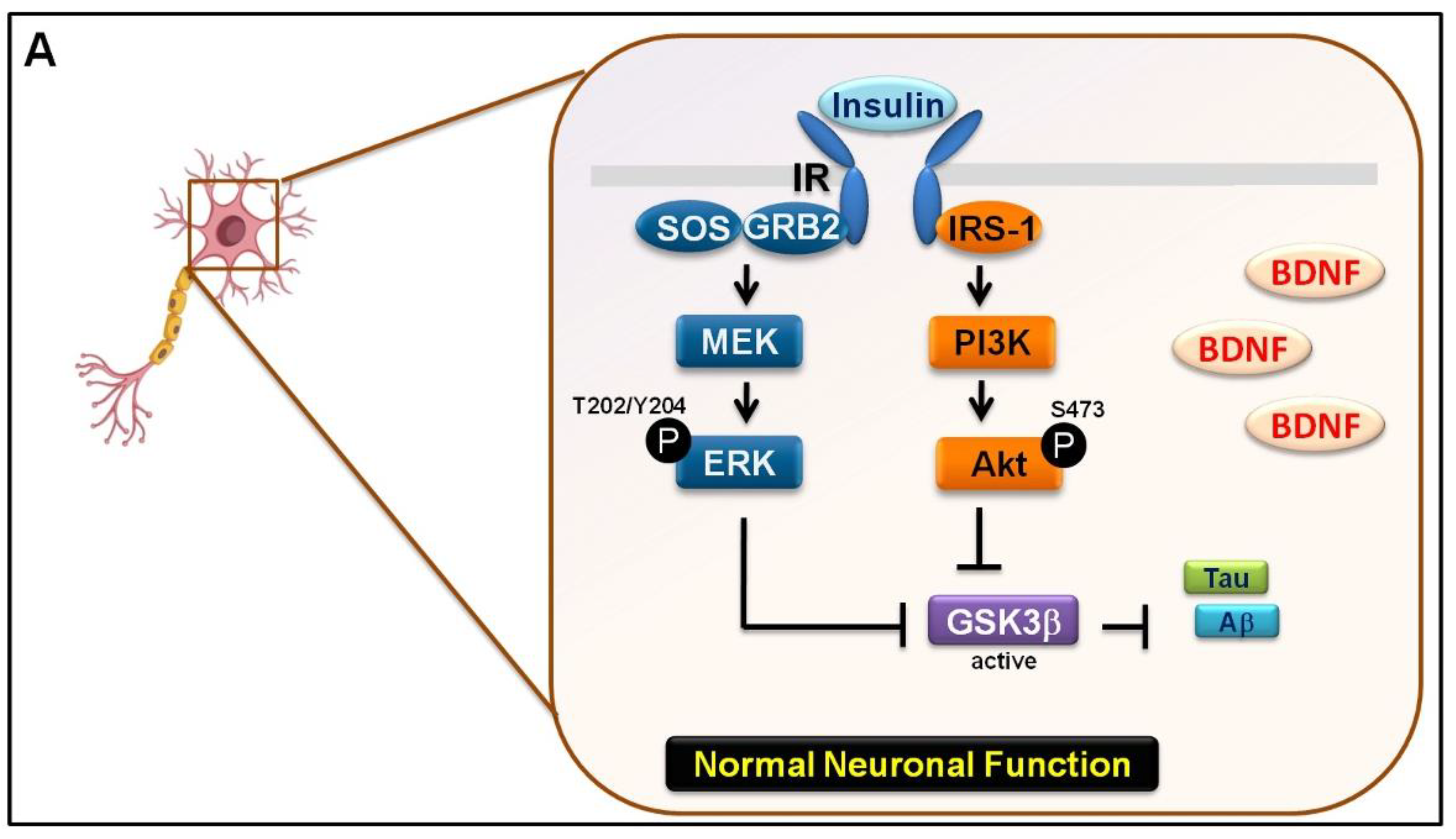
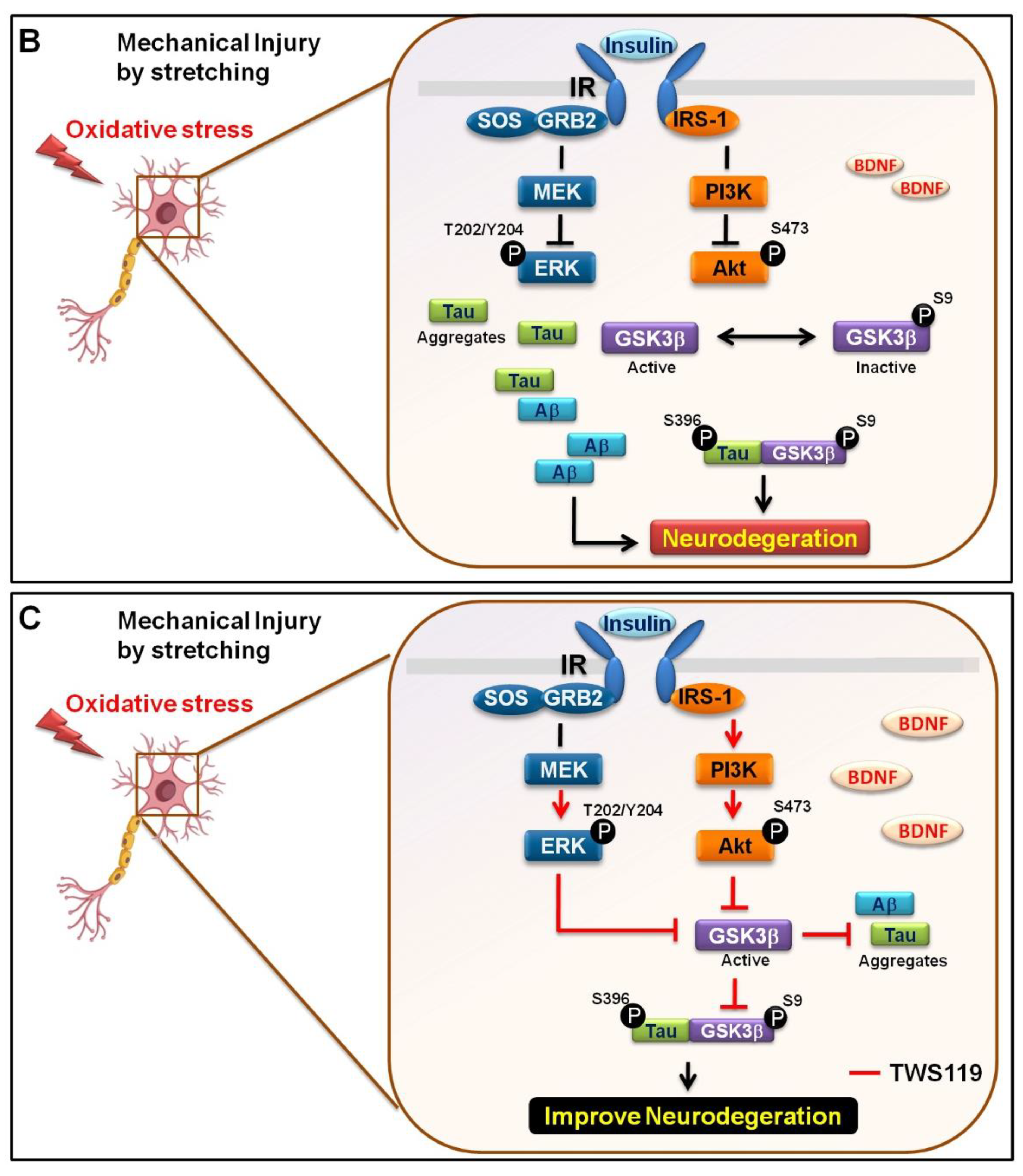
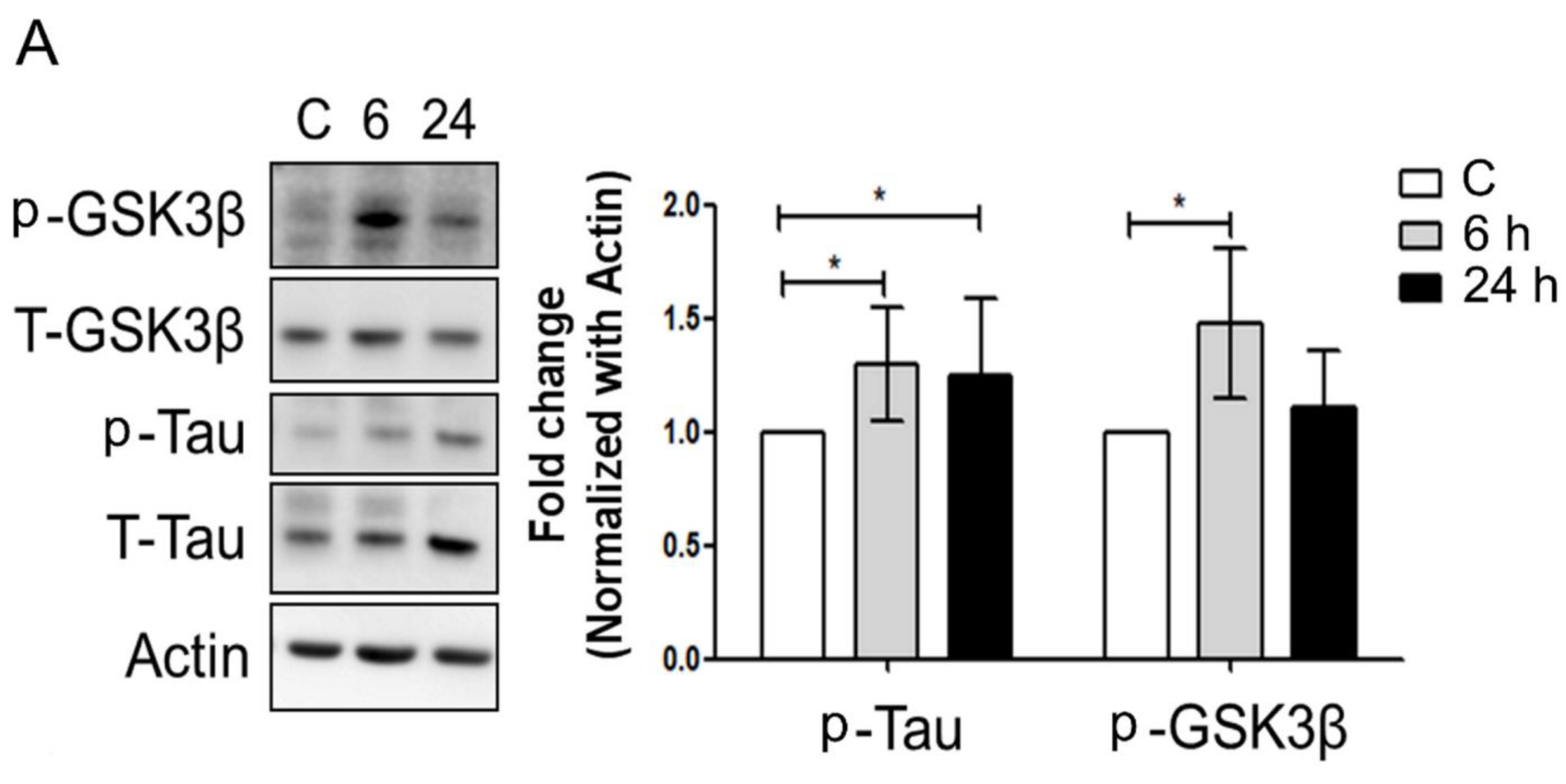
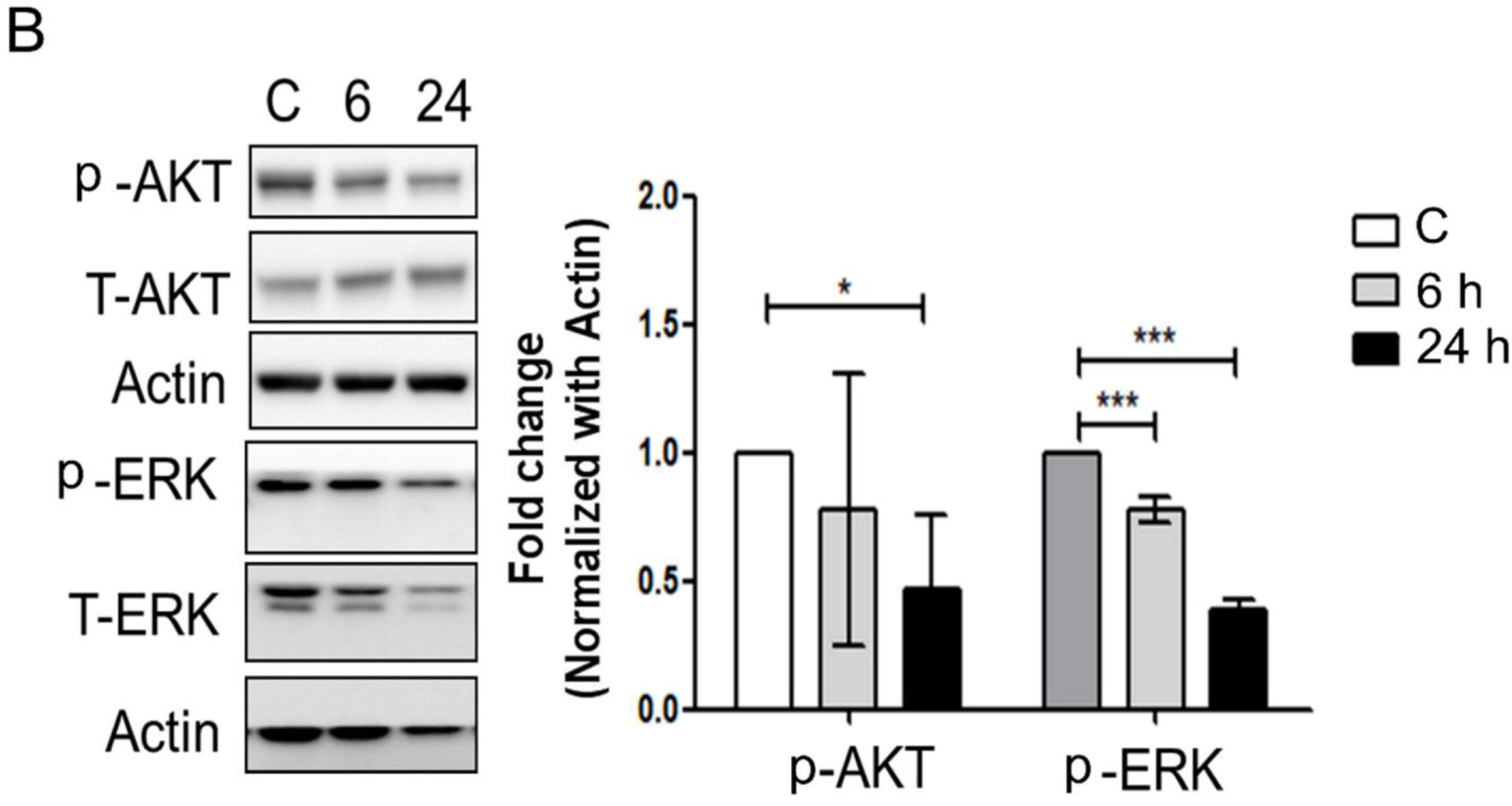
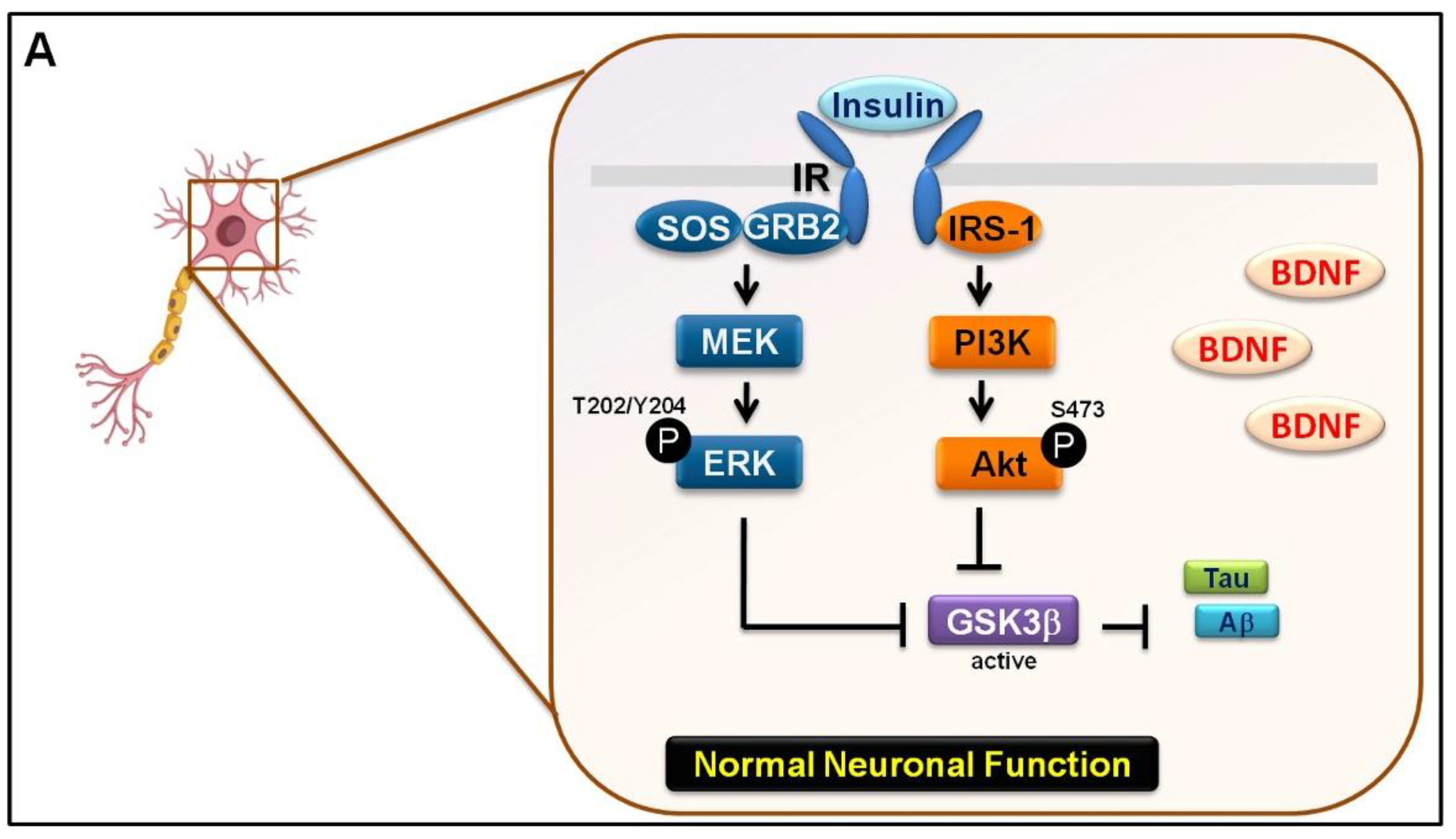
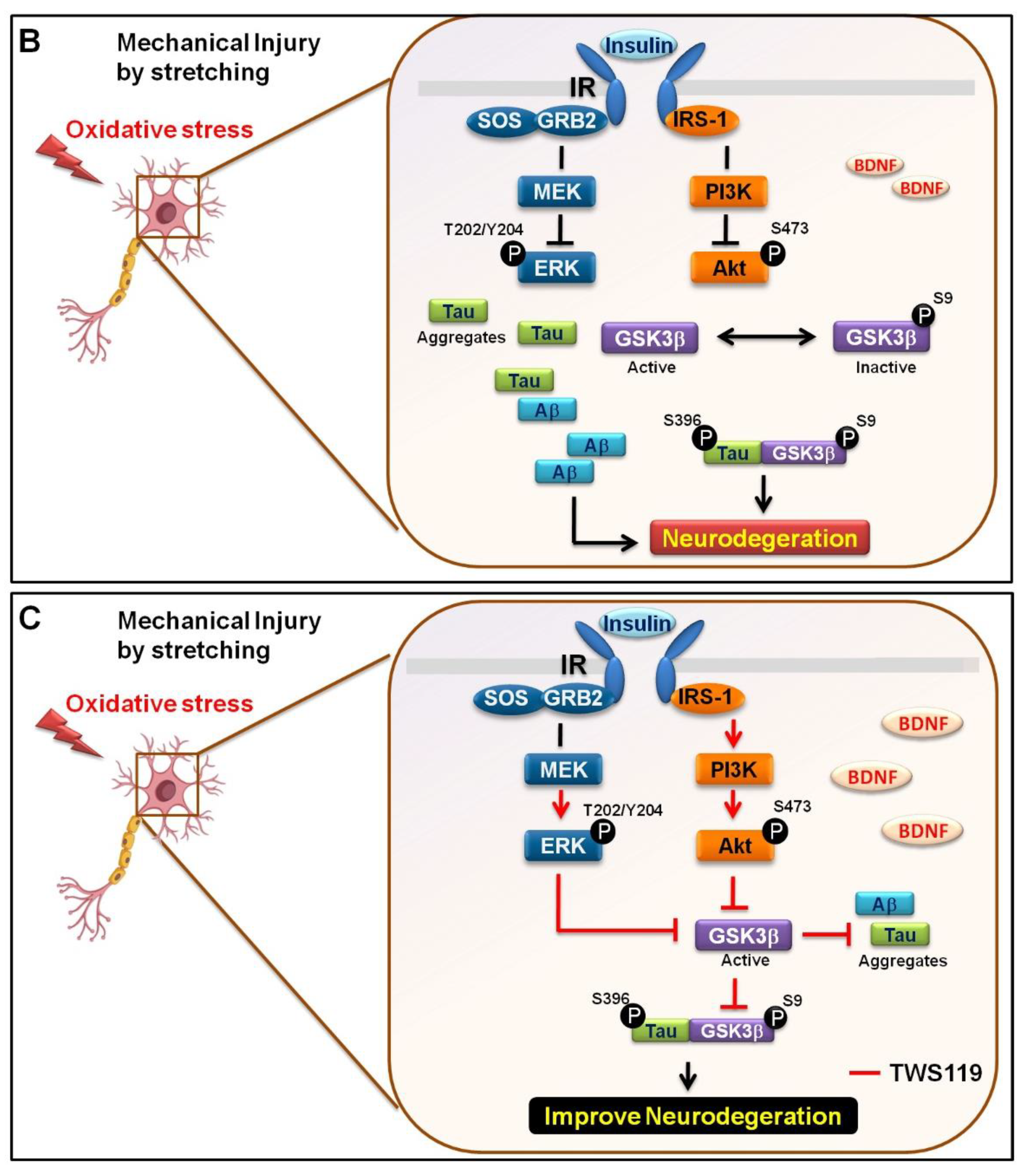
3.3. Mechanical Injury through Stretching Upregulated p-GSK3β/p-Tau Protein Levels and Are Associated with a Reduction of the Insulin Pathway
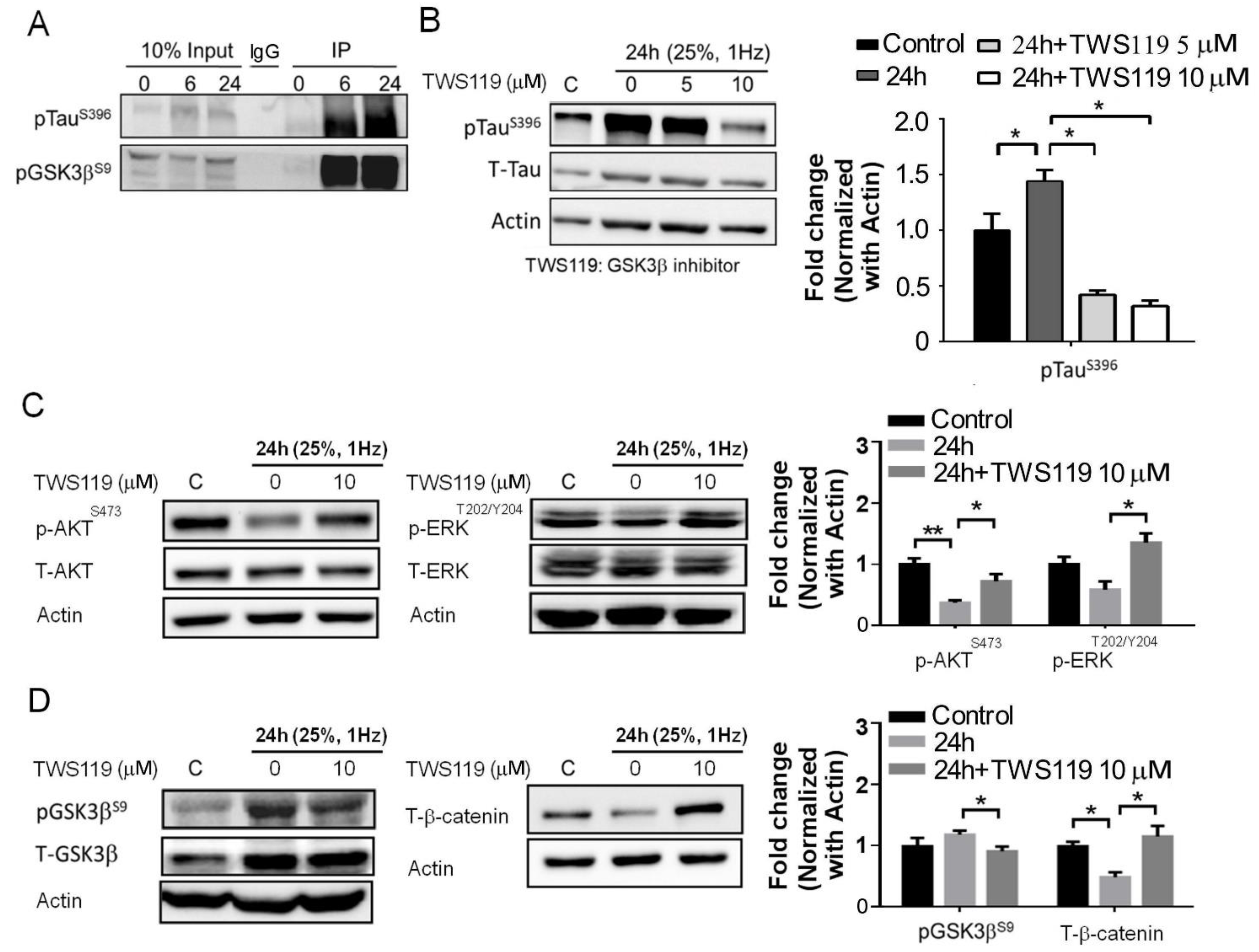
3.4. The Interaction between GSK-3β and p-Tau Plays a Crucial Role and Is Associated with the Reduction of the Insulin Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhandapani, S.; Manju, D.; Sharma, B.; Mahapatra, A. Prognostic significance of age in traumatic brain injury. J. Neurosci. Rural Pract. 2012, 3, 131–135. [Google Scholar] [CrossRef]
- Fleminger, S.; Ponsford, J. Long term outcome after traumatic brain injury: More attention needs to be paid to neuropsychiatric functioning. BMJ Br. Med. J. 2005, 331, 1419–1420. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head injury and the risk of ad in the mirage study. Neurology 2000, 54, 1316–1323. [Google Scholar] [CrossRef]
- Nordstrom, P.; Michaelsson, K.; Gustafson, Y.; Nordstrom, A. Traumatic brain injury and young onset dementia: A nationwide cohort study. Ann. Neurol. 2014, 75, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Kaup, A.; Barnes, D.E.; Yaffe, K. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurol. 2014, 71, 1490–1497. [Google Scholar] [CrossRef]
- Hall, E.D.; Wang, J.A.; Bosken, J.M.; Singh, I.N. Lipid peroxidation in brain or spinal cord mitochondria after injury. J. Bioenerg. Biomembr. 2016, 48, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Freund, R.K.; Gibson, E.S.; Potter, H.; Dell’Acqua, M.L. Inhibition of the motor protein eg5/kinesin-5 in amyloid β-mediated impairment of hippocampal long-term potentiation and dendritic spine loss. Mol. Pharmacol. 2016, 89, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodiere, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olczak, M.; Niderla-Bielinska, J.; Kwiatkowska, M.; Samojlowicz, D.; Tarka, S.; Wierzba-Bobrowicz, T. Tau protein (mapt) as a possible biochemical marker of traumatic brain injury in postmortem examination. Forensic Sci. Int. 2017, 280, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, J.C.; Bitow, F.; Haack, J.; d’Hedouville, Z.; Zhang, J.N.; Tönges, L.; Michel, U.; Oliveira, L.M.A.; Jovin, T.M.; Liman, J.; et al. Alpha-synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in cns neurons. Cell Death Dis. 2015, 6, e1811. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Sasaki, K.; Akiyama, K. Increased insulin levels after ogtt load in peripheral blood and cerebrospinal fluid of patients with dementia of alzheimer type. Biol. Psychiatry 1991, 30, 1219–1228. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Ganju, N.; Banerjee, K.; Brown, N.V.; Luong, T.; Wands, J.R. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol. Clin. Exp. Res. 2000, 24, 716–726. [Google Scholar] [CrossRef]
- Xu, J.; Yeon, J.E.; Chang, H.; Tison, G.; Chen, G.J.; Wands, J.; de la Monte, S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: Role of pten phosphatase. J. Biol. Chem. 2003, 278, 26929–26937. [Google Scholar] [CrossRef] [Green Version]
- De Ferrari, G.V.; Inestrosa, N.C. Wnt signaling function in alzheimer’s disease. Brain Res. Brain Res. Rev. 2000, 33, 1–12. [Google Scholar] [CrossRef]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-beta: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-beta. Brain Res. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Neselius, S.; Zetterberg, H.; Blennow, K.; Randall, J.; Wilson, D.; Marcusson, J.; Brisby, H. Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj. 2013, 27, 425–433. [Google Scholar] [CrossRef]
- Gill, J.; Merchant-Borna, K.; Jeromin, A.; Livingston, W.; Bazarian, J. Acute plasma tau relates to prolonged return to play after concussion. Neurology 2017, 88, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. Nadph oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Cao, S.; Chao, H.; Liu, Y.; Ji, J. Sex-related differences in striatal dopaminergic system after traumatic brain injury. Brain Res. Bull. 2016, 124, 214–221. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin resistance as a link between amyloid-beta and tau pathologies in alzheimer’s disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Wang, T.; Xie, C.; Yu, P.; Fang, F.; Zhu, J.; Cheng, J.; Gu, A.; Wang, J.; Xiao, H. Involvement of insulin signaling disturbances in bisphenol a-induced alzheimer’s disease-like neurotoxicity. Sci. Rep. 2017, 7, 7497. [Google Scholar] [CrossRef]
- Morrison, B., 3rd; Cater, H.L.; Benham, C.D.; Sundstrom, L.E. An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J. Neurosci. Methods 2006, 150, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Yang, D.; Fu, Y.; Wang, W.; Yang, G.; Yuan, F.; Chen, H.; Ding, J.; Chen, S.; Tian, H. Neuroprotective effects of serpina3k in traumatic brain injury. Front. Neurol. 2019, 10, 1215. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, M.A.; Dauth, S.; Yu, C.J.; Dabiri, B.E.; Parker, K.K. Traumatic brain injury and the neuronal microenvironment: A potential role for neuropathological mechanotransduction. Neuron 2015, 85, 1177–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy following repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B., 3rd; Elkin, B.S.; Dolle, J.P.; Yarmush, M.L. In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 2011, 13, 91–126. [Google Scholar] [CrossRef] [PubMed]
- Pfister, B.J.; Weihs, T.P.; Betenbaugh, M.; Bao, G. An in vitro uniaxial stretch model for axonal injury. Ann. Biomed. Eng. 2003, 31, 589–598. [Google Scholar] [CrossRef]
- Bar-Kochba, E.; Scimone, M.T.; Estrada, J.B.; Franck, C. Strain and rate-dependent neuronal injury in a 3d in vitro compression model of traumatic brain injury. Sci. Rep. 2016, 6, 30550. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Hernandez, H.; Burks, S.M.; Cuevas, E.; Ali, S.F. Stretch-induced deformation as a model to study dopaminergic dysfunction in traumatic brain injury. Neurochem. Res. 2019, 44, 2546–2555. [Google Scholar] [CrossRef]
- Shin, S.S.; Bray, E.R.; Zhang, C.Q.; Dixon, C.E. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 2011, 1369, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Liu, Y.; Yang, D.; Yuan, F.; Ding, J.; Chen, H.; Tian, H. Sesamin protects sh-sy5y cells against mechanical stretch injury and promoting cell survival. BMC Neurosci. 2017, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Medina, M.; Avila, J. Glycogen synthase kinase-3 (gsk-3) inhibitors for the treatment of alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2790–2798. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Ansari, J.A.; Ansari, Z.E.; Alam, Q.; Gan, S.H.; Kamal, M.A.; Ahmad, E. A molecular bridge: Connecting type 2 diabetes and alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 312–321. [Google Scholar] [CrossRef]
- Leng, S.; Zhang, W.; Zheng, Y.; Liberman, Z.; Rhodes, C.J.; Eldar-Finkelman, H.; Sun, X.J. Glycogen synthase kinase 3 beta mediates high glucose-induced ubiquitination and proteasome degradation of insulin receptor substrate 1. J. Endocrinol. 2010, 206, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, R.; Bucht, G.; Lithner, F.; Winblad, B. Hypoglycemia in alzheimer’s disease. Acta Med. Scand. 1980, 208, 387–388. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Boyd, G.W. The role of insulin-like growth factors and insulin-like growth factor-binding proteins in the nervous system. Biochem. Insights 2019, 12, 1178626419842176. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-derived neurotrophic factor: A key molecule for memory in the healthy and the pathological brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.H.; Yao, Y.; Lin, Y.M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis p-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.S.; Stutzmann, G.E. Inhibition of glycogen synthase kinase-3: An emerging target in the treatment of traumatic brain injury. J. Neurotrauma 2016, 33, 2065–2076. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Rzigalinski, B.A.; Willoughby, K.A.; Sitterding, H.A.; Ellis, E.F. Stretch-induced injury alters mitochondrial membrane potential and cellular atp in cultured astrocytes and neurons. J. Neurochem. 2000, 74, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Arundine, M.; Aarts, M.; Lau, A.; Tymianski, M. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J. Neurosci. 2004, 24, 8106–8123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Franco, R.; Skotak, M.; Hu, G.; Chandra, N. Mechanical stretch exacerbates the cell death in sh-sy5y cells exposed to paraquat: Mitochondrial dysfunction and oxidative stress. Neurotoxicology 2014, 41, 54–63. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, P.-W.; Wu, Y.-C.; Wong, T.-Y.; Sun, G.-C.; Tseng, C.-J. Mechanical Stretching-Induced Traumatic Brain Injury Is Mediated by the Formation of GSK-3β-Tau Complex to Impair Insulin Signaling Transduction. Biomedicines 2021, 9, 1650. https://doi.org/10.3390/biomedicines9111650
Cheng P-W, Wu Y-C, Wong T-Y, Sun G-C, Tseng C-J. Mechanical Stretching-Induced Traumatic Brain Injury Is Mediated by the Formation of GSK-3β-Tau Complex to Impair Insulin Signaling Transduction. Biomedicines. 2021; 9(11):1650. https://doi.org/10.3390/biomedicines9111650
Chicago/Turabian StyleCheng, Pei-Wen, Yi-Chung Wu, Tzyy-Yue Wong, Gwo-Ching Sun, and Ching-Jiunn Tseng. 2021. "Mechanical Stretching-Induced Traumatic Brain Injury Is Mediated by the Formation of GSK-3β-Tau Complex to Impair Insulin Signaling Transduction" Biomedicines 9, no. 11: 1650. https://doi.org/10.3390/biomedicines9111650
APA StyleCheng, P.-W., Wu, Y.-C., Wong, T.-Y., Sun, G.-C., & Tseng, C.-J. (2021). Mechanical Stretching-Induced Traumatic Brain Injury Is Mediated by the Formation of GSK-3β-Tau Complex to Impair Insulin Signaling Transduction. Biomedicines, 9(11), 1650. https://doi.org/10.3390/biomedicines9111650