
DNA Methylation Signature in Mononuclear Cells and Proinflammatory Cytokines May Define Molecular Subtypes in Sporadic Meniere Disease

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Clinical Data
2.3. DNA Extraction
2.4. WGBS Library Preparation
2.5. WGBS Data Analysis
2.6. Undermetlylated Regions
2.7. Inner Ear Gene Sets
2.8. Functional Analysis
2.9. Visualizations
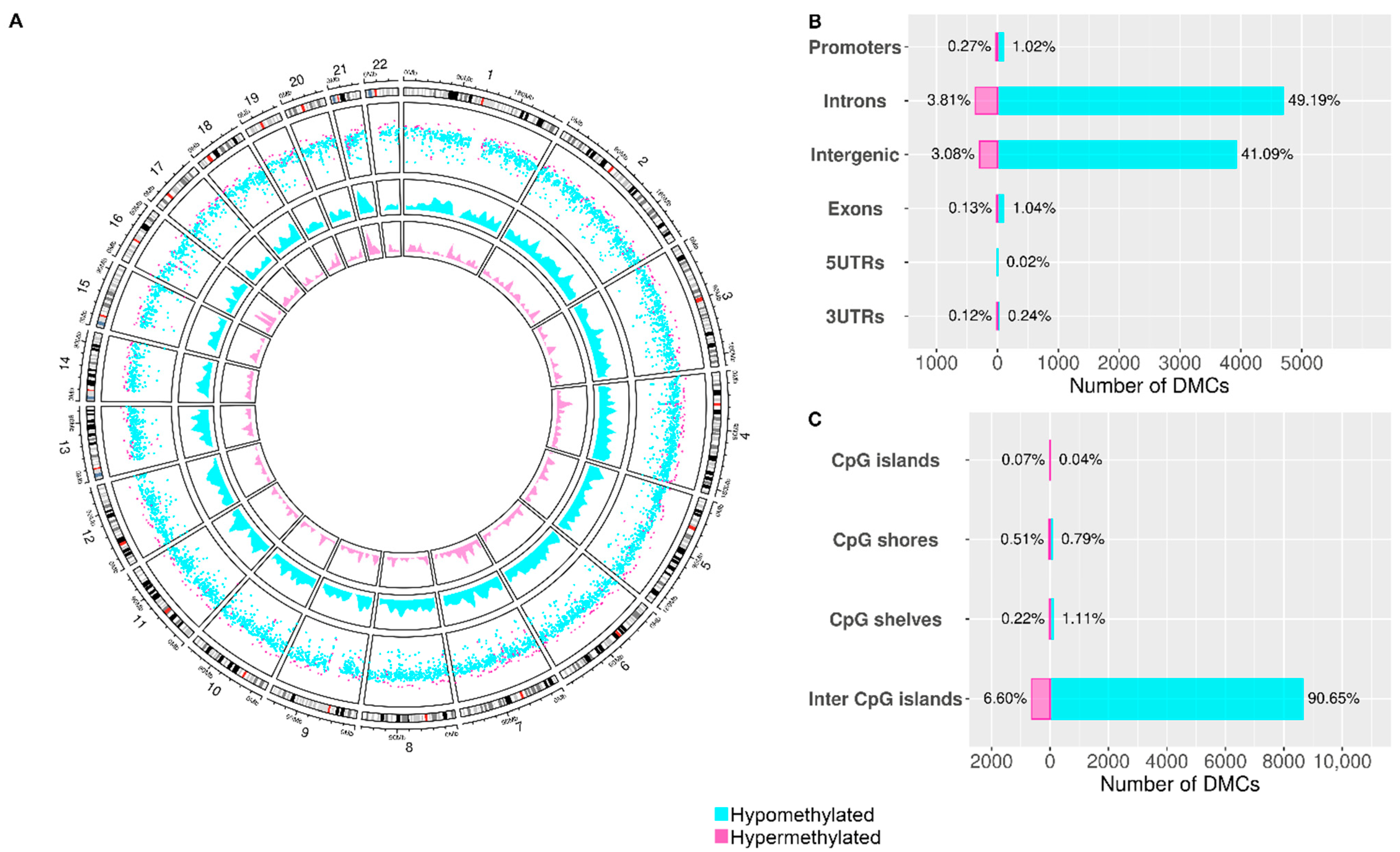
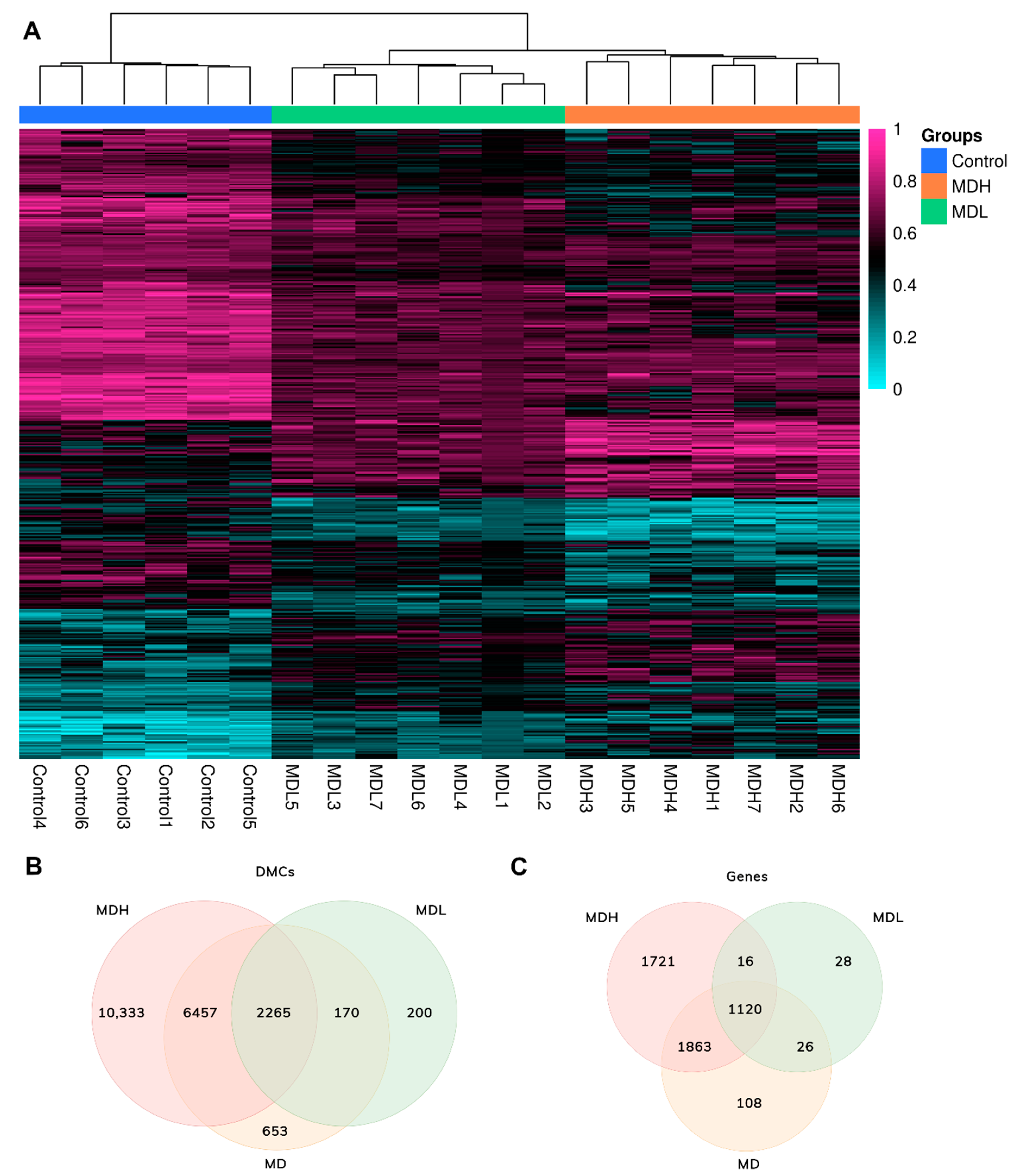
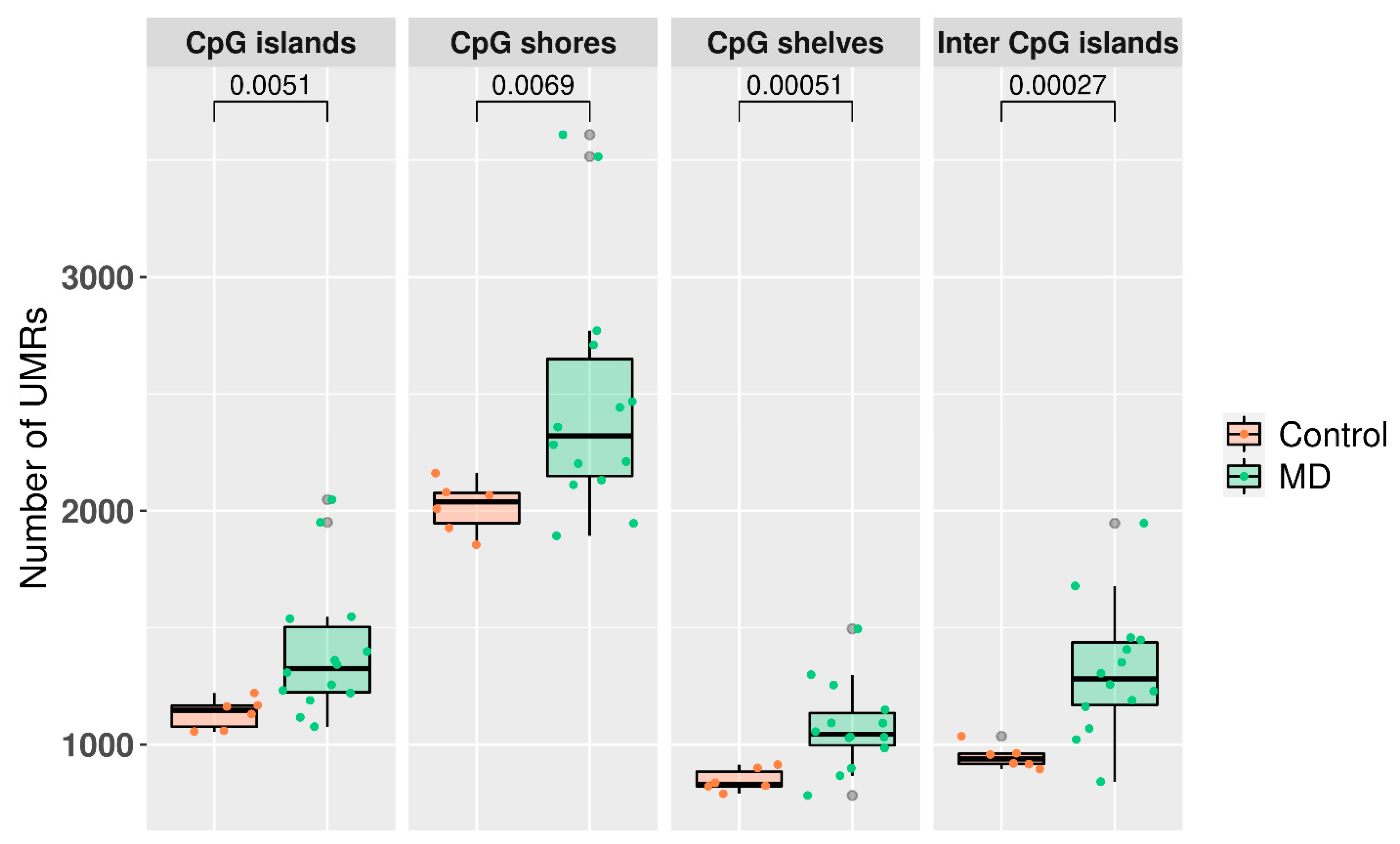
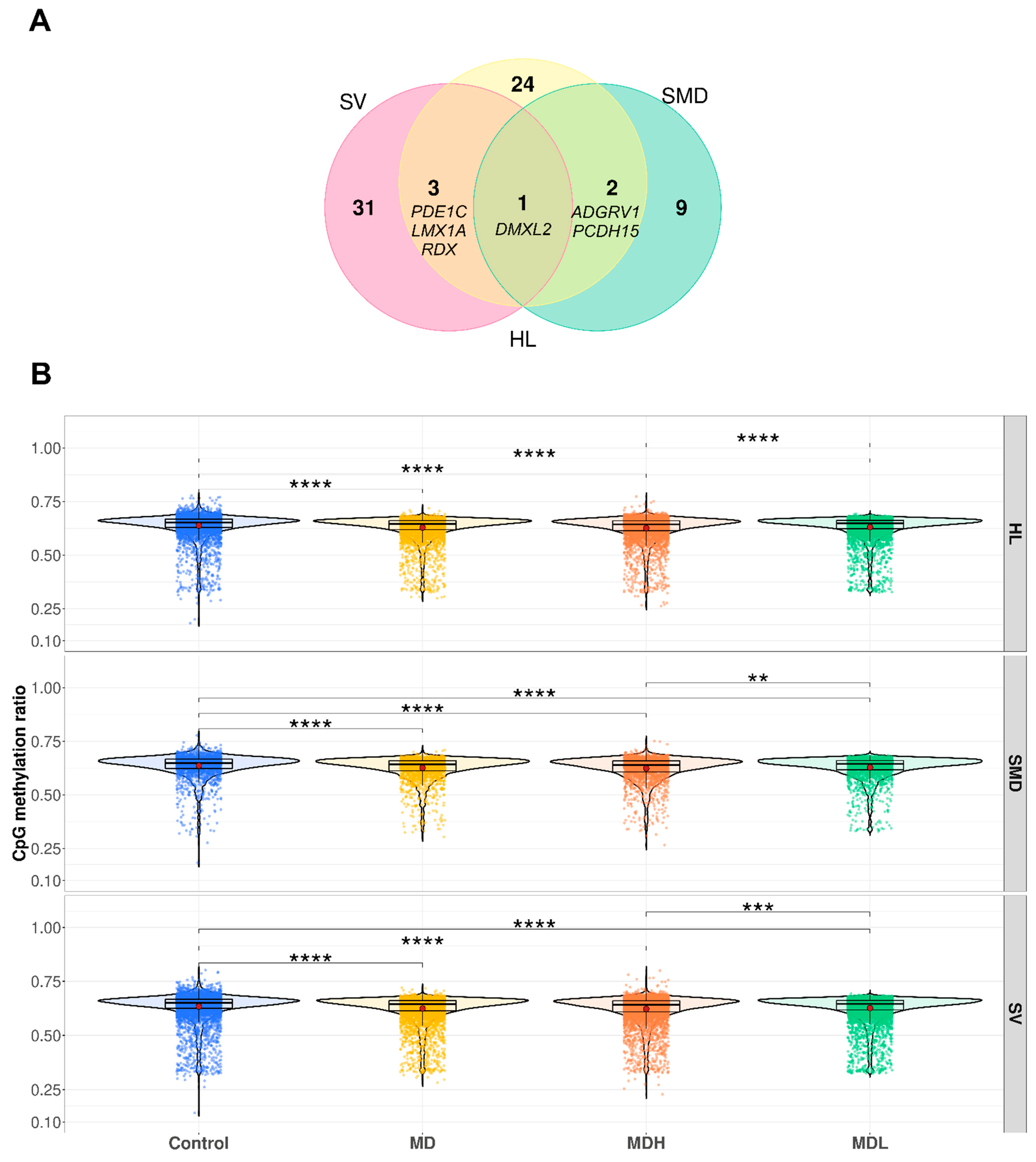
3. Results
3.1. Patient Clinical History
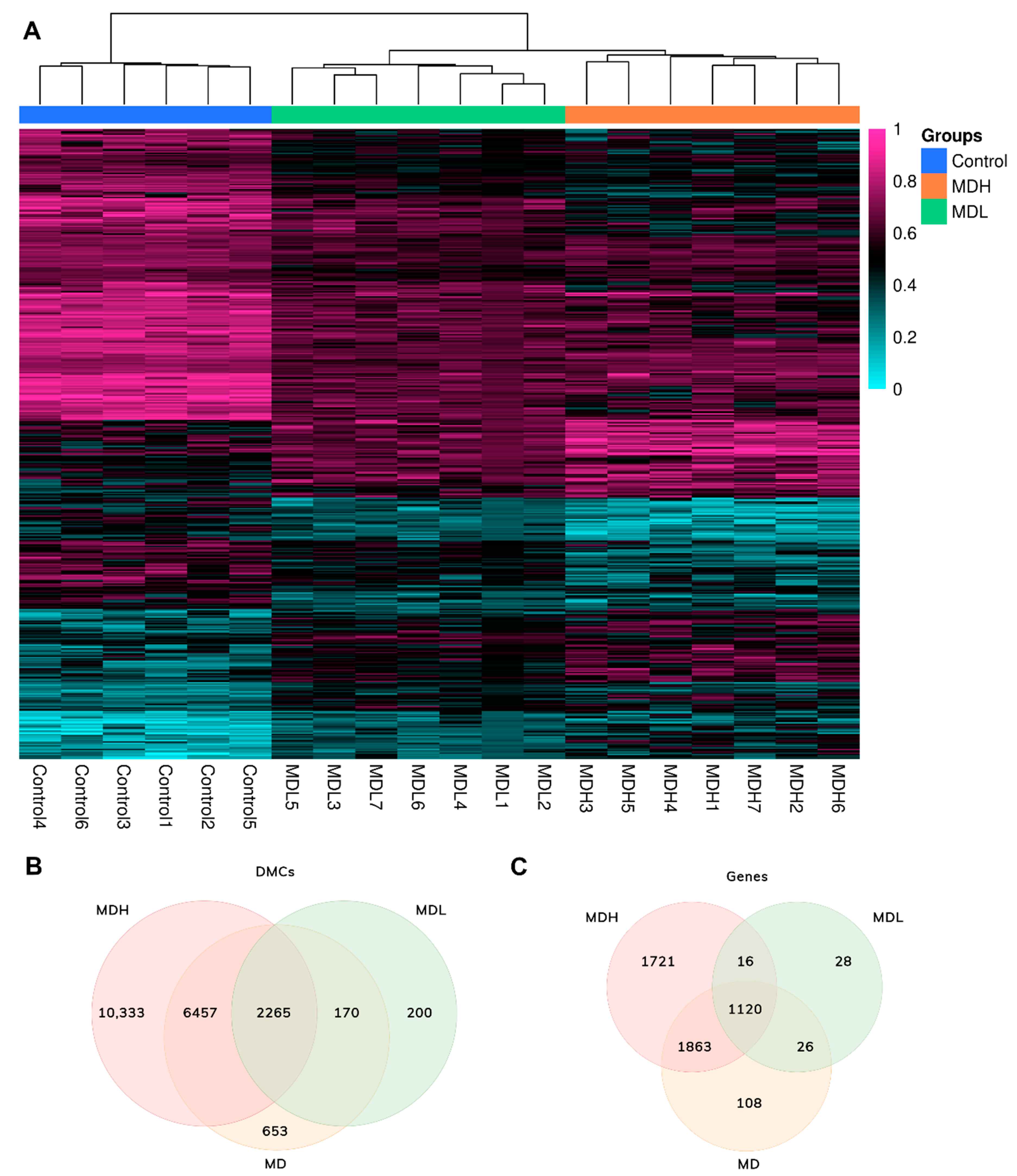
3.2. Screening DNA Methylation in Mononuclear Cells in Sporadic Meniere Disease
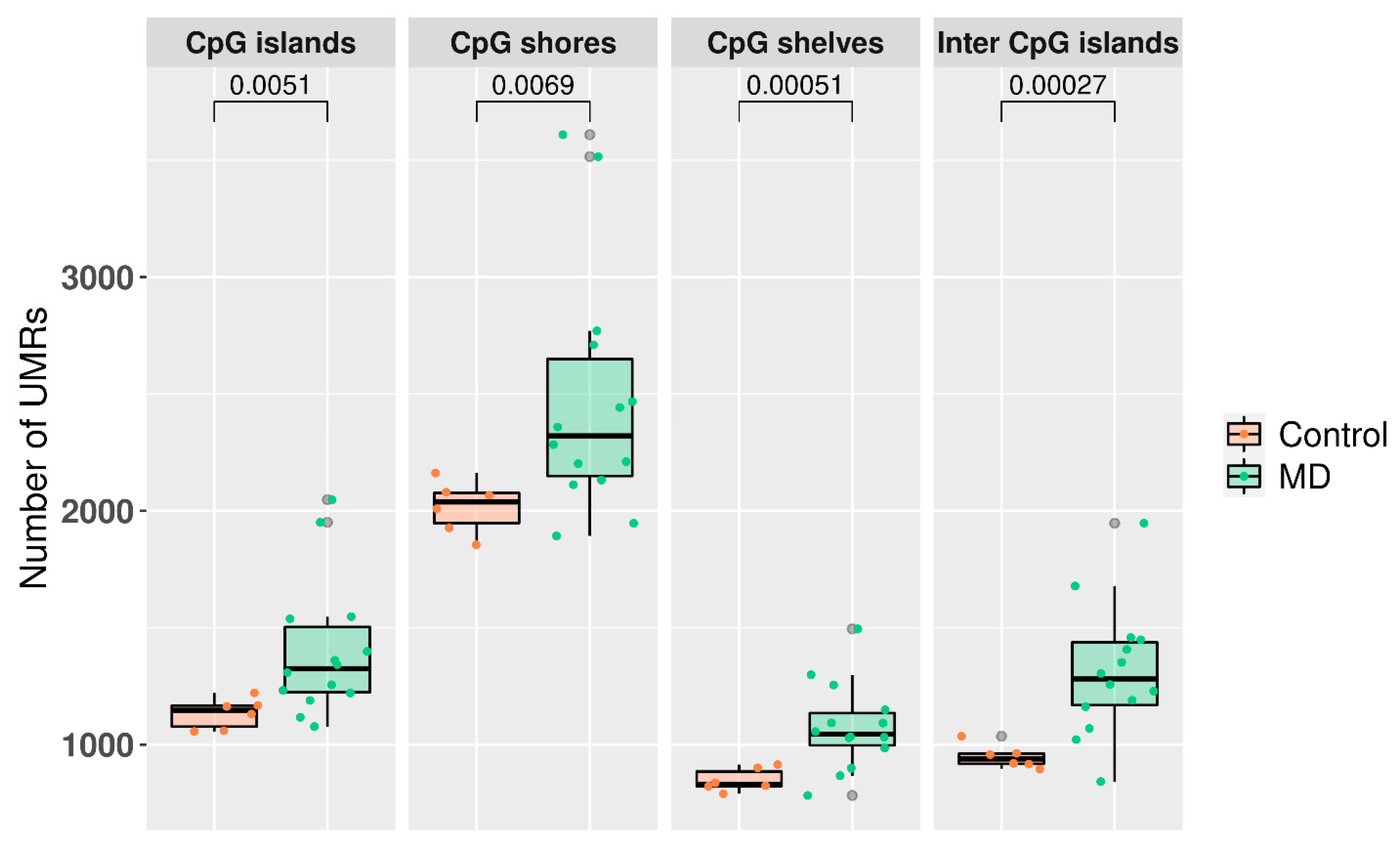
3.3. Undermethylated Regions in Meniere Disease
3.4. Mapping Differential Methylated Sites
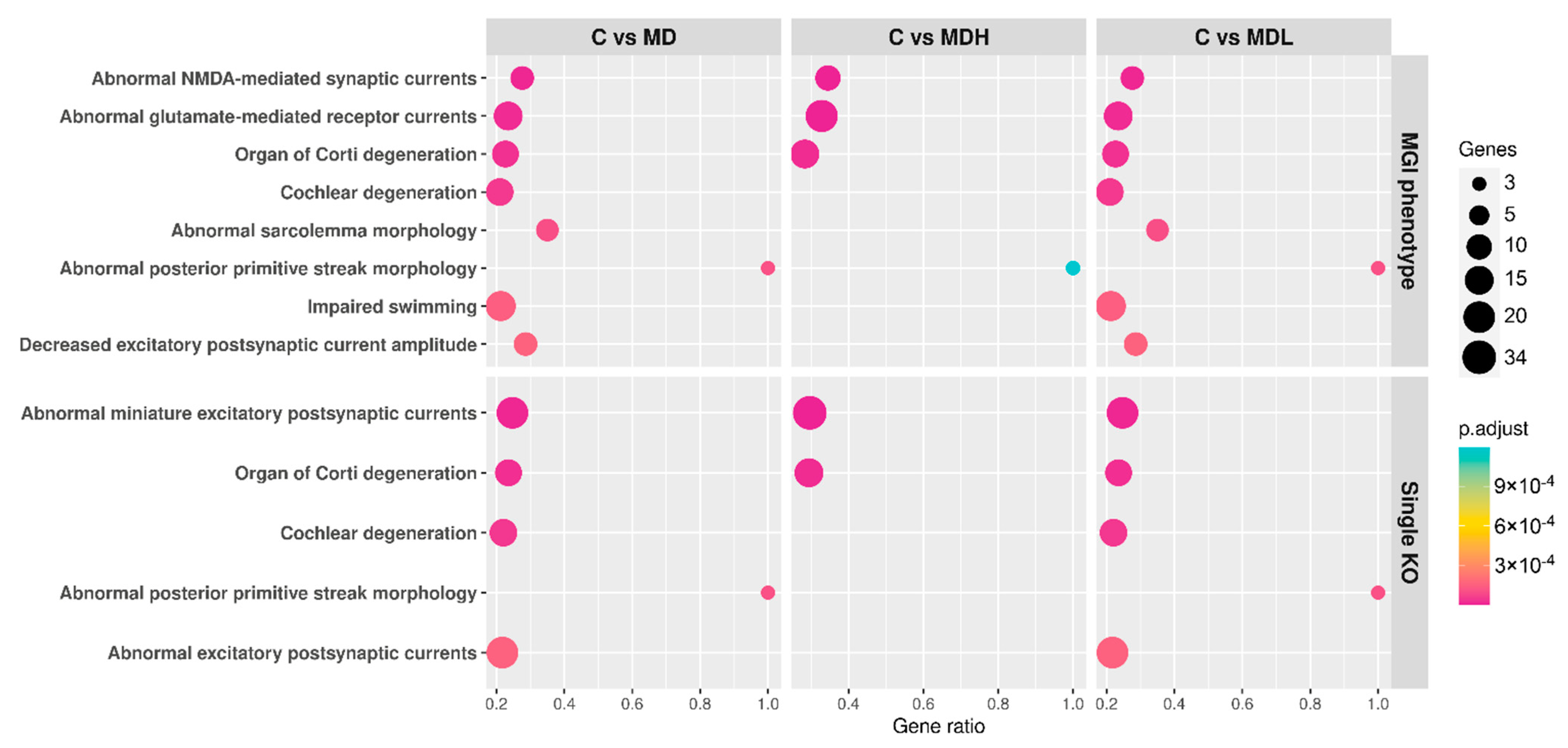
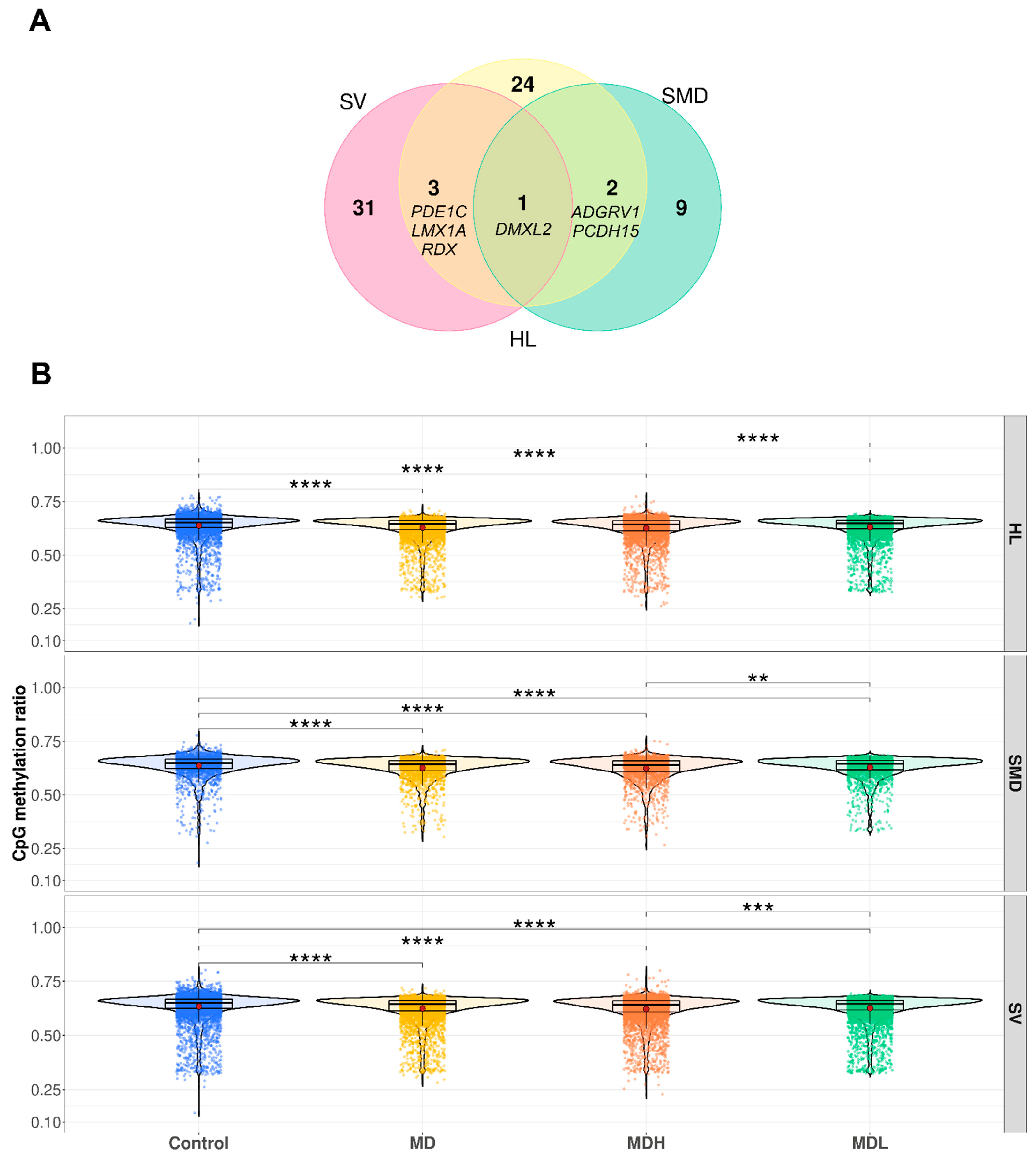
3.5. Hearing Loss Gene Sets
3.6. Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez-Escamez, J.A.; Carey, J.; Chung, W.-H.; Goebel, J.A.; Magnusson, M.; Mandalà, M.; Newman-Toker, D.E.; Strupp, M.; Suzuki, M.; Trabalzini, F.; et al. Diagnostic Criteria for Menière’s Disease. J. Vestib. Res. 2015, 25, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Escamez, J.A.; Viciana, D.; Garrido-Fernandez, P. Impact of Bilaterality and Headache on Health-Related Quality of Life in Meniere’s Disease. Ann. Otol. Rhinol. Laryngol. 2009, 118, 409–416. [Google Scholar] [CrossRef]
- Requena, T.; Espinosa-Sanchez, J.M.; Cabrera, S.; Trinidad, G.; Soto-Varela, A.; Santos-Perez, S.; Teggi, R.; Perez, P.; Batuecas-Caletrio, A.; Fraile, J.; et al. Familial Clustering and Genetic Heterogeneity in Meniere’s Disease. Clin. Genet. 2014, 85, 245–252. [Google Scholar] [CrossRef]
- Gallego-Martinez, A.; Lopez-Escamez, J.A. Genetic Architecture of Meniere’s Disease. Hear. Res. 2020, 397, 107872. [Google Scholar] [CrossRef] [PubMed]
- Roman-Naranjo, P.; Gallego-Martinez, A.; Soto-Varela, A.; Aran, I.; Moleon, M.D.C.; Espinosa-Sanchez, J.M.; Amor-Dorado, J.C.; Batuecas-Caletrio, A.; Perez-Vazquez, P.; Lopez-Escamez, J.A. Burden of Rare Variants in the OTOG Gene in Familial Meniere’s Disease. Ear Hear. 2020, 41, 1598–1605. [Google Scholar] [CrossRef]
- Flook, M.; Lopez Escamez, J.A. Meniere’s Disease: Genetics and the Immune System. Curr. Otorhinolaryngol. Rep. 2018, 6, 24–31. [Google Scholar] [CrossRef]
- Frejo, L.; Gallego-Martinez, A.; Requena, T.; Martin-Sanz, E.; Amor-Dorado, J.C.; Soto-Varela, A.; Santos-Perez, S.; Espinosa-Sanchez, J.M.; Batuecas-Caletrio, A.; Aran, I.; et al. Proinflammatory Cytokines and Response to Molds in Mononuclear Cells of Patients with Meniere Disease. Sci. Rep. 2018, 8, 5974. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Morison, I.M.; Eccles, M.R.; Stockwell, P.A. Tools and Strategies for Analysis of Genome-Wide and Gene-Specific DNA Methylation Patterns. In Oral Biology: Molecular Techniques and Applications; Methods in Molecular Biology; Seymour, G.J., Cullinan, M.P., Heng, N.C.K., Eds.; Springer: New York, NY, USA, 2017; pp. 249–277. ISBN 978-1-4939-6685-1. [Google Scholar]
- Provenzano, M.J.; Domann, F.E. A Role for Epigenetics in Hearing: Establishment and Maintenance of Auditory Specific Gene Expression Patterns. Hear. Res. 2007, 233, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzid, A.; Smeti, I.; Dhouib, L.; Roche, M.; Achour, I.; Khalfallah, A.; Gibriel, A.A.; Charfeddine, I.; Ayadi, H.; Lachuer, J.; et al. Down-Expression of P2RX2, KCNQ5, ERBB3 and SOCS3 through DNA Hypermethylation in Elderly Women with Presbycusis. Biomarkers 2018, 23, 347–356. [Google Scholar] [CrossRef]
- Hao, J.; Hua, L.; Fu, X.; Zhang, X.; Zou, Q.; Li, Y. Genome-Wide DNA Methylation Analysis of Human Peripheral Blood Reveals Susceptibility Loci of Diabetes-Related Hearing Loss. J. Hum. Genet. 2018, 63, 1241–1250. [Google Scholar] [CrossRef]
- Wolber, L.E.; Steves, C.J.; Tsai, P.-C.; Deloukas, P.; Spector, T.D.; Bell, J.T.; Williams, F.M.K. Epigenome-Wide DNA Methylation in Hearing Ability: New Mechanisms for an Old Problem. PLoS ONE 2014, 9, e105729. [Google Scholar] [CrossRef]
- Lassaletta, L.; Bello, M.J.; Del Río, L.; Alfonso, C.; Roda, J.M.; Rey, J.A.; Gavilan, J. DNA Methylation of Multiple Genes in Vestibular Schwannoma: Relationship with Clinical and Radiological Findings. Otol. Neurotol. 2006, 27, 1180–1185. [Google Scholar] [CrossRef]
- Yizhar-Barnea, O.; Valensisi, C.; Jayavelu, N.D.; Kishore, K.; Andrus, C.; Koffler-Brill, T.; Ushakov, K.; Perl, K.; Noy, Y.; Bhonker, Y.; et al. DNA Methylation Dynamics during Embryonic Development and Postnatal Maturation of the Mouse Auditory Sensory Epithelium. Sci. Rep. 2018, 8, 17348. [Google Scholar] [CrossRef] [Green Version]
- Szczepek, A.J.; Frejo, L.; Vona, B.; Trpchevska, N.; Cederroth, C.R.; Caria, H.; Lopez-Escamez, J.A. Recommendations on Collecting and Storing Samples for Genetic Studies in Hearing and Tinnitus Research. Ear Hear. 2019, 40, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.; Koeman, J.M.; Johnson, B.K.; Foy, K.K.; Beddows, I.; Zhou, W.; Chesla, D.W.; Rossell, L.L.; Siegwald, E.J.; Adams, M.; et al. Evaluation of Whole-Genome DNA Methylation Sequencing Library Preparation Protocols. Epigenet. Chromatin 2021, 14, 28. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A Reference Methylome Database and Analysis Pipeline to Facilitate Integrative and Comparative Epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [Green Version]
- Matoba, S.; Wang, H.; Jiang, L.; Lu, F.; Iwabuchi, K.A.; Wu, X.; Inoue, K.; Yang, L.; Press, W.; Lee, J.T.; et al. Loss of H3K27me3 Imprinting in Somatic Cell Nuclear Transfer Embryos Disrupts Post-Implantation Development. Cell Stem Cell 2018, 23, 343–354.e5. [Google Scholar] [CrossRef] [Green Version]
- Howe, C.G.; Zhou, M.; Wang, X.; Pittman, G.S.; Thompson, I.J.; Campbell, M.R.; Bastain, T.M.; Grubbs, B.H.; Salam, M.T.; Hoyo, C.; et al. Associations between Maternal Tobacco Smoke Exposure and the Cord Blood CD4+ DNA Methylome. Environ. Health Perspect. 2019, 127, 047009. [Google Scholar] [CrossRef] [PubMed]
- Burger, L.; Gaidatzis, D.; Schübeler, D.; Stadler, M.B. Identification of Active Regulatory Regions from DNA Methylation Data. Nucleic Acids Res. 2013, 41, e155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman-Naranjo, P. Análisis Agregado de Variantes En El Exoma de Pacientes Con Enfermedad de Meniere Familiar e Inicio Precoz; Universidad de Granada: Granada, Spain, 2020. [Google Scholar]
- Gu, S.; Olszewski, R.; Nelson, L.; Gallego-Martinez, A.; Lopez-Escamez, J.A.; Hoa, M. Identification of Potential Meniere’s Disease Targets in the Adult Stria Vascularis. Front. Neurol. 2021, 12, 61. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- García-Moreno, A.; López-Domínguez, R.; Ramirez-Mena, A.; Pascual-Montano, A.; Aparicio-Puerta, E.; Hackenberg, M.; Carmona-Saez, P. GeneCodis 4: Expanding the Modular Enrichment Analysis to Regulatory Elements. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, R.G.; Sartor, M.A. Annotatr: Genomic Regions in Context. Bioinformatics 2017, 33, 2381–2383. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 10 September 2021).
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wickham, H.; François, R.; Henry, L.; Muller, K. dplyr: A Grammar of Data Manipulation. 2020. Available online: https://cran.r-project.org/web/packages/dplyr/index.html (accessed on 10 September 2021).
- Wickham, H. Forcats: Tools for Working with Categorical Variables (Factors). 2020. Available online: https://cran.r-project.org/web/packages/forcats/index.html (accessed on 10 September 2021).
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. 2020. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 10 September 2021).
- Narui, Y.; Sotomayor, M. Tuning Inner-Ear Tip-Link Affinity Through Alternatively Spliced Variants of Protocadherin-15. Biochemistry 2018, 57, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.P.; Petit, C. Hair-Bundle Links: Genetics as the Gateway to Function. Cold Spring Harb. Perspect. Med. 2019, 9, a033142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, N.; Michel, V.; Bahloul, A.; Lefèvre, G.; Barral, J.; Yagi, H.; Chardenoux, S.; Weil, D.; Martin, P.; Hardelin, J.-P.; et al. Molecular Characterization of the Ankle-Link Complex in Cochlear Hair Cells and Its Role in the Hair Bundle Functioning. J. Neurosci. 2007, 27, 6478–6488. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Hansen, K.D.; Meissner, A.; Aryee, M.J. Coverage Recommendations for Methylation Analysis by Whole-Genome Bisulfite Sequencing. Nat. Methods 2015, 12, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA Methylome of Human Peripheral Blood Mononuclear Cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef] [PubMed]
- Zi Xu, Y.X.; Ande, S.R.; Mishra, S. Prohibitin: A New Player in Immunometabolism and in Linking Obesity and Inflammation with Cancer. Cancer Lett. 2018, 415, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Theiss, A.L.; Obertone, T.S.; Merlin, D.; Sitaraman, S.V. Interleukin-6 Transcriptionally Regulates Prohibitin Expression in Intestinal Epithelial Cells. J. Biol. Chem. 2007, 282, 12804–12812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.-L.; Chen, P.; Xun, Y.-P.; Yang, W.-K.; Yang, C.-H.; Chen, G.-Y.; Du, H.-W. Prohibitin as a Novel Autoantigen in Rheumatoid Arthritis. Cent. Eur. J. Immunol. 2015, 40, 78–82. [Google Scholar] [CrossRef]
- Yu, X.; Guan, M.; Shang, H.; Teng, Y.; Gao, Y.; Wang, B.; Ma, Z.; Cao, X.; Li, Y. The Expression of PHB2 in the Cochlea: Possible Relation to Age-Related Hearing Loss. Cell Biol. Int. 2021. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic Regulation of Inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human Cytochromes P450 in Health and Disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Erkelens, M.N.; Mebius, R.E. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 2017, 38, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Fransén, K.; Franzén, P.; Magnuson, A.; Elmabsout, A.A.; Nyhlin, N.; Wickbom, A.; Curman, B.; Törkvist, L.; D’Amato, M.; Bohr, J.; et al. Polymorphism in the Retinoic Acid Metabolizing Enzyme CYP26B1 and the Development of Crohn’s Disease. PLoS ONE 2013, 8, e72739. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Wari, N.; Singh, A.K.; Das, U.; Bopanna, S.; Gupta, V.; Nayak, B.; Velapandian, T.; Kedia, S.; Kumar, D.; et al. Retinoic Acid Is Elevated in the Mucosa of Patients with Active Ulcerative Colitis and Displays a Proinflammatory Role by Augmenting IL-17 and IFNγ Production. Inflamm. Bowel Dis. 2021, 27, 74–83. [Google Scholar] [CrossRef]
- Morita, H.; Kubo, T.; Rückert, B.; Ravindran, A.; Soyka, M.B.; Rinaldi, A.O.; Sugita, K.; Wawrzyniak, M.; Wawrzyniak, P.; Motomura, K.; et al. Induction of Human Regulatory Innate Lymphoid Cells from Group 2 Innate Lymphoid Cells by Retinoic Acid. J. Allergy Clin. Immunol. 2019, 143, 2190–2201.e9. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Keller, J.; López Ramírez, O.; González Garrido, A.; Zobeiri, O.A.; Chang, H.H.V.; Vijayakumar, S.; Ayiotis, A.; Duester, G.; Della Santina, C.C.; et al. Retinoic Acid Degradation Shapes Zonal Development of Vestibular Organs and Sensitivity to Transient Linear Accelerations. Nat. Commun. 2020, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Aass, K.R.; Kastnes, M.H.; Standal, T. Molecular Interactions and Functions of IL-32. J. Leukoc. Biol. 2021, 109, 143–159. [Google Scholar] [CrossRef]
- Meyer, B.; Chavez, R.A.; Munro, J.E.; Chiaroni-Clarke, R.C.; Akikusa, J.D.; Allen, R.C.; Craig, J.M.; Ponsonby, A.-L.; Saffery, R.; Ellis, J.A. DNA Methylation at IL32 in Juvenile Idiopathic Arthritis. Sci. Rep. 2015, 5, 11063. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Müller, U. Mechanically Gated Ion Channels in Mammalian Hair Cells. Front. Cell. Neurosci. 2018, 12, 100. [Google Scholar] [CrossRef] [Green Version]
- Bouzid, A.; Smeti, I.; Chakroun, A.; Loukil, S.; Gibriel, A.A.; Grati, M.; Ghorbel, A.; Masmoudi, S. CDH23 Methylation Status and Presbycusis Risk in Elderly Women. Front. Aging Neurosci. 2018, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Krey, J.F.; Barr-Gillespie, P.G. Molecular Composition of Vestibular Hair Bundles. Cold Spring Harb. Perspect. Med. 2019, 9, a033209. [Google Scholar] [CrossRef]
- Coate, T.M.; Scott, M.K.; Gurjar, M. Current Concepts in Cochlear Ribbon Synapse Formation. Synapse 2019, 73, e22087. [Google Scholar] [CrossRef]
- Sanchez, J.T.; Ghelani, S.; Otto-Meyer, S. From Development to Disease: Diverse Functions of NMDA-Type Glutamate Receptors in the Lower Auditory Pathway. Neuroscience 2015, 285, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Goutman, J.D.; Elgoyhen, A.B.; Gómez-Casati, M.E. Cochlear Hair Cells: The Sound-Sensing Machines. FEBS Lett. 2015, 589, 3354–3361. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Martinez, A.; Requena, T.; Roman-Naranjo, P.; May, P.; Lopez-Escamez, J.A. Enrichment of Damaging Missense Variants in Genes Related with Axonal Guidance Signalling in Sporadic Meniere’s Disease. J. Med. Genet. 2020, 57, 82–88. [Google Scholar] [CrossRef]
- Alvizi, L.; Ke, X.; Brito, L.A.; Seselgyte, R.; Moore, G.E.; Stanier, P.; Passos-Bueno, M.R. Differential Methylation Is Associated with Non-Syndromic Cleft Lip and Palate and Contributes to Penetrance Effects. Sci. Rep. 2017, 7, 2441. [Google Scholar] [CrossRef] [PubMed]
- Bend, E.G.; Aref-Eshghi, E.; Everman, D.B.; Rogers, R.C.; Cathey, S.S.; Prijoles, E.J.; Lyons, M.J.; Davis, H.; Clarkson, K.; Gripp, K.W.; et al. Gene Domain-Specific DNA Methylation Episignatures Highlight Distinct Molecular Entities of ADNP Syndrome. Clin. Epigenet. 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, L.C.; Aref-Eshghi, E.; Rooney, K.; Kerkhof, J.; Levy, M.A.; McConkey, H.; Rogers, R.C.; Phelan, K.; Sarasua, S.M.; Jain, L.; et al. DNA Methylation Epi-Signature Is Associated with Two Molecularly and Phenotypically Distinct Clinical Subtypes of Phelan-McDermid Syndrome. Clin. Epigenet. 2021, 13, 2. [Google Scholar] [CrossRef]
- Grimm, S.A.; Shimbo, T.; Takaku, M.; Thomas, J.W.; Auerbach, S.; Bennett, B.D.; Bucher, J.R.; Burkholder, A.B.; Day, F.; Du, Y.; et al. DNA Methylation in Mice Is Influenced by Genetics as Well as Sex and Life Experience. Nat. Commun. 2019, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of Cytosine Methylation on DNA Binding Specificities of Human Transcription Factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
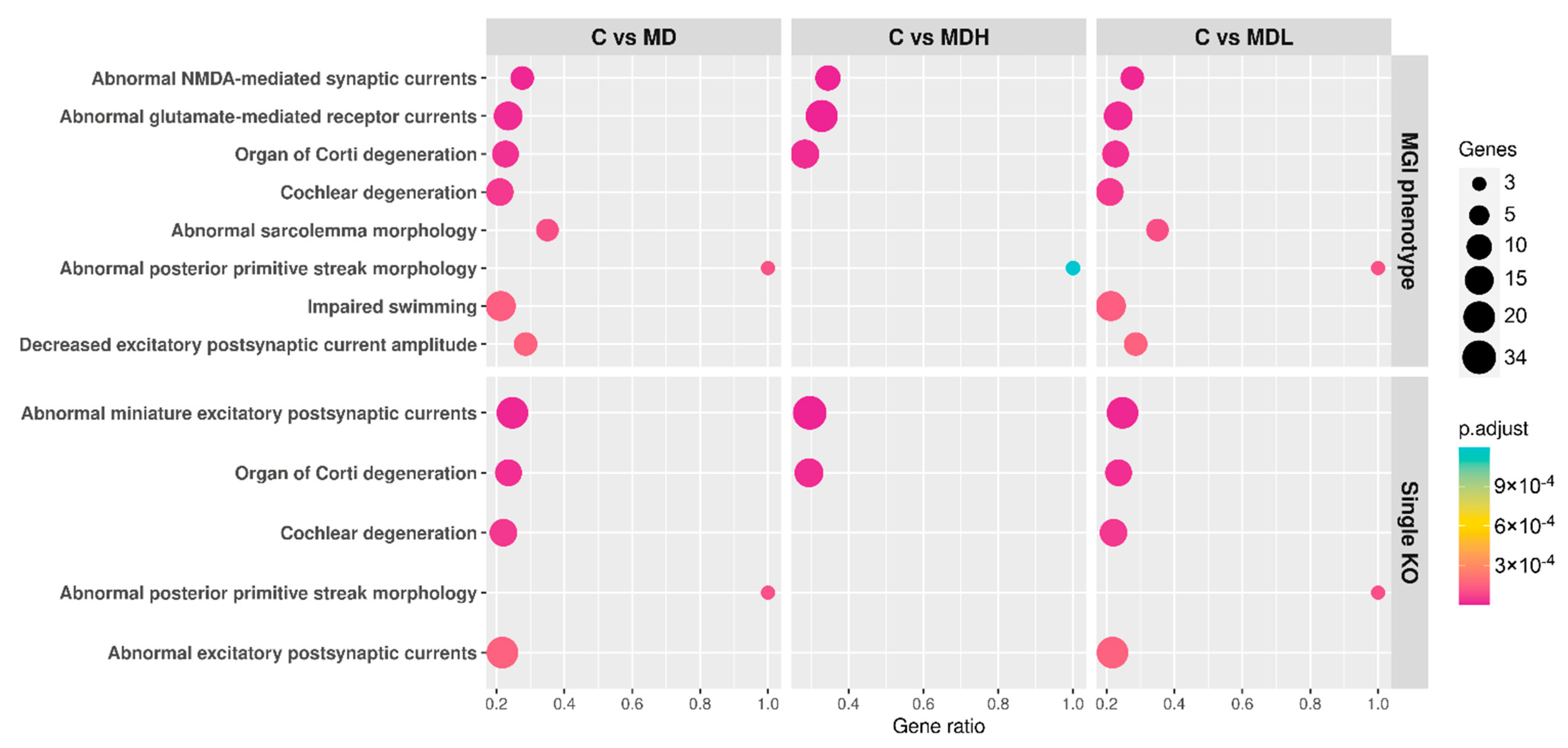{kind=link}
| Variable | MDH (n = 7) | MDL (n = 7) | Controls (n = 6) | p-Value |
|---|---|---|---|---|
| Age (mean ± SD) | 59.6 ± 11.4 | 46.0 ± 11.8 | 51.2 ± 13.8 | 0.11 |
| Age of onset (mean ± SD) | 50.2 ± 9.9 | 37.6 ± 12.4 | - | 0.07 |
| Sex (% female) | 42.9 (3) | 71.4 (5) | 33.3 (2) | 0.35 |
| Laterality (% unilateral) | 28.6 (2) | 57.1 (4) | - | 0.39 |
| Ear Family History (%) | 0 (0) | 14.3 (1) | - | 0.36 |
| Migraine (%) | 14.3 (1) | 14.3 (1) | - | 0.91 |
| History of autoimmune disease (%) | 0 (0) | 14.3 (1) | - | 0.30 |
| Clinical Subtype (%) | ||||
| 1 (no autoimmune disorder) | 83.3 (5) | 71.4 (5) | - | 0.40 |
| 2 (delayed MD) | 0 (0) | 14.3 (1) | - | |
| 3 (familial history of MD) | 0 (0) | 0 (0) | - | |
| 4 (MD and migraine) | 16.7 (1) | 0 (0) | - | |
| 5 (MD with autoimmune disorder) | 0 (0) | 14.3 (1) | - |
| Gene Set | Gene | Protein Activity or Function/Location | Position | ∆Mean | p-Value |
|---|---|---|---|---|---|
| HL | MSRB3 | Reduction of methionine sulfoxide to methionine | chr12:65397684 | −0.20 | 5.77 × 10−3 |
| PTPRQ | Plasma membrane tyrosine phosphatase receptor | chr12:80550423 | −0.18 | 6.49 × 10−3 | |
| ADGRV1 | G-protein coupled receptor, binds calcium | chr5:90721360 | −0.15 | 5.73 × 10−3 | |
| ADGRV1 | G-protein coupled receptor, binds calcium | chr5:90665789 | −0.15 | 2.64 × 10−2 | |
| MSRB3 | Reduction of methionine sulfoxide to methionine | chr12:65440113 | −0.15 | 9.32 × 10−3 | |
| CACNA1D | Voltage-dependent calcium channel | chr3:53684153 | −0.14 | 9.09 × 10−4 | |
| USH2A | Usherin—maintenance of the hair bundle ankle formation | chr1:215677582 | −0.13 | 6.81 × 10−5 | |
| LMX1A | Transcriptional activator | chr1:165321950 | −0.13 | 2.04 × 10−4 | |
| PCDH15 | Membrane protein that mediates calcium-dependent cell-cell adhesion | chr10:54924915 | −0.12 | 1.83 × 10−4 | |
| ATP2B2 | Intracellular calcium homeostasis | chr3:10545443 | −0.12 | 1.85 × 10−5 | |
| SMD | ADGRV1 | G-protein coupled receptor, binds calcium | chr5:90721360 | −0.15 | 5.73 × 10−3 |
| ADGRV1 | G-protein coupled receptor, binds calcium | chr5:90665789 | −0.15 | 2.64 × 10−2 | |
| ADAM12 | Cell-cell and cell-matrix interactions | chr10:126355102 | −0.13 | 3.83 × 10−4 | |
| PCDH15 | Membrane protein that mediates calcium-dependent cell-cell adhesion | chr10:54924915 | −0.12 | 1.83 × 10−4 | |
| TPTE | Signal transduction | chr21:10561174 | −0.12 | 1.78 × 10−2 | |
| MPDZ | AMPAR potentiation and synaptic plasticity in excitatory synapses | chr9:13106557 | −0.10 | 8.14 × 10−3 | |
| PCDH15 | Membrane protein that mediates calcium-dependent cell-cell adhesion | chr10:54280633 | −0.10 | 2.93 × 10−4 | |
| CFTR | Chloride channel | chr7:117360906 | −0.10 | 2.56 × 10−4 | |
| ATM | Cell cycle checkpoint kinase | chr11:108237615 | 0.10 | 1.29 × 10−2 | |
| PCDH15 | Membrane protein that mediates calcium-dependent cell-cell adhesion | chr10:55026981 | −0.10 | 5.23 × 10−3 | |
| SV | ROBO2 | Axon guidance and cell migration | chr3:76840338 | 0.25 | 9.46 × 10−3 |
| ROBO2 | Axon guidance and cell migration | chr3:76611689 | −0.20 | 2.71 × 10−4 | |
| NFKB1 | Pleiotropic transcription factor | chr4:102589956 | 0.19 | 4.21 × 10−2 | |
| DLC1 | Regulation of small GTP-binding proteins | chr8:13480274 | 0.16 | 1.79 × 10−4 | |
| BMPR1B | Transmembrane serine/threonine kinases receptor | chr4:95037875 | −0.16 | 3.75 × 10−2 | |
| DLC1 | Regulation of small GTP-binding proteins | chr8:13446699 | −0.16 | 8.60 × 10−5 | |
| ROBO1 | Mediates cellular responses to molecular guidance cues | chr3:79605304 | −0.16 | 3.69 × 10−3 | |
| DLC1 | Regulation of small GTP-binding proteins | chr8:13522539 | −0.15 | 1.82 × 10−2 | |
| PARD3 | Asymmetrical cell division and cell polarization processes | chr10:34349620 | −0.14 | 3.23 × 10−3 | |
| ROBO1 | Mediates cellular responses to molecular guidance cues | chr3:79389106 | −0.14 | 4.67 × 10−2 |
| DMC | Term | Category | nDMInCat | nInCat | Ratio | p, Adjust | Genes |
|---|---|---|---|---|---|---|---|
| All | Retinol metabolism | 830 | 16 | 40 | 0.40 | 2.03 × 10−2 | ALDH1A1, ALDH1A2, CYP2B6, CYP2C19, CYP2C9, CYP3A5, LRAT, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 |
| Metabolism of xenobiotics by cytochrome P450 | 980 | 15 | 43 | 0.35 | 2.97 × 10−2 | ALDH1A3, CYP2B6, CYP2C19, CYP2C9, CYP2F1, CYP3A5, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Hypomethylated | Retinol metabolism | 830 | 16 | 40 | 0.40 | 6.20 × 10−3 | ALDH1A1, ALDH1A2, CYP2B6, CYP2C19, CYP2C9, CYP3A5, LRAT, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 |
| Metabolism of xenobiotics by cytochrome P450 | 980 | 15 | 43 | 0.35 | 9.38 × 10−3 | ALDH1A3, CYP2B6, CYP2C19, CYP2C9, CYP2F1, CYP3A5, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Drug metabolism—cytochrome P450 | 982 | 14 | 45 | 0.31 | 2.71 × 10−2 | ALDH1A3, CYP2B6, CYP2C19, CYP2C9, CYP3A5, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Ascorbate and aldarate metabolism | 53 | 10 | 21 | 0.48 | 2.82 × 10−2 | ALDH2, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Steroid hormone biosynthesis | 140 | 13 | 40 | 0.33 | 3.11 × 10−2 | CYP3A5, CYP7B1, HSD17B3, HSD17B6, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Pentose and glucuronate interconversions | 40 | 10 | 22 | 0.45 | 3.11 × 10−2 | ALDH2, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 | |
| Starch and sucrose metabolism | 500 | 14 | 39 | 0.36 | 3.11 × 10−2 | ENPP1, ENPP3, HK1, MGAM, SI, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2B4 |
| Motif Name | Consensus | p-Value |
|---|---|---|
| Hoxa9 (Homeobox) | RGCAATNAAA | 1.00 × 10−4 |
| ZFX (Zf) | AGGCCTRG | 1.00 × 10−3 |
| Sox10 (HMG) | CCWTTGTYYB | 1.00 × 10−3 |
| ZNF711 (Zf) | AGGCCTAG | 1.00 × 10−3 |
| Sox6 (HMG) | CCATTGTTNY | 1.00 × 10−3 |
| Hoxd11 (Homeobox) | VGCCATAAAA | 1.00 × 10−3 |
| MYB (HTH) | GGCVGTTR | 1.00 × 10−3 |
| Foxa3 (Forkhead) | BSNTGTTTACWYWGN | 1.00 × 10−3 |
| Hoxa11 (Homeobox) | TTTTATGGCM | 1.00 × 10−2 |
| BMYB (HTH) | NHAACBGYYV | 1.00 × 10−2 |
| AMYB (HTH) | TGGCAGTTGG | 1.00 × 10−2 |
| Zic (Zf) | CCTGCTGAGH | 1.00 × 10−2 |
| NFY (CCAAT) | RGCCAATSRG | 1.00 × 10−2 |
| Foxo3 (Forkhead) | DGTAAACA | 1.00 × 10−2 |
| Sox15 (HMG) | RAACAATGGN | 1.00 × 10−2 |
| NPAS2 (bHLH) | KCCACGTGAC | 1.00 × 10−2 |
| Hoxd10 (Homeobox) | GGCMATGAAA | 1.00 × 10−2 |
| Bcl6 (Zf) | NNNCTTTCCAGGAAA | 1.00 × 10−2 |
| STAT1 (Stat) | NATTTCCNGGAAAT | 1.00 × 10−2 |
| Hoxa13 (Homeobox) | CYHATAAAAN | 1.00 × 10−2 |
| CDX4 (Homeobox) | NGYCATAAAWCH | 1.00 × 10−2 |
| TFE3 (bHLH) | GTCACGTGACYV | 1.00 × 10−2 |
| Smad4 (MAD) | VBSYGTCTGG | 1.00 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flook, M.; Escalera-Balsera, A.; Gallego-Martinez, A.; Espinosa-Sanchez, J.M.; Aran, I.; Soto-Varela, A.; Lopez-Escamez, J.A. DNA Methylation Signature in Mononuclear Cells and Proinflammatory Cytokines May Define Molecular Subtypes in Sporadic Meniere Disease. Biomedicines 2021, 9, 1530. https://doi.org/10.3390/biomedicines9111530
Flook M, Escalera-Balsera A, Gallego-Martinez A, Espinosa-Sanchez JM, Aran I, Soto-Varela A, Lopez-Escamez JA. DNA Methylation Signature in Mononuclear Cells and Proinflammatory Cytokines May Define Molecular Subtypes in Sporadic Meniere Disease. Biomedicines. 2021; 9(11):1530. https://doi.org/10.3390/biomedicines9111530
Chicago/Turabian StyleFlook, Marisa, Alba Escalera-Balsera, Alvaro Gallego-Martinez, Juan Manuel Espinosa-Sanchez, Ismael Aran, Andres Soto-Varela, and Jose Antonio Lopez-Escamez. 2021. "DNA Methylation Signature in Mononuclear Cells and Proinflammatory Cytokines May Define Molecular Subtypes in Sporadic Meniere Disease" Biomedicines 9, no. 11: 1530. https://doi.org/10.3390/biomedicines9111530
APA StyleFlook, M., Escalera-Balsera, A., Gallego-Martinez, A., Espinosa-Sanchez, J. M., Aran, I., Soto-Varela, A., & Lopez-Escamez, J. A. (2021). DNA Methylation Signature in Mononuclear Cells and Proinflammatory Cytokines May Define Molecular Subtypes in Sporadic Meniere Disease. Biomedicines, 9(11), 1530. https://doi.org/10.3390/biomedicines9111530