
Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs

 ,
,  and
and 
Abstract
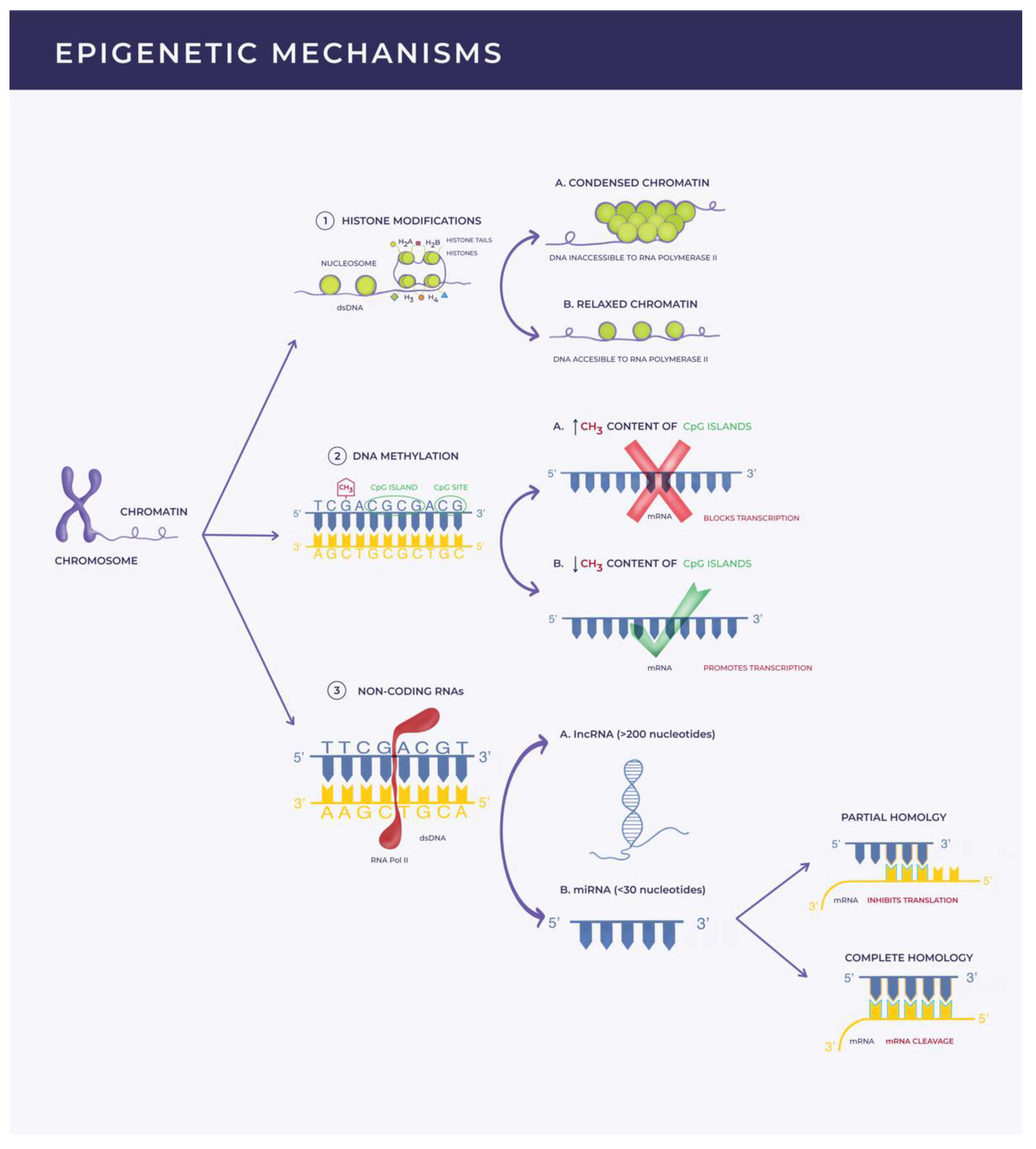
1. Introduction
2. Serum- and Tissue-Specific miRNA Signatures in SSc
3. MiRNAs: Culprits in SSc Pathogenesis
3.1. Profibrotic miRNA Transcripts
3.2. Antifibrotic miRNA Transcripts
3.3. Apoptosis and miRNAs
3.4. Microangiopathy and miRNAs
3.5. Immune Dysfunction and miRNAs
4. MiRNAs: Diagnostic and Prognostic Biomarkers
5. Role of miRNAs in SSc Interstitial Lung Disease (SSc-ILD) Pathogenesis
6. Future Directions
7. Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leroy, E.C.; Black, C.; Fleischmajer, R.; Jablonska, S.; Krieg, T.; Medsger, T.A.; Rowell, N.; Wollheim, F. Scleroderma (systemic sclerosis): Classification, subsets and pathogenesis. J. Rheumatol. 1988, 15, 202–205. [Google Scholar] [PubMed]
- Steen, V.D. Autoantibodies in Systemic Sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Prim. 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Molberg, Ø.; Midtvedt, Ø.; Garen, T.; Gran, J.T. Survival and Causes of Death in an Unselected and Complete Cohort of Norwegian Patients with Systemic Sclerosis. J. Rheumatol. 2013, 40, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Trojanowska, M.; Kuwana, M. Pathogenesis of systemic sclerosis: Recent insights of molecular and cellular mechanisms and therapeutic opportunities. J. Scleroderma Relat. Disord. 2017, 2, 137–152. [Google Scholar] [CrossRef]
- Salazar, G.; Mayes, M.D. Genetics, Epigenetics, and Genomics of Systemic Sclerosis. Rheum. Dis. Clin. North Am. 2015, 41, 345–366. [Google Scholar] [CrossRef] [PubMed]
- Aslani, S.; Sobhani, S.; Gharibdoost, F.; Jamshidi, A.; Mahmoudi, M. Epigenetics and pathogenesis of systemic sclerosis; the ins and outs. Hum. Immunol. 2018, 79, 178–187. [Google Scholar] [CrossRef]
- Tsou, P.-S.; Sawalha, A.H. Unfolding the pathogenesis of scleroderma through genomics and epigenomics. J. Autoimmun. 2017, 83, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of systemic sclerosis: Current understanding and new insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y. Systemic sclerosis. J. Dermatol. 2018, 45, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Epattanaik, D.; Ebrown, M.; Postlethwaite, B.C.; Postlethwaite, A.E. Pathogenesis of Systemic Sclerosis. Front. Immunol. 2015, 6, 272. [Google Scholar] [CrossRef]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence That Systemic Sclerosis Is a Vascular Disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Capece, D.; Zazzeroni, F.; Liakouli, V.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Pecetti, G.; et al. The Endothelial-mesenchymal Transition in Systemic Sclerosis Is Induced by Endothelin-1 and Transforming Growth Factor-β and May Be Blocked by Macitentan, a Dual Endothelin-1 Receptor Antagonist. J. Rheumatol. 2015, 42, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S.; Sundberg, C.; Abraham, D.J.; Rubin, K.; Black, C.M. Activation of microvascular pericytes in autoimmune Raynaud’s phenomenon and systemic sclerosis. Arthritis Rheum. 1999, 42, 930–941. [Google Scholar] [CrossRef]
- Dowson, C.; Simpson, N.; Duffy, L.; O’Reilly, S. Innate Immunity in Systemic Sclerosis. Curr. Rheumatol. Rep. 2017, 19, 2. [Google Scholar] [CrossRef]
- Chizzolini, C.; Boin, F. The role of the acquired immune response in systemic sclerosis. Semin. Immunopathol. 2015, 37, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Guggino, G.; Zazzeroni, F.; Alesse, E.; et al. Perivascular Cells in Diffuse Cutaneous Systemic Sclerosis Overexpress Activated ADAM12 and Are Involved in Myofibroblast Transdifferentiation and Development of Fibrosis. J. Rheumatol. 2016, 43, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; O’Reilly, S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin. Exp. Immunol. 2018, 195, 310–321. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2007, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Altorok, N.; Almeshal, N.; Wang, Y.; Kahaleh, B. Epigenetics, the holy grail in the pathogenesis of systemic sclerosis. Rheumatology 2015, 54, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Broen, J.C.A.; Radstake, T.R.D.J.; Rossato, M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; Distler, J.; O’Reilly, S. The Role of Epigenetic Modifications in Systemic Sclerosis: A Druggable Target. Trends Mol. Med. 2019, 25, 395–411. [Google Scholar] [CrossRef]
- Bergmann, C.; Distler, J.H. Epigenetic factors as drivers of fibrosis in systemic sclerosis. Epigenomics 2017, 9, 463–477. [Google Scholar] [CrossRef]
- Altorok, N.; Kahaleh, B. Epigenetics and systemic sclerosis. Semin. Immunopathol. 2015, 37, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Yokoyama, Y.; Hattori, T.; Motegi, S.-I.; Amano, H.; Hatada, I.; Ishikawa, O. Global DNA hypomethylation and hypoxia-induced expression of the ten eleven translocation (TET) family, TET1, in scleroderma fibroblasts. Exp. Dermatol. 2015, 24, 841–846. [Google Scholar] [CrossRef]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4+ and CD8+ T Cells. J. Investig. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shu, Y.; Xiao, Y.; Wang, Q.; Kanekura, T.; Li, Y.; Wang, J.; Zhao, M.; Lu, Q.; Xiao, R. Hypomethylation and overexpression of ITGAL (CD11a) in CD4+ T cells in systemic sclerosis. Clin. Epigenet. 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Luo, Y.; Yan, K.; Zhao, S.; Li, Y.; Qiu, X.; Zhou, Y.; Long, H.; Zhao, M.; Liang, Y.; et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand. J. Rheumatol. 2009, 38, 369–374. [Google Scholar] [CrossRef]
- Dees, C.; Schlottmann, I.; Funke, R.; Distler, A.; Palumbo-Zerr, K.; Zerr, P.; Lin, N.-Y.; Beyer, C.; Distler, O.; Schett, G.; et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann. Rheum. Dis. 2013, 73, 1232–1239. [Google Scholar] [CrossRef]
- Krämer, M.; Dees, C.; Huang, J.; Schlottmann, I.; Palumbo-Zerr, K.; Zerr, P.; Gelse, K.; Beyer, C.; Distler, A.; Marquez, V.E.; et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Ann. Rheum. Dis. 2012, 72, 614–620. [Google Scholar] [CrossRef]
- Deng, Q.; Luo, Y.; Chang, C.; Wu, H.; Ding, Y.; Xiao, R. The Emerging Epigenetic Role of CD8+T Cells in Autoimmune Diseases: A Systematic Review. Front. Immunol. 2019, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, Y.; Qu, S.; Luo, H.; Zhou, Y.; Wang, Y.; Zhao, H.; You, Y.; Xiao, X.; Zuo, X. MicroRNA Expression Abnormalities in Limited Cutaneous Scleroderma and Diffuse Cutaneous Scleroderma. J. Clin. Immunol. 2012, 32, 514–522. [Google Scholar] [CrossRef]
- Henry, T.W.; Mendoza, F.A.; Jimenez, S.A. Role of microRNA in the pathogenesis of systemic sclerosis tissue fibrosis and vasculopathy. Autoimmun. Rev. 2019, 18, 102396. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, B.S.; Rosa, I.; Romano, E.; Wang, Y.; Guiducci, S.; Zhang, G.; Manetti, M.; Matucci-Cerinic, M. The contribution of epigenetics to the pathogenesis and gender dimorphism of systemic sclerosis: A comprehensive overview. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720–20918456. [Google Scholar] [CrossRef]
- Ramahi, A.; Altorok, N.; Kahaleh, B. Epigenetics and systemic sclerosis: An answer to disease onset and evolution? Eur. J. Rheumatol. 2020, 7, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Perera, B.; Faulk, C.; Svoboda, L.K.; Goodrich, J.M.; Dolinoy, D.C. The role of environmental exposures and the epigenome in health and disease. Environ. Mol. Mutagen. 2020, 61, 176–192. [Google Scholar] [CrossRef]
- Toraño, E.G.; García, M.G.; Fernández-Morera, J.L.; Niño-García, P.; Fernandez, A. The Impact of External Factors on the Epigenome:In Uteroand over Lifetime. BioMed Res. Int. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Fernandez, A.; Munoz, A.; Fraga, M. Epigenetics and environment: A complex relationship. J. Appl. Physiol. 2010, 109, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.-S.; Campbell, P.; Amin, M.A.; Coit, P.; Miller, S.; Fox, D.A.; Khanna, D.; Sawalha, A.H. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc. Natl. Acad. Sci. USA 2019, 116, 3695–3702. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, Y.; Shi, Y.; Luo, Y.; Li, Y.; Zhao, M.; Lu, Q.; Xiao, R. Overexpression of JMJD3 may contribute to demethylation of H3K27me3 in CD4 + T cells from patients with systemic sclerosis. Clin. Immunol. 2015, 161, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Chouri, E.; Servaas, N.H.; Bekker, C.P.; Affandi, A.J.; Cossu, M.; Hillen, M.; Angiolilli, C.; Mertens, J.S.; Hoogen, L.L.V.D.; Silva-Cardoso, S.; et al. Serum microRNA screening and functional studies reveal miR-483-5p as a potential driver of fibrosis in systemic sclerosis. J. Autoimmun. 2018, 89, 162–170. [Google Scholar] [CrossRef]
- Rusek, M.; Michalska-Jakubus, M.; Kowal, M.; Bełtowski, J.; Krasowska, D. A novel miRNA-4484 is up-regulated on microarray and associated with increased MMP-21 expression in serum of systemic sclerosis patients. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. In Systemic Sclerosis, a Unique Long Non Coding RNA Regulates Genes and Pathways Involved in the Three Main Features of the Disease (Vasculopathy, Fibrosis and Autoimmunity) and in Carcinogenesis. J. Clin. Med. 2019, 8, 320. [Google Scholar] [CrossRef]
- Messemaker, T.C.; Chadli, L.; Cai, G.; Goelela, V.S.; Boonstra, M.; Dorjée, A.L.; Andersen, S.N.; Mikkers, H.M.; Hof, P.V.T.; Mei, H.; et al. Antisense Long Non-Coding RNAs Are Deregulated in Skin Tissue of Patients with Systemic Sclerosis. J. Investig. Dermatol. 2018, 138, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenet. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and function. Thromb. Haemost. 2012, 107, 605–610. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Steen, S.O.; Iversen, L.V.; Carlsen, A.L.; Burton, M.; Nielsen, C.T.; Jacobsen, S.; Heegaard, N.H. The Circulating Cell-free microRNA Profile in Systemic Sclerosis Is Distinct from Both Healthy Controls and Systemic Lupus Erythematosus. J. Rheumatol. 2015, 42, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, H.; Zhao, M.; Lu, Q. Meta-analysis of differentially expressed microRNAs in systemic sclerosis. Int. J. Rheum. Dis. 2020, 23, 1297–1304. [Google Scholar] [CrossRef]
- Tsou, P.-S. Epigenetic Control of Scleroderma: Current Knowledge and Future Perspectives. Curr. Rheumatol. Rep. 2019, 21, 69. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, R.; Fan, X.; Gu, T.; Zhao, Z.; Chang, D.; Wang, W.; Wang, C. MicroRNA array analysis of microRNAs related to systemic scleroderma. Rheumatol. Int. 2012, 32, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wuttge, D.M.; Carlsen, A.L.; Teku, G.; Steen, S.O.; Wildt, M.; Vihinen, M.; Hesselstrand, R.; Heegaard, N.H.H. Specific autoantibody profiles and disease subgroups correlate with circulating micro-RNA in systemic sclerosis. Rheumatology 2015, 54, 2100–2107. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, P.; Piera-Velazquez, S.; Jimenez, S.A. Exosomes isolated from serum of systemic sclerosis patients display alterations in their content of profibrotic and antifibrotic microRNA and induce a profibrotic phenotype in cultured normal dermal fibroblasts. Clin. Exp. Rheumatol. 2017, 35, 21–30. [Google Scholar]
- Zhu, H.; Luo, H.; Li, Y.; Zhou, Y.; Jiang, Y.; Chai, J.; Xiao, X.; You, Y.; Zuo, X. MicroRNA-21 in Scleroderma Fibrosis and its Function in TGF-β- Regulated Fibrosis-Related Genes Expression. J. Clin. Immunol. 2013, 33, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Jafarinejad-Farsangi, S.; Gharibdoost, F.; Farazmand, A.; Kavosi, H.; Jamshidi, A.; Karimizadeh, E.; Noorbakhsh, F.; Mahmoudi, M. MicroRNA-21 and microRNA-29a modulate the expression of collagen in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity 2019, 52, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.-D.; Riedel, L.; Fischer, B.; Schmidt, V.; Hendig, D.; Distler, J.; Kuhn, J.; Knabbe, C.; Faust, I. microRNA-145 mediates xylosyltransferase-I induction in myofibroblasts via suppression of transcription factor KLF4. Biochem. Biophys. Res. Commun. 2020, 523, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Sing, T.; Jinnin, M.; Yamane, K.; Honda, N.; Makino, K.; Kajihara, I.; Makino, T.; Sakai, K.; Masuguchi, S.; Fukushima, S.; et al. microRNA-92a expression in the sera and dermal fibroblasts increases in patients with scleroderma. Rheumatology 2012, 51, 1550–1556. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, H.; Luo, H.; Gao, S.; Dai, X.; Li, Y.; Zuo, X. MicroRNA-202-3p regulates scleroderma fibrosis by targeting matrix metalloproteinase 1. Biomed. Pharmacother. 2017, 87, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, W.; Jinnin, M.; Tomizawa, Y.; Nakamura, K.; Kudo, H.; Inoue, K.; Makino, K.; Honda, N.; Kajihara, I.; Fukushima, S.; et al. Dysregulated interleukin-23 signalling contributes to the increased collagen production in scleroderma fibroblasts via balancing microRNA expression. Rheumatology 2017, 56, 145–155. [Google Scholar] [CrossRef]
- Yan, Q.; Chen, J.; Li, W.; Bao, C.; Fu, Q. Targeting miR-155 to Treat Experimental Scleroderma. Sci. Rep. 2016, 6, 20314. [Google Scholar] [CrossRef]
- Christmann, R.B.; Wooten, A.; Sampaio-Barros, P.; Borges, C.L.; Carvalho, C.R.R.; Kairalla, R.A.; Feghali-Bostwick, C.; Ziemek, J.; Mei, Y.; Goummih, S.; et al. miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res. 2016, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Sassi-Gaha, S.; Hope, J.L.; Feghali-Bostwick, C.A.; Katsikis, P.D. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res. 2017, 19, 1–8. [Google Scholar] [CrossRef]
- Henderson, J.; Wilkinson, S.; Przyborski, S.; Stratton, R.; O’Reilly, S. microRNA27a-3p mediates reduction of the Wnt antagonist sFRP-1 in systemic sclerosis. Epigenetics 2021, 16, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; Pryzborski, S.; Stratton, R.; O’Reilly, S. Wnt antagonist DKK-1 levels in systemic sclerosis are lower in skin but not in blood and are regulated by microRNA33a-3p. Exp. Dermatol. 2021, 30, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Derk, C.T. Transforming Growth Factor-β (TGF-β) and its Role in the Pathogenesis of Systemic Sclerosis: A Novel Target for Therapy? Recent Patents Inflamm. Allergy Drug Discov. 2007, 1, 142–145. [Google Scholar] [CrossRef] [PubMed]
- La, J.; Reed, E.; Chan, L.; Smolyaninova, L.V.; Akomova, O.A.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Downregulation of TGF-β Receptor-2 Expression and Signaling through Inhibition of Na/K-ATPase. PLoS ONE 2016, 11, e0168363. [Google Scholar] [CrossRef]
- Meng, J.; Li, L.; Zhao, Y.; Zhou, Z.; Zhang, M.; Li, D.; Zhang, C.-Y.; Zen, K.; Liu, Z. MicroRNA-196a/b Mitigate Renal Fibrosis by Targeting TGF-β Receptor 2. J. Am. Soc. Nephrol. 2016, 27, 3006–3021. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Chen, B.; Fan, X.; Li, G.; Dong, P.; Zheng, J. Epigenetically-Regulated MicroRNA-9-5p Suppresses the Activation of Hepatic Stellate Cells via TGFBR1 and TGFBR2. Cell. Physiol. Biochem. 2017, 43, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Liu, Q.; Li, N.; Tu, W.; Luo, R.; Mei, X.; Ma, Y.; Xu, W.; Chu, H.; Jiang, S.; et al. MiR-3606-3p inhibits systemic sclerosis through targeting TGF-β type II receptor. Cell Cycle 2018, 17, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Jinnin, M.; Hirano, A.; Yamane, K.; Eto, M.; Kusano, T.; Honda, N.; Kajihara, I.; Makino, T.; Sakai, K.; et al. The Downregulation of microRNA let-7a Contributes to the Excessive Expression of Type I Collagen in Systemic and Localized Scleroderma. J. Immunol. 2013, 190, 3905–3915. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Stanczyk, J.; Jüngel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.W.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheumatol. 2010, 62, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; O’Reilly, S.; Suwara, M.; Bogunia-Kubik, K.; Van Laar, J.M. MiR-29a Reduces TIMP-1 Production by Dermal Fibroblasts via Targeting TGF-β Activated Kinase 1 Binding Protein 1, Implications for Systemic Sclerosis. PLoS ONE 2014, 9, e115596. [Google Scholar] [CrossRef]
- Honda, N.; Jinnin, M.; Kira-Etoh, T.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Okamoto, Y.; Hasegawa, M.; et al. miR-150 Down-Regulation Contributes to the Constitutive Type I Collagen Overexpression in Scleroderma Dermal Fibroblasts via the Induction of Integrin β3. Am. J. Pathol. 2013, 182, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Suto, A.; Ikeda, K.; Sanayama, Y.; Nakagomi, D.; Iwamoto, T.; Suzuki, K.; Kambe, N.; Matsue, H.; Matsumura, R.; et al. Alteration of circulating miRNAs in SSc: miR-30b regulates the expression of PDGF receptor β. Rheumatol. 2013, 52, 1963–1972. [Google Scholar] [CrossRef]
- O’Reilly, S.; Ciechomska, M.; Fullard, N.; Przyborski, S.; Van Laar, J.M. IL-13 mediates collagen deposition via STAT6 and microRNA-135b: A role for epigenetics. Sci. Rep. 2016, 6, 25066. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Jinnin, M.; Aoi, J.; Hirano, A.; Kajihara, I.; Makino, T.; Sakai, K.; Fukushima, S.; Inoue, Y.; Ihn, H. Discoidin Domain Receptor 2–microRNA 196a—Mediated Negative Feedback against Excess Type I Collagen Expression Is Impaired in Scleroderma Dermal Fibroblasts. J. Investig. Dermatol. 2013, 133, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Edbauer, D. Good guy or bad guy: The opposing roles of microRNA 125b in cancer. Cell Commun. Signal. 2014, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, A.; Pachera, E.; Maurer, B.; Jüngel, A.; Distler, J.H.W.; Kania, G.; Distler, O. Regulation of Fibroblast Apoptosis and Proliferation by Micro RNA -125b in Systemic Sclerosis. Arthritis Rheumatol. 2019, 71, 2068–2080. [Google Scholar] [CrossRef]
- Yao, Q.; Xing, Y.; Wang, Z.; Liang, J.; Lin, Q.; Huang, M.; Chen, Y.; Lin, B.; Xu, X.; Chen, W. MiR-16-5p suppresses myofibroblast activation in systemic sclerosis by inhibiting NOTCH signaling. Aging 2021, 13, 2640–2654. [Google Scholar] [CrossRef] [PubMed]
- Kissin, E.; Korn, J.H. Apoptosis and myofibroblasts in the pathogenesis of systemic sclerosis. Curr. Rheumatol. Rep. 2002, 4, 129–135. [Google Scholar] [CrossRef]
- Jafarinejad-Farsangi, S.; Farazmand, A.; Mahmoudi, M.; Gharibdoost, F.; Karimizadeh, E.; Noorbakhsh, F.; Faridani, H.; Jamshidi, A.R. MicroRNA-29a induces apoptosis via increasing the Bax:Bcl-2 ratio in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity 2015, 48, 369–378. [Google Scholar] [CrossRef]
- Jafarinejad-Farsangi, S.; Farazmand, A.; Gharibdoost, F.; Karimizadeh, E.; Noorbakhsh, F.; Faridani, H.; Mahmoudi, M.; Jamshidi, A.R. Inhibition of MicroRNA-21 induces apoptosis in dermal fibroblasts of patients with systemic sclerosis. Int. J. Dermatol. 2016, 55, 1259–1267. [Google Scholar] [CrossRef]
- Iwamoto, N.; Vettori, S.; Maurer, B.; Brock, M.; Pachera, E.; Jüngel, A.; Calcagni, M.; Gay, R.E.; Whitfield, M.L.; Distler, J.H.; et al. Downregulation of miR-193b in systemic sclerosis regulates the proliferative vasculopathy by urokinase-type plasminogen activator expression. Ann. Rheum. Dis. 2016, 75, 303–310. [Google Scholar] [CrossRef]
- Liakouli, V.; Cipriani, P.; Di Benedetto, P.; Panzera, N.; Ruscitti, P.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Esteves, F.; Mavria, G.; et al. Epidermal Growth Factor Like-domain 7 and miR-126 are abnormally expressed in diffuse Systemic Sclerosis fibroblasts. Sci. Rep. 2019, 9, 4589. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, G.; Francois, A.; Philippe, L.; Gong, Y.-Z.; Bahram, S.; Cetin, S.; Pfeffer, S.; Gottenberg, J.-E.; Wachsmann, D.; Georgel, P.; et al. MiR-30a-3p Negatively Regulates BAFF Synthesis in Systemic Sclerosis and Rheumatoid Arthritis Fibroblasts. PLoS ONE 2014, 9, e111266. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; Wojtas, B.; Swacha, M.; Olesinska, M.; Benes, V.; Maslinski, W. Global miRNA and mRNA expression profiles identify miRNA-26a-2-3p-dependent repression of IFN signature in systemic sclerosis human monocytes. Eur. J. Immunol. 2020, 50, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Jinnin, M.; Kajihara, I.; Honda, N.; Sakai, K.; Masuguchi, S.; Fukushima, S.; Inoue, Y.; Ihn, H. Circulating miR-142-3p levels in patients with systemic sclerosis. Clin. Exp. Dermatol. 2012, 37, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Jinnn, M.; Kimura, Y.; Wang, Z.; Onoue, Y.; Hanatani, S.; Araki, S.; Ihn, H.; Ogawa, H. Expression of Let-7 family microRNAs in skin correlates negatively with severity of pulmonary hypertension in patients with systemic scleroderma. Int. J. Cardiol. Heart Vasc. 2015, 8, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Akiyama, Y.; Yuasa, Y. Multiple-to-Multiple Relationships between MicroRNAs and Target Genes in Gastric Cancer. PLoS ONE 2013, 8, e62589. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Pelosi, A.; Fiore, P.F.; Patuzzo, G.; Tinazzi, E.; Lunardi, C.; Puccetti, A. Gene Profiling in Patients with Systemic Sclerosis Reveals the Presence of Oncogenic Gene Signatures. Front. Immunol. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Y.; Xie, Y.; Wei, S.; Liu, Y. Identification of Potential ceRNA Network and Patterns of Immune Cell Infiltration in Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Cell Dev. Biol. 2021, 9, 622021. [Google Scholar] [CrossRef]
- Mullenbrock, S.; Liu, F.; Szak, S.; Hronowski, X.; Gao, B.; Juhasz, P.; Sun, C.; Liu, M.; McLaughlin, H.; Xiao, Q.; et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes 2018, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Tao, J.-H.; Zuo, T.; Li, X.-M.; Wang, G.-S.; Fang, X.; Xu, X.-L. The correlation between miR-200c and the severity of interstitial lung disease associated with different connective tissue diseases. Scand. J. Rheumatol. 2017, 46, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Shu, E.; Niwa, H.; Seishima, M.; Ozaki, K.-I. MicroRNA-30c attenuates fibrosis progression and vascular dysfunction in systemic sclerosis model mice. Mol. Biol. Rep. 2021, 48, 3431–3437. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Hu, C.; Zhou, J.; Xu, D.; Hou, Y.; Wu, C.; Zhao, J.; Li, M.; Zeng, X.; et al. MicroRNA-320a: An important regulator in the fibrotic process in interstitial lung disease of systemic sclerosis. Arthritis Res. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; Van Rooij, E. Micro RNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S. Epigenetic modulation as a therapy in systemic sclerosis. Rheumatology 2018, 58, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Colletti, M.; Galardi, A.; De Santis, M.; Guidelli, G.M.; Di Giannatale, A.; Di Luigi, L.; Antinozzi, C. Exosomes in Systemic Sclerosis: Messengers Between Immune, Vascular and Fibrotic Components? Int. J. Mol. Sci. 2019, 20, 4337. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| miRNA | Expression | Tissue Specimen(s) | Target Gene(s) | Reference(s) |
|---|---|---|---|---|
| miR-21 | Upregulated | Fibroblasts Skin Bleomycin-treated mice skin samples | SMAD7 | Zhu et al. [34,57] Jafarinejad-Farsangi et al. [58] |
| miR-145 | Upregulated | Fibroblasts TGF-β1-stimulated fibroblasts | KLF4 | Ly et al. [59] |
| miR-92a | Upregulated | Fibroblasts Serum TGF-β-stimulated fibroblasts | MMP1 | Sing et al. [60] |
| miR-202-3p | Upregulated | Fibroblasts Skin | MMP1 | Zhou et al. [61] |
| miR-4458 | Upregulated | Fibroblasts | Unknown | Nakayama et al. [62] |
| miR-155 | Upregulated | Fibroblasts Skin Serum | CSNK1A1 SHIP1 | Yan et al. [63] Christmann et al. [64] Artlett et al. [65] |
| miR-27a-3p | Upregulated | Fibroblasts Skin Serum | sFRP-1 | Henderson et al. [66] |
| miR-33a-3p | Upregulated | Fibroblasts | DKK-1 | Henderson et al. [67] |
| miRNA | Expression | Tissue Specimen(s) | Target Gene(s) | Reference(s) |
|---|---|---|---|---|
| miR-145 | Downregulated | Fibroblasts Skin | SMAD3 | Zhu et al. [34] |
| miR-29b | Downregulated | Fibroblasts Skin | COL1A1 | Zhu et al. [34] |
| miR-let-7a | Downregulated | Fibroblasts Skin Serum | Unknown | Makino et al. [74] |
| miR-29a | Downregulated | Fibroblasts Skin Bleomycin-treated mice skin samples | COL1A1 COL3A1 TAB1 | Maurer et al. [75] Jafarinejad-Farsangi et al. [58] Ciechomska et al. [76] |
| miR-3606-3p | Downregulated | Fibroblasts Skin | TGFBR2 | Shi et al. [73] |
| miR-18a | Downregulated | Fibroblasts | Unknown | Nakayama et al. [62] |
| miR-150 | Downregulated | Fibroblasts | ITGB3 | Honda et al. [77] |
| miR-30b | Downregulated | Skin Serum Experimental mouse model | Unknown | Tanaka et al. [78] |
| miR-135b | Downregulated | Fibroblasts Serum Monocytes | STAT6 | O’Reilly et al. [79] |
| miR-16-5p | Downregulated | Fibroblasts Serum | NOTCH2 | Yao et al. [84] |
| miRNAs | Expression | Tissue Sample(s) | Regulatory Effect | Consequence | Reference |
|---|---|---|---|---|---|
| miR-29a | Downregulated | Fibroblasts TGF-β-stimulated fibroblasts | Increased Bax:Bcl2 ratio | Proapoptotic | Jafarinejad-Farsangi et al. [86] |
| miR-21 | Upregulated | Fibroblasts | Decreased Bax:Bcl2 ratio | Antiapoptotic | Jafarinejad-Farsangi et al. [87] |
| miRNA | Tissue Samples | Regulatory Effect | Reference |
|---|---|---|---|
| miR-193b | Fibroblasts Skin HPASMCs cultures | uPA expression | Iwamoto et al. [88] |
| miR-126 | Fibroblasts Skin HUVECs | EGFL7 expression | Liakouli et al. [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabo, I.; Muntean, L.; Crisan, T.; Rednic, V.; Sirbe, C.; Rednic, S. Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs. Biomedicines 2021, 9, 1471. https://doi.org/10.3390/biomedicines9101471
Szabo I, Muntean L, Crisan T, Rednic V, Sirbe C, Rednic S. Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs. Biomedicines. 2021; 9(10):1471. https://doi.org/10.3390/biomedicines9101471
Chicago/Turabian StyleSzabo, Iulia, Laura Muntean, Tania Crisan, Voicu Rednic, Claudia Sirbe, and Simona Rednic. 2021. "Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs" Biomedicines 9, no. 10: 1471. https://doi.org/10.3390/biomedicines9101471
APA StyleSzabo, I., Muntean, L., Crisan, T., Rednic, V., Sirbe, C., & Rednic, S. (2021). Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs. Biomedicines, 9(10), 1471. https://doi.org/10.3390/biomedicines9101471