
Bis-thiobarbiturates as Promising Xanthine Oxidase Inhibitors: Synthesis and Biological Evaluation

 , ,
, ,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Instrumentation
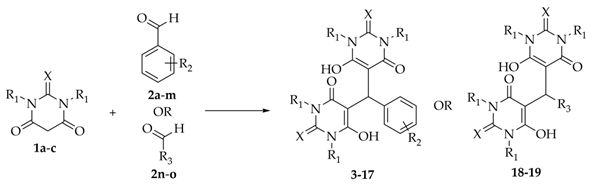
2.2. Synthesis of Bis-thiobarbiturates 3–19
- 5,5′-(Phenylmethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (3)
- 5,5′-(p-Tolymethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (4)
- 4-(Bis(1,3-diethyl-6-hydroxy-4-oxo-2-thioxo-1,2,3,4-tetrtahydropyrimidin-5-yl)methyl)benzonitrile (5)
- 5,5′-((4-Nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropirimidin-4(1H)-one) (6)
- 5,5′-((4-Nitrophenyl)methylene)bis(6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (7)
- 5,5′-((4-Nitrophenyl)methylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) (8)
- N-(4-(Bis(1,3-diethyl-6-hydroxy-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)phenyl)acetamide (9)
- 5,5′-((4-Methoxyphenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (10)
- 5,5′-((4-Bromophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (11)
- 5,5′-((3-Hydroxyphenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (12)
- 5,5′-((2-Nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (13)
- 5,5′-((4-(Dimethylamino)-2-nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (14)
- 5,5′-((2,4-Dinitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (15)
- 5,5′-((5-Hydroxy-2-nitrophenyl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (16)
- 5,5′-((6-Nitrobenzo[d][1,3]dioxol-5-yl)methylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (17)
- 5,5′-(2-Methyl-3-phenylprop-2-ene-1,1-diyl)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (18)
- 5,5′-(Pyridin-3-ylmethylene)bis(1,3-diethyl-6-hydroxy-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (19)
2.3. In Vitro Studies
2.3.1. Solutions Preparation
2.3.2. XO Inhibitory Assay
2.3.3. Antioxidant Assay
2.3.4. Cytotoxicity Assay
2.3.5. Statistics
2.4. In Silico Studies
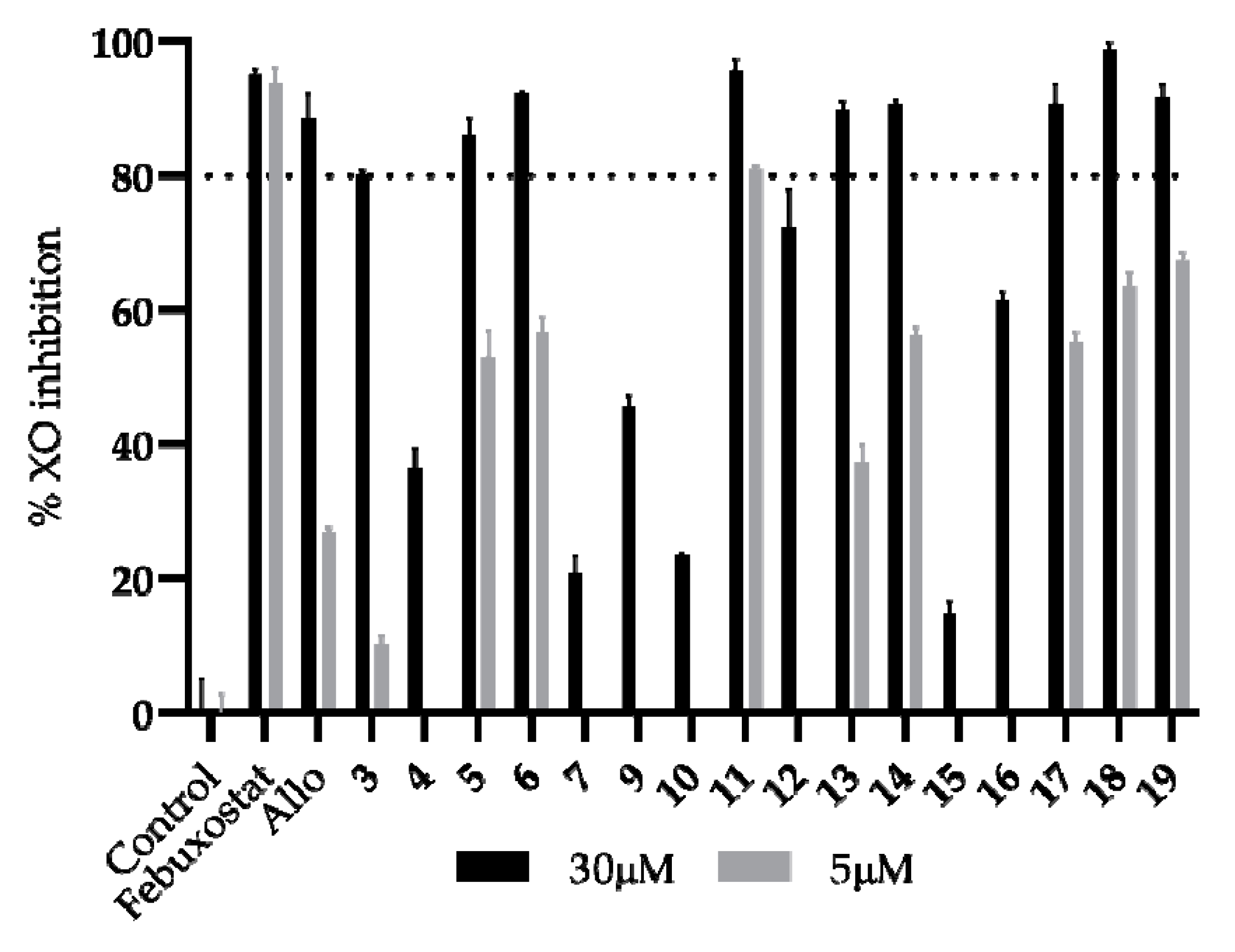
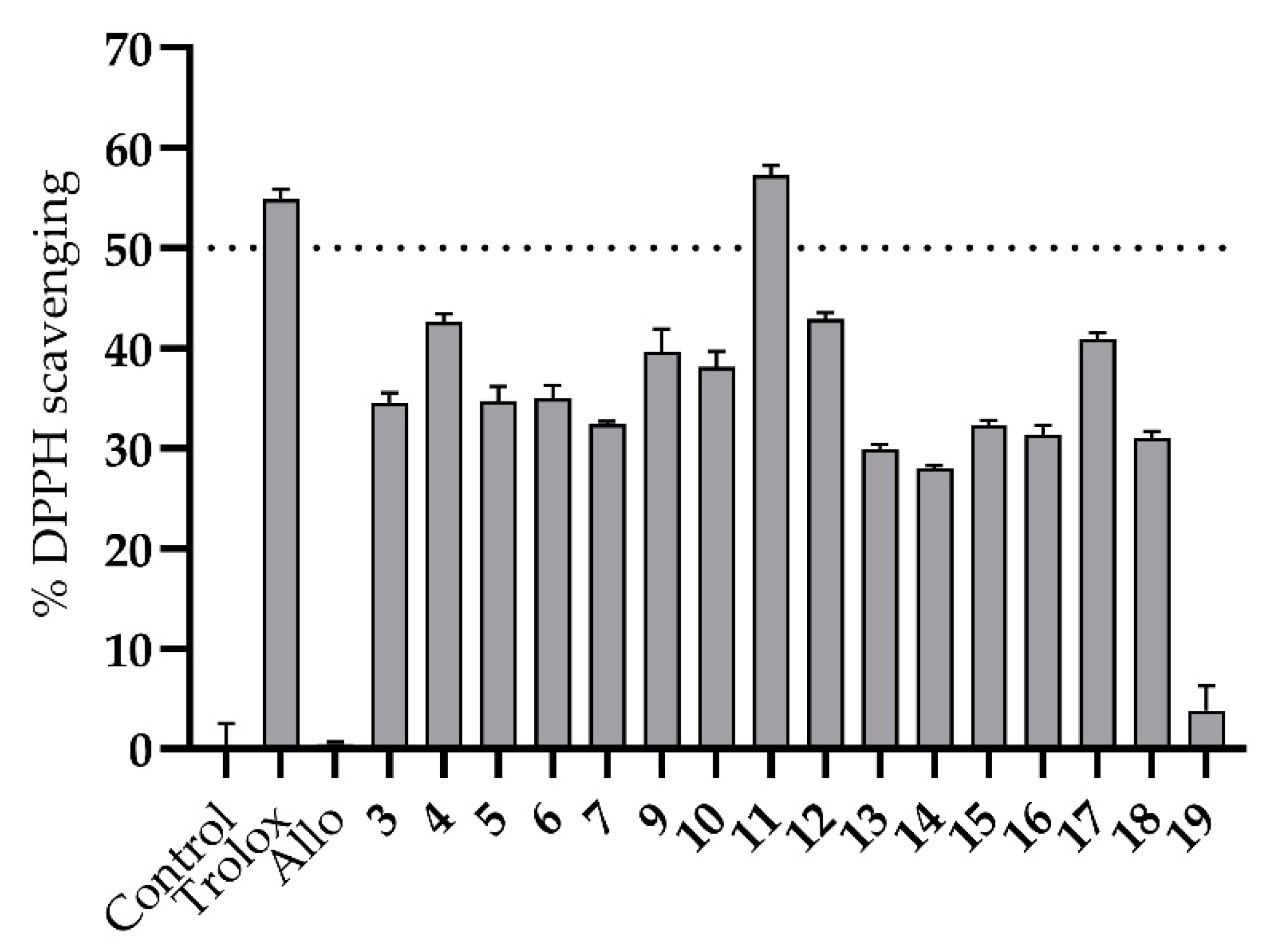
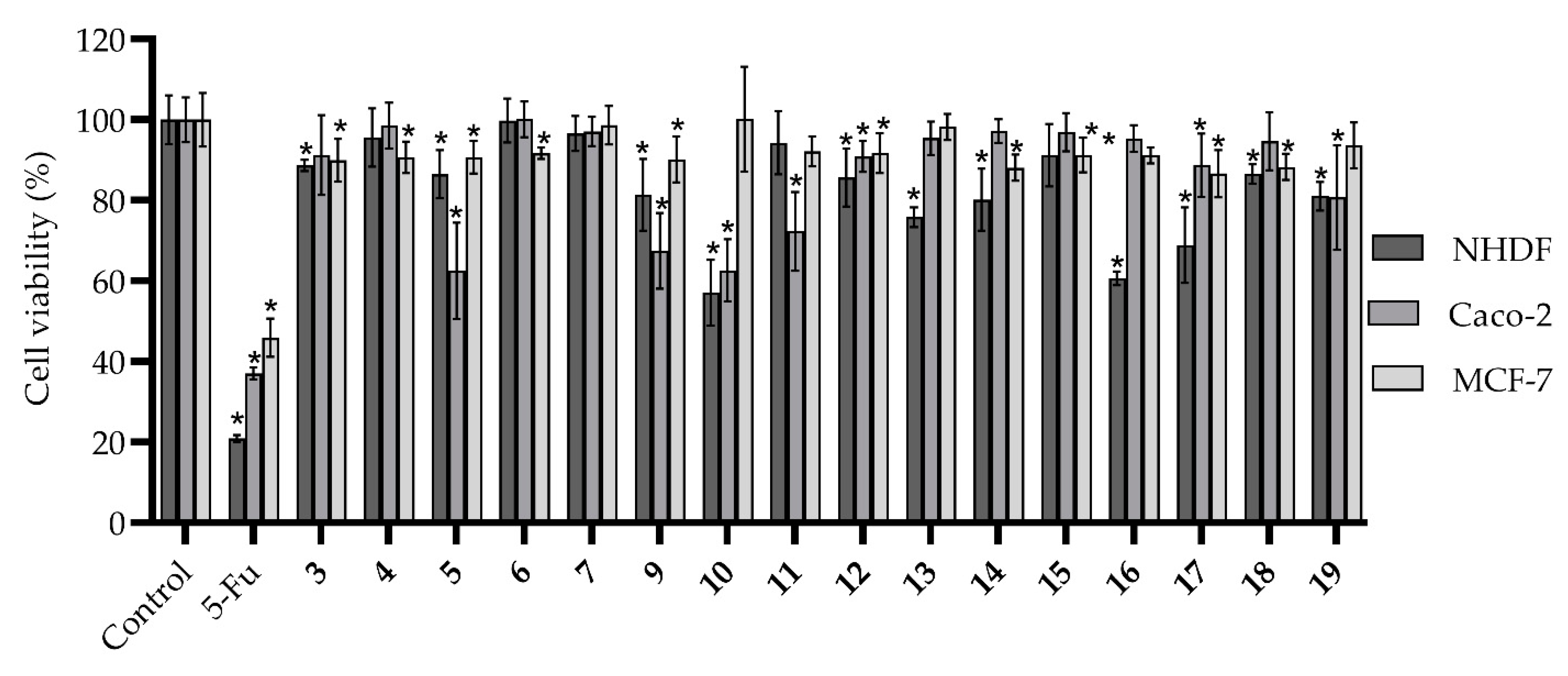
3. Results and Discussion
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Šmelcerović, A.; Tomović, K.; Šmelcerović, Ž.; Petronijević, Ž.; Kocić, G.; Tomašič, T.; Jakopin, Ž.; Anderluh, M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017, 135, 491–516. [Google Scholar] [CrossRef]
- Cos, P.; Ying, L.; Calomme, M.; Hu, J.P.; Cimanga, K.; Van Poel, B.; Pieters, L.; Vlietinck, A.J.; Berghe, D.V. Structure−activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Martillo, M.A.; Nazzal, L.; Crittenden, D.B. The crystallization of monosodium urate. Curr. Rheumat. Rep. 2013, 16, 400. [Google Scholar] [CrossRef]
- Benn, C.L.; Dua, P.; Gurrell, R.; Loudon, P.; Pike, A.; Storer, R.I.; Vangjeli, C. Physiology of hyperuricemia and urate-lowering treatments. Front. Med. 2018, 5, 160. [Google Scholar] [CrossRef] [Green Version]
- Serrano, J.L.; Figueiredo, J.; Almeida, P.; Silvestre, S. From xanthine oxidase inhibition to in vivo hypouricemic effect: An integrated overview of in vitro and in vivo studies with focus on natural molecules and analogues. Evid-Based. Complement. Alternat. Med. 2020, 2020, 9531725. [Google Scholar] [CrossRef]
- Singh, J.V.; Bedi, P.M.S.; Singh, H.; Sharma, S. Xanthine oxidase inhibitors: Patent landscape and clinical development (2015–2020). Expert Opin. Ther. Pat. 2020, 30, 769–780. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Nusrat, S.; Uzma, A.; Gul, Z.; Shagufta, P.; Irum, J.; Aisha, A. A comprehensive review: Bio-potential of barbituric acid and its analogues. Curr. Org. Chem. 2020, 24, 129–161. [Google Scholar]
- Prasher, P.; Sharma, M.; Singh, S.P.; Rawat, D.S. Barbiturate derivatives for managing multifaceted oncogenic pathways: A mini review. Drug Develop. Res. 2021, 82, 364–373. [Google Scholar] [CrossRef]
- Shabeer, M.; Barbosa, L.C.A.; Karak, M.; Coelho, A.C.S.; Takahashi, J.A. Thiobarbiturates as potential antifungal agents to control human infections caused by Candida and Cryptococcus species. Med. Chem. Res. 2018, 27, 1043–1049. [Google Scholar] [CrossRef]
- Figueiredo, J.; Serrano, J.L.; Cavalheiro, E.; Keurulainen, L.; Yli-Kauhaluoma, J.; Moreira, V.M.; Ferreira, S.; Domingues, F.C.; Silvestre, S.; Almeida, P. Trisubstituted barbiturates and thiobarbiturates: Synthesis and biological evaluation as xanthine oxidase inhibitors, antioxidants, antibacterial and anti-proliferative agents. Eur. J. Med. Chem. 2018, 143, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, J.; Serrano, J.L.; Soares, M.; Ferreira, S.; Domingues, F.C.; Almeida, P.; Silvestre, S. 5-Hydrazinylethylidenepyrimidines effective against multidrug-resistant Acinetobacter baumannii: Synthesis and in vitro biological evaluation of antibacterial, radical scavenging and cytotoxic activities. Eur. J. Pharm. Sci. 2019, 137, 104964. [Google Scholar] [CrossRef]
- Sharma, A.; Noki, S.; Zamisa, S.J.; Hazzah, H.A.; Almarhoon, Z.M.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploiting the thiobarbituric acid scaffold for antibacterial activity. ChemMedChem 2018, 13, 1923–1930. [Google Scholar] [CrossRef]
- Ortega, J.A.; Riccardi, L.; Minniti, E.; Borgogno, M.; Arencibia, J.M.; Greco, M.L.; Minarini, A.; Sissi, C.; De Vivo, M. Pharmacophore hybridization to discover novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 2018, 61, 1375–1379. [Google Scholar] [CrossRef]
- Bhatt, P.; Kumar, M.; Jha, A. Design, synthesis and anticancer evaluation of oxa/thiadiazolylhydrazones of barbituric and thiobarbituric acid: A collective in vitro and in silico approach. ChemistrySelect 2018, 3, 7060–7065. [Google Scholar] [CrossRef]
- Cagno, V.; Tintori, C.; Civra, A.; Cavalli, R.; Tiberi, M.; Botta, L.; Brai, A.; Poli, G.; Tapparel, C.; Lembo, D.; et al. Novel broad spectrum virucidal molecules against enveloped viruses. PLoS ONE 2018, 13, e0208333. [Google Scholar]
- Rauf, A.; Shahzad, S.; Bajda, M.; Yar, M.; Ahmed, F.; Hussain, N.; Akhtar, M.N.; Khan, A.; Jończyk, J. Design and synthesis of new barbituric- and thiobarbituric acid derivatives as potent urease inhibitors: Structure activity relationship and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Marecki, J.C.; Aarattuthodiyil, S.; Byrd, A.K.; Penthala, N.R.; Crooks, P.A.; Raney, K.D. N-Naphthoyl-substituted indole thio-barbituric acid analogs inhibit the helicase activity of the hepatitis C virus NS3. Bioorg. Med. Chem. Lett. 2019, 29, 430–434. [Google Scholar] [CrossRef]
- Adamson, J.; Coe, J.B.; Grassam, L.H.; Jeffery, C.J.; Coles, J.S.; Hursthouse, B.M. Reactions of 1,3-diethyl-2-thiobarbituric acid with aldehydes: Formation of arylbis(1,3-diethyl-2-thiobarbitur-5-yl)methanes † and crystallographic evidence for ground state polarisation in 1,3-diethyl-5-[4-(dimethylamino)benzylidene]-2-thiobarbituric acid. J. Chem. Soc. Perkin Trans. I 1999, 1, 2483–2488. [Google Scholar] [CrossRef]
- Rahim, F.; Ali, M.; Ullah, S.; Rashid, U.; Ullah, H.; Taha, M.; Javed, M.T.; Rehman, W.; Khan, A.A.; Abid, O.U.R.; et al. Development of bis-thiobarbiturates as successful urease inhibitors and their molecular modeling studies. Chin. Chem. Lett. 2016, 27, 693–697. [Google Scholar] [CrossRef]
- Sachar, A.; Gupta, P.; Gupta, S.; Sharma, R.L. A novel approach towards the synthesis of tricyclic systems based on pyridine, pyran, thiopyran, azepine, oxepin, thiepin, and pyrimidine rings under different solvent conditions. Can. J. Chem. 2010, 88, 478–484. [Google Scholar] [CrossRef]
- Matias, M.; Campos, G.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. Potential antitumoral 3,4-dihydropyrimidin-2-(1H)-ones: Synthesis, in vitro biological evaluation and QSAR studies. RSC Adv. 2016, 6, 84943–84958. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Jursic, B.S.; Neumann, D.M. Preparation of 5,5′-pyrilidene and 5,5′-quinolidene bis-barbituric acid derivatives. J. Het. Chem. 2003, 40, 465–474. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019, 21, 101070. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase in cancer: More than a differentiation marker. Cancer Med. 2016, 5, 546–557. [Google Scholar] [CrossRef]
- Joshi, G.; Sharma, M.; Kalra, S.; Gavande, N.S.; Singh, S.; Kumar, R. Design, synthesis, biological evaluation of 3,5-diaryl-4,5-dihydro-1H-pyrazole carbaldehydes as non-purine xanthine oxidase inhibitors: Tracing the anticancer mechanism via xanthine oxidase inhibition. Bioorg. Chem. 2021, 107, 104620. [Google Scholar] [CrossRef]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In silico methods for drug design and discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Gessner, A.; König, J.; Fromm, M.F. Clinical aspects of transporter-mediated drug–drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Bis-(thio) Barbiturate | Starting Material | X | R1 | R2 | R3 | Yield (%) |
| 3 | 1a + 2a | S | Et | H | - | 89 |
| 4 | 1a + 2b | S | Et | 4-CH3 | - | 79 |
| 5 | 1a + 2c | S | Et | 4-CN | - | 78 |
| 6 | 1a + 2d | S | Et | 4-NO2 | - | 78 |
| 7 | 1b + 2d | S | H | 4-NO2 | - | 72 |
| 8 | 1c + 2d | O | H | 4-NO2 | - | 82 |
| 9 | 1a + 2e | S | Et | 4-NHCOCH3 | - | 89 |
| 10 | 1a + 2f | S | Et | 4-OCH3 | - | 68 |
| 11 | 1a + 2g | S | Et | 4-Br | - | 71 |
| 12 | 1a + 2h | S | Et | 3-OH | - | 73 |
| 13 | 1a + 2i | S | Et | 2-NO2 | - | 78 |
| 14 | 1a + 2j | S | Et | 2-NO2, 4-N(CH3)2 | - | 85 |
| 15 | 1a + 2k | S | Et | 2,4-NO2 | - | 75 |
| 16 | 1a + 2l | S | Et | 2-NO2, 5-OH | - | 92 |
| 17 | 1a + 2m | S | Et | 2-NO2, 4,5-OCH2O | - | 77 |
| 18 | 1a + 2n | S | Et | - | 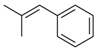 | 50 |
| 19 | 1a + 2o | S | Et | - | 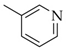 | 54 |
| XO Inhibition | DPPH Scavenging | Cytotoxicity on NHDF | ||||
|---|---|---|---|---|---|---|
| IC50 | R2 | IC50 | R2 | IC50 | R2 | |
| Febuxostat | 0.03 ± 0.01 | 0.9732 | n.d. b | - | n.d. b | - |
| Allo | 10.73 ± 0.81 | 0.9959 | n.d. b | - | n.d. b | - |
| Trolox | n.d. b | - | 23.82 ± 2.13 | 0.9947 | n.d. b | - |
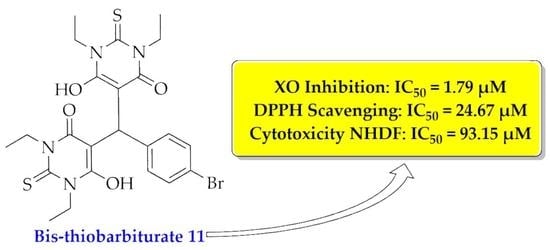
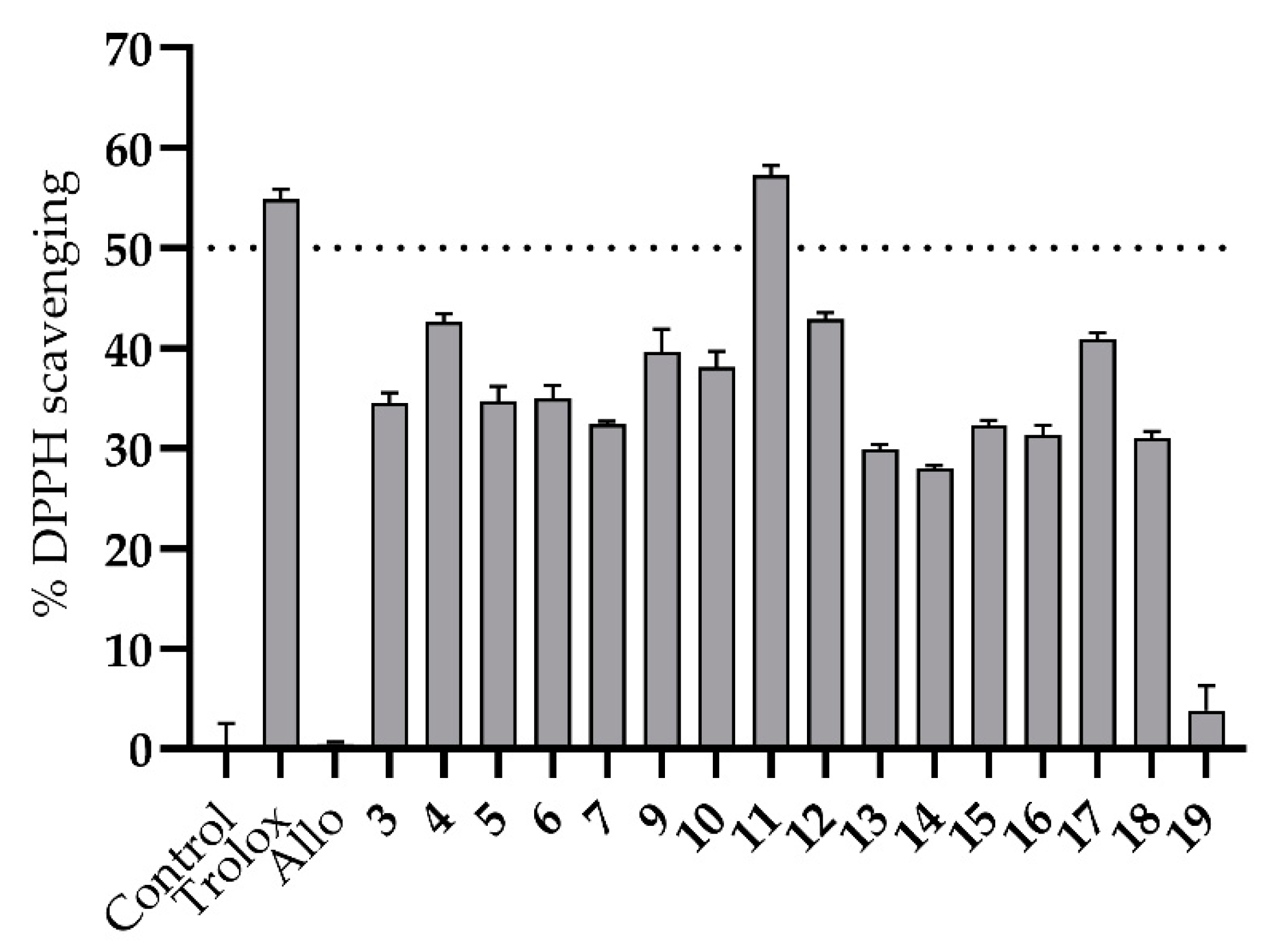
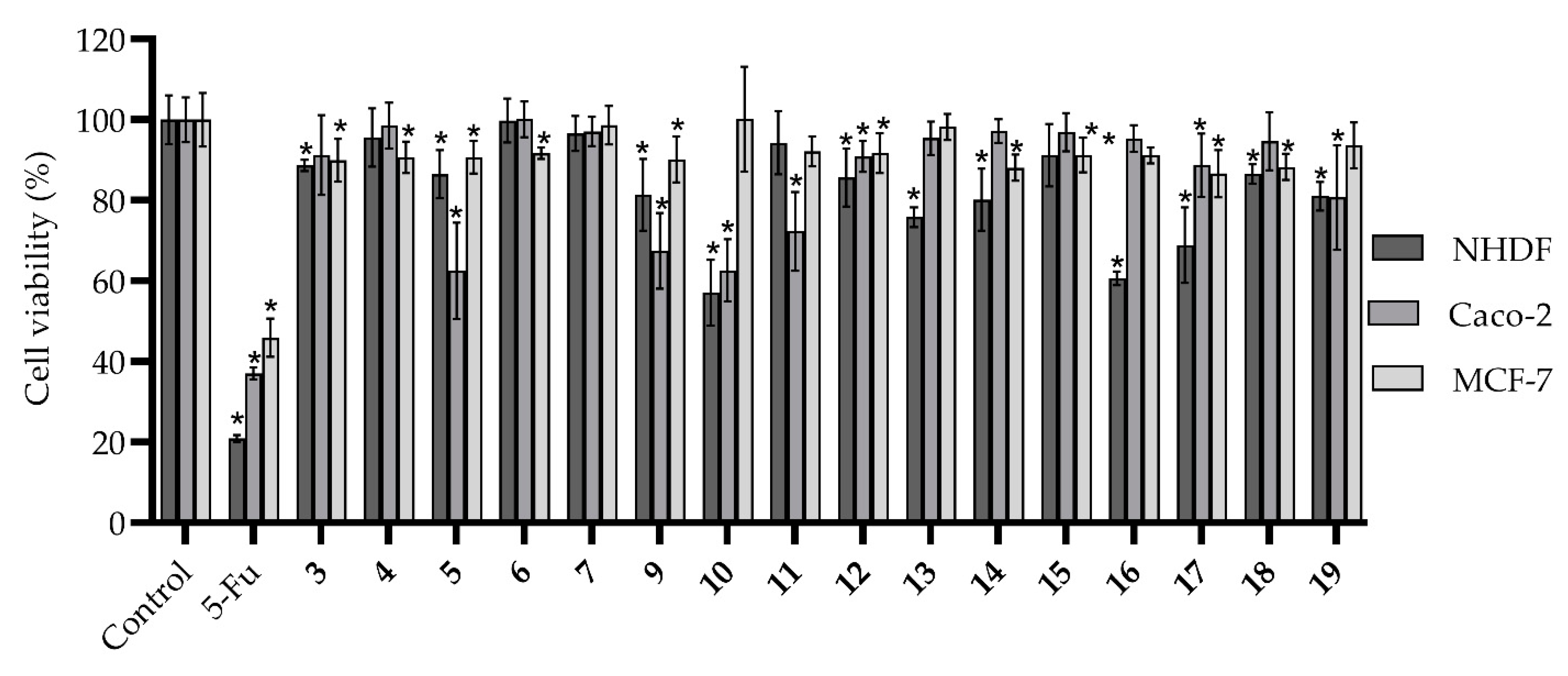
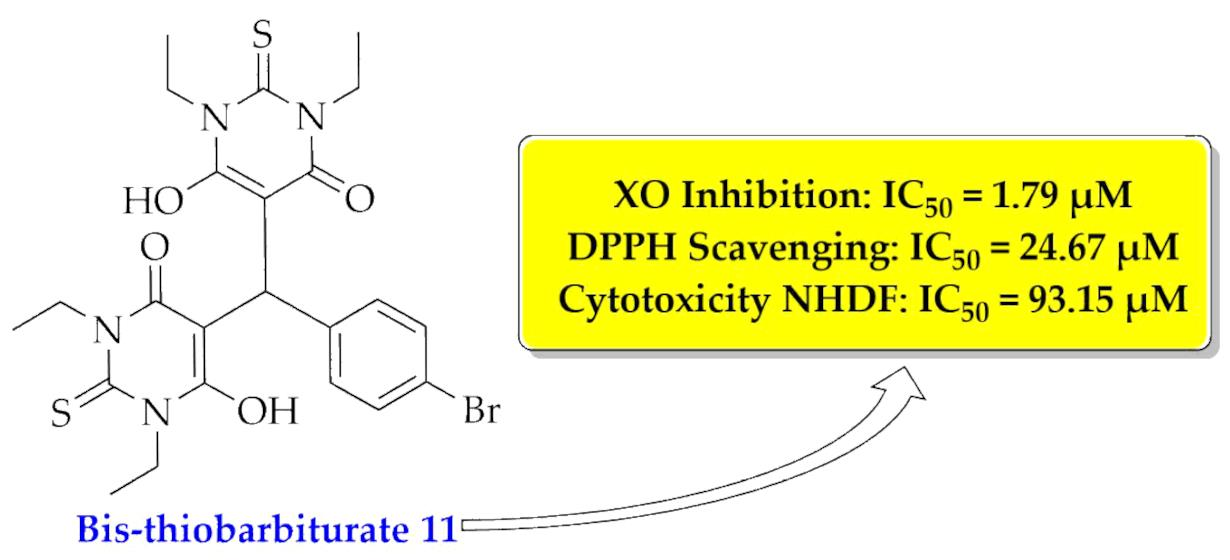
| 11 | 1.79 ± 0.07 | 0.9986 | 24.67 ± 0.88 | 0.9992 | 93.15 ± 5.54 | 0.8913 |
| Descriptor | Value | |
|---|---|---|
| Molecular weight | 567.52 g/mol | |
| Log P | 4.01 | |
| nON | 6 | |
| nOHNH | 2 | |
| n-rot | 7 | |
| TPSA | 158.50 Å2 | |
| Drug-likeness | Lipinski | Yes; 1 violation: MW > 500 |
| Veber | Yes | |
| Medicinal Chemistry | PAINS | 0 alert |
| Property | Model Name | Predicted Value |
|---|---|---|
| Absorption | Water solubility | −4.66 b (Moderately soluble) |
| Intestinal absorption (human) | 71.66% | |
| P-glycoprotein substrate | No | |
| Distribution | BBB permeant | No |
| LogBB | −1.38 | |
| LogPS | −2.42 | |
| Metabolism | CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No | |
| CYP2C9 inhibitor | Yes | |
| CYP2D6 inhibitor | No | |
| CYP3A4 inhibitor | Yes | |
| Excretion | Log total clearance | −0.36 c |
| Renal OCT2 substrate | No | |
| Toxicity | AMES toxicity | No |
| hERG I inhibitor | No | |
| hERG II inhibitor | No | |
| Hepatotoxicity | Yes | |
| Skin Sensitisation | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano, J.L.; Lopes, D.; Reis, M.J.A.; Boto, R.E.F.; Silvestre, S.; Almeida, P. Bis-thiobarbiturates as Promising Xanthine Oxidase Inhibitors: Synthesis and Biological Evaluation. Biomedicines 2021, 9, 1443. https://doi.org/10.3390/biomedicines9101443
Serrano JL, Lopes D, Reis MJA, Boto REF, Silvestre S, Almeida P. Bis-thiobarbiturates as Promising Xanthine Oxidase Inhibitors: Synthesis and Biological Evaluation. Biomedicines. 2021; 9(10):1443. https://doi.org/10.3390/biomedicines9101443
Chicago/Turabian StyleSerrano, João L., Diana Lopes, Melani J. A. Reis, Renato E. F. Boto, Samuel Silvestre, and Paulo Almeida. 2021. "Bis-thiobarbiturates as Promising Xanthine Oxidase Inhibitors: Synthesis and Biological Evaluation" Biomedicines 9, no. 10: 1443. https://doi.org/10.3390/biomedicines9101443
APA StyleSerrano, J. L., Lopes, D., Reis, M. J. A., Boto, R. E. F., Silvestre, S., & Almeida, P. (2021). Bis-thiobarbiturates as Promising Xanthine Oxidase Inhibitors: Synthesis and Biological Evaluation. Biomedicines, 9(10), 1443. https://doi.org/10.3390/biomedicines9101443