
Bilateral Adrenal Hyperplasia: Pathogenesis and Treatment


Abstract
1. Introduction
2. Clinical Features
2.1. Micronodular Adrenal Hyperplasia
2.1.1. PPNAD
2.1.2. Carney Complex
2.2. Macronodular Adrenal Hyperplasia
2.2.1. PBMAH
2.2.2. Multiple Tumor Syndromes Associated with Macronodular Adrenal Hyperplasia
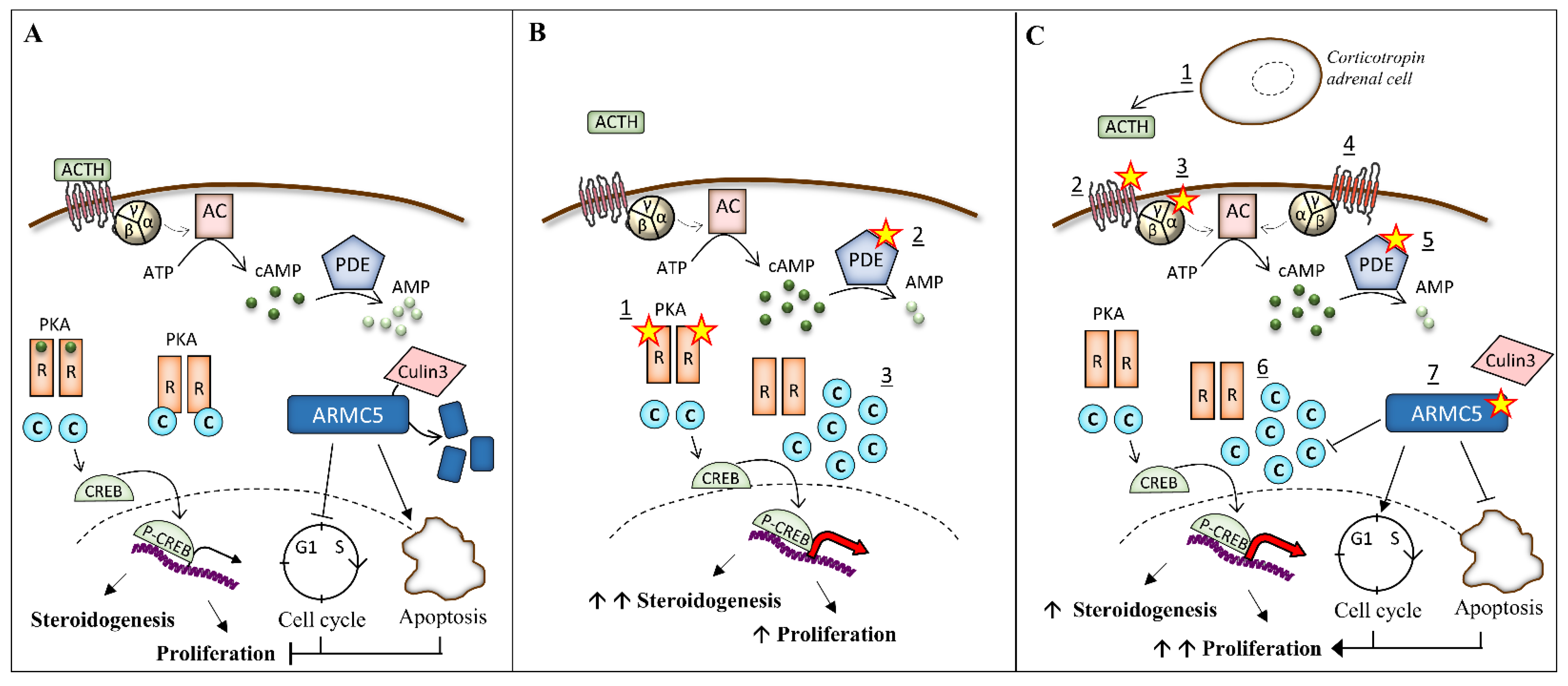
3. Pathogenesis of Bilateral Adrenal Hyperplasia
3.1. Alteration of the PKA Pathway
3.1.1. Alteration of PRKAR1A in PPNAD
- Mutations leading to the creation of a mutant protein are associated with a higher number of CNC manifestations.
- Mutations on the exons are more often associated with acromegaly, cardiac myxomas, lentigines, and schwannomas.
- Mutations on the intronic splice sites are associated with a milder phenotype.
- The c.491–492delTG hotspot mutation is more often associated with cardiac myxomas, lentigines, and thyroid tumors.
- The c.709(−7–2)del6 hotspot mutation and the c.1A > G mutation are associated with isolated PPNADs.
- Patients without the PRKAR1A mutation have fewer tumors that appear later in life.
3.1.2. Alteration of Other Genes Involved in the PKA Pathway
3.1.3. Aberrant Expression of G-Coupled Protein Receptor in PBMAH
- Eutopic receptors (normally expressed in adrenocortical cells), such as the vasopressin V1 receptor, the luteinizing hormone/human chorionic gonadotropin (LH/HCG) receptor, the serotonin 5-HT4 receptor, and the leptin receptor.
- Ectopic receptors (absent in normal adrenocortical cells), such as the GIP receptor, the vasopressin V2 and V3 receptors, the serotonin 5-HT7 receptor, the glucagon receptor, the beta-adrenergic receptor, and the angiotensin II AT1-receptor.
3.2. Mutation of ARMC5 in PBMAH
3.2.1. Genetic Mutations of ARMC5
3.2.2. Function of ARMC5
3.3. Paracrine and Autocrine Factors in PBMAH
4. Treatment
4.1. Decision for Treatment in Bilateral Adrenal Hyperplasia
4.2. Surgical Treatment
4.2.1. Surgical Treatment of PPNAD
4.2.2. Surgical Treatment of PBMAH
4.3. Medical Treatment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lodish, M.; Stratakis, C.A. A Genetic and Molecular Update on Adrenocortical Causes of Cushing Syndrome. Nat. Rev. Endocrinol. 2016, 12, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Ragazzon, B.; Rizk-Rabin, M.; Bertherat, J. Protein Kinase A Alterations in Endocrine Tumors. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Métabolisme 2012, 44, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A.; Gordon, H.; Carpenter, P.C.; Shenoy, B.V.; Go, V.L. The Complex of Myxomas, Spotty Pigmentation, and Endocrine Overactivity. Medicine (Baltimore) 1985, 64, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Sarlis, N.; Kirschner, L.S.; Carney, J.A.; Doppman, J.L.; Nieman, L.K.; Chrousos, G.P.; Papanicolaou, D.A. Paradoxical Response to Dexamethasone in the Diagnosis of Primary Pigmented Nodular Adrenocortical Disease. Ann. Intern. Med. 1999, 131, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, B.V.; Carpenter, P.C.; Carney, J.A. Bilateral Primary Pigmented Nodular Adrenocortical Disease. Rare Cause of the Cushing Syndrome. Am. J. Surg. Pathol. 1984, 8, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; René-Corail, F.; Stergiopoulos, S.; Bourdeau, I.; et al. Mutations in Regulatory Subunit Type 1A of Cyclic Adenosine 5′-Monophosphate-Dependent Protein Kinase (PRKAR1A): Phenotype Analysis in 353 Patients and 80 Different Genotypes. J. Clin. Endocrinol. Metab. 2009, 94, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Boikos, S.A. Genetics of Adrenal Tumors Associated with Cushing’s Syndrome: A New Classification for Bilateral Adrenocortical Hyperplasias. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 748–757. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Clinical and Molecular Features of the Carney Complex: Diagnostic Criteria and Recommendations for Patient Evaluation. J. Clin. Endocrinol. Metab. 2001, 86, 4041–4046. [Google Scholar] [CrossRef]
- Gunther, D.F.; Bourdeau, I.; Matyakhina, L.; Cassarino, D.; Kleiner, D.E.; Griffin, K.; Courkoutsakis, N.; Abu-Asab, M.; Tsokos, M.; Keil, M.; et al. Cyclical Cushing Syndrome Presenting in Infancy: An Early Form of Primary Pigmented Nodular Adrenocortical Disease, or a New Entity? J. Clin. Endocrinol. Metab. 2004, 89, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Espiard, S.; Bertherat, J. Carney Complex. Front. Horm. Res. 2013, 41, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Espiard, S.; Vantyghem, M.-C.; Assié, G.; Cardot-Bauters, C.; Raverot, G.; Brucker-Davis, F.; Archambeaud-Mouveroux, F.; Lefebvre, H.; Nunes, M.-L.; Tabarin, A.; et al. Frequency and Incidence of Carney Complex Manifestations: A Prospective Multicenter Study With a Three-Year Follow-Up. J. Clin. Endocrinol. Metab. 2020, 105, dgaa002. [Google Scholar] [CrossRef] [PubMed]
- Courcoutsakis, N.A.; Tatsi, C.; Patronas, N.J.; Lee, C.-C.R.; Prassopoulos, P.K.; Stratakis, C.A. The Complex of Myxomas, Spotty Skin Pigmentation and Endocrine Overactivity (Carney Complex): Imaging Findings with Clinical and Pathological Correlation. Insights Imaging 2013, 4, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Groussin, L.; Jullian, E.; Perlemoine, K.; Louvel, A.; Leheup, B.; Luton, J.P.; Bertagna, X.; Bertherat, J. Mutations of the PRKAR1A Gene in Cushing’s Syndrome Due to Sporadic Primary Pigmented Nodular Adrenocortical Disease. J. Clin. Endocrinol. Metab. 2002, 87, 4324–4329. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, J.; Medeiros, S.; Carneiro, V.; Greene, E.; Levy, I.; Nesterova, M.; Lyssikatos, C.; Horvath, A.; Carney, J.A.; Stratakis, C.A. A Large Family with Carney Complex Caused by the S147G PRKAR1A Mutation Shows a Unique Spectrum of Disease Including Adrenocortical Cancer. J. Clin. Endocrinol. Metab. 2012, 97, 351–359. [Google Scholar] [CrossRef]
- Morin, E.; Mete, O.; Wasserman, J.D.; Joshua, A.M.; Asa, S.L.; Ezzat, S. Carney Complex with Adrenal Cortical Carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E202–E206. [Google Scholar] [CrossRef]
- Hofland, J.; de Herder, W.W.; Derks, L.; Hofland, L.J.; van Koetsveld, P.M.; de Krijger, R.R.; van Nederveen, F.H.; Horvath, A.; Stratakis, C.A.; de Jong, F.H.; et al. Regulation of Steroidogenesis in a Primary Pigmented Nodular Adrenocortical Disease-Associated Adenoma Leading to Virilization and Subclinical Cushing’s Syndrome. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2013, 168, 67–74. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Carney Complex: Diagnosis and Management of the Complex of Spotty Skin Pigmentation, Myxomas, Endocrine Overactivity, and Schwannomas. Am. J. Med. Genet. 1998, 80, 183–185. [Google Scholar] [CrossRef]
- Rothenbuhler, A.; Stratakis, C.A. Clinical and Molecular Genetics of Carney Complex. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 389–399. [Google Scholar] [CrossRef]
- Carney, J.A.; Hruska, L.S.; Beauchamp, G.D.; Gordon, H. Dominant Inheritance of the Complex of Myxomas, Spotty Pigmentation, and Endocrine Overactivity. Mayo Clin. Proc. Mayo Clin. 1986, 61, 165–172. [Google Scholar] [CrossRef]
- Kirschner, L.S. PRKAR1A and the Evolution of Pituitary Tumors. Mol. Cell. Endocrinol. 2010, 326, 3–7. [Google Scholar] [CrossRef]
- Kirschner, M.A.; Powell, R.D.; Lipsett, M.B. Cushing’s syndrome: Nodular cortical hyperplasia of adrenal glands with clinical and pathological features suggesting adrenocortical tumor. J. Clin. Endocrinol. Metab. 1964, 24, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Espiard, S.; Benomar, K.; Loyer, C.; Vahé, C.; Vantyghem, M.-C. European Recommendations for the Management of Adrenal Incidentalomas: A Debate on Patients Follow-Up. Ann. Endocrinol. 2018, 79, 45–48. [Google Scholar] [CrossRef]
- Espiard, S.; Drougat, L.; Libé, R.; Assié, G.; Perlemoine, K.; Guignat, L.; Barrande, G.; Brucker-Davis, F.; Doullay, F.; Lopez, S.; et al. ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J. Clin. Endocrinol. Metab. 2015, jc20144204. [Google Scholar] [CrossRef]
- Sohaib, S.A.; Hanson, J.A.; Newell-Price, J.D.; Trainer, P.J.; Monson, J.P.; Grossman, A.B.; Besser, G.M.; Reznek, R.H. CT Appearance of the Adrenal Glands in Adrenocorticotrophic Hormone-Dependent Cushing’s Syndrome. AJR Am. J. Roentgenol. 1999, 172, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Vitellius, G.; Trabado, S.; Hoeffel, C.; Bouligand, J.; Bennet, A.; Castinetti, F.; Decoudier, B.; Guiochon-Mantel, A.; Lombes, M.; Delemer, B.; et al. Significant Prevalence of NR3C1 Mutations in Incidentally Discovered Bilateral Adrenal Hyperplasia: Results of the French MUTA-GR Study. Eur. J. Endocrinol. 2018, 178, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of Adrenal Incidentalomas: European Society of Endocrinology Clinical Practice Guideline in Collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K. Multiple Endocrine Neoplasia Type 1. Front. Horm. Res. 2013, 41, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gatta-Cherifi, B.; Chabre, O.; Murat, A.; Niccoli, P.; Cardot-Bauters, C.; Rohmer, V.; Young, J.; Delemer, B.; Du Boullay, H.; Verger, M.F.; et al. Adrenal Involvement in MEN1. Analysis of 715 Cases from the Groupe d’etude Des Tumeurs Endocrines Database. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2012, 166, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Langer, P.; Cupisti, K.; Bartsch, D.K.; Nies, C.; Goretzki, P.E.; Rothmund, M.; Röher, H.D. Adrenal Involvement in Multiple Endocrine Neoplasia Type 1. World J. Surg. 2002, 26, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, J.; Bartsch, D.K.; Kann, P.H.; Fendrich, V.; Rothmund, M.; Langer, P. Adrenal Involvement in Multiple Endocrine Neoplasia Type 1: Results of 7 Years Prospective Screening. Langenbecks Arch. Surg. Dtsch. Ges. Für Chir. 2007, 392, 437–443. [Google Scholar] [CrossRef]
- Yoshida, M.; Hiroi, M.; Imai, T.; Kikumori, T.; Himeno, T.; Nakamura, Y.; Sasano, H.; Yamada, M.; Murakami, Y.; Nakamura, S.; et al. A Case of ACTH-Independent Macronodular Adrenal Hyperplasia Associated with Multiple Endocrine Neoplasia Type 1. Endocr. J. 2011, 58, 269–277. [Google Scholar] [CrossRef]
- Harding, B.; Lemos, M.C.; Reed, A.A.C.; Walls, G.V.; Jeyabalan, J.; Bowl, M.R.; Tateossian, H.; Sullivan, N.; Hough, T.; Fraser, W.D.; et al. Multiple Endocrine Neoplasia Type 1 Knockout Mice Develop Parathyroid, Pancreatic, Pituitary and Adrenal Tumours with Hypercalcaemia, Hypophosphataemia and Hypercorticosteronaemia. Endocr. Relat. Cancer 2009, 16, 1313–1327. [Google Scholar] [CrossRef]
- Matyakhina, L.; Freedman, R.J.; Bourdeau, I.; Wei, M.-H.; Stergiopoulos, S.G.; Chidakel, A.; Walther, M.; Abu-Asab, M.; Tsokos, M.; Keil, M.; et al. Hereditary Leiomyomatosis Associated with Bilateral, Massive, Macronodular Adrenocortical Disease and Atypical Cushing Syndrome: A Clinical and Molecular Genetic Investigation. J. Clin. Endocrinol. Metab. 2005, 90, 3773–3779. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Ricketts, C.J.; Vocke, C.D.; Valera, V.A.; Chen, C.C.; Gautam, R.; Gupta, G.N.; Gomez Macias, G.S.; Merino, M.J.; Bratslavsky, G.; et al. Adrenal Nodular Hyperplasia in Hereditary Leiomyomatosis and Renal Cell Cancer. J. Urol. 2013, 189, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.-P.; Kirschner, L.S.; Bourdeau, I.; Keil, M.F.; Boikos, S.A.; Verma, S.; Robinson-White, A.J.; Nesterova, M.; Lacroix, A.; Stratakis, C.A. Clinical and Genetic Heterogeneity, Overlap with Other Tumor Syndromes, and Atypical Glucocorticoid Hormone Secretion in Adrenocorticotropin-Independent Macronodular Adrenal Hyperplasia Compared with Other Adrenocortical Tumors. J. Clin. Endocrinol. Metab. 2009, 94, 2930–2937. [Google Scholar] [CrossRef]
- Half, E.; Bercovich, D.; Rozen, P. Familial Adenomatous Polyposis. Orphanet J. Rare Dis. 2009, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Pinson, S.; Gimenez-Roqueplo, A.-P.; Amar, L.; Ragazzon, B.; Launay, P.; Meatchi, T.; Libé, R.; Bertagna, X.; Audebourg, A.; et al. Inactivation of the APC Gene Is Constant in Adrenocortical Tumors from Patients with Familial Adenomatous Polyposis but Not Frequent in Sporadic Adrenocortical Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5133–5141. [Google Scholar] [CrossRef]
- Hosogi, H.; Nagayama, S.; Kanamoto, N.; Yoshizawa, A.; Suzuki, T.; Nakao, K.; Sakai, Y. Biallelic APC Inactivation Was Responsible for Functional Adrenocortical Adenoma in Familial Adenomatous Polyposis with Novel Germline Mutation of the APC Gene: Report of a Case. Jpn. J. Clin. Oncol. 2009, 39, 837–846. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert Consensus Document: Clinical and Molecular Diagnosis, Screening and Management of Beckwith-Wiedemann Syndrome: An International Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Carney, J.A.; Ho, J.; Kitsuda, K.; Young, W.F.J.; Stratakis, C.A. Massive Neonatal Adrenal Enlargement Due to Cytomegaly, Persistence of the Transient Cortex, and Hyperplasia of the Permanent Cortex: Findings in Cushing Syndrome Associated with Hemihypertrophy. Am. J. Surg. Pathol. 2012, 36, 1452–1463. [Google Scholar] [CrossRef]
- Matyakhina, L.; Pack, S.; Kirschner, L.S.; Pak, E.; Mannan, P.; Jaikumar, J.; Taymans, S.E.; Sandrini, F.; Carney, J.A.; Stratakis, C.A. Chromosome 2 (2p16) Abnormalities in Carney Complex Tumours. J. Med. Genet. 2003, 40, 268–277. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the Gene Encoding the Protein Kinase A Type I-Alpha Regulatory Subunit in Patients with the Carney Complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Casey, M.; Vaughan, C.J.; He, J.; Hatcher, C.J.; Winter, J.M.; Weremowicz, S.; Montgomery, K.; Kucherlapati, R.; Morton, C.C.; Basson, C.T. Mutations in the Protein Kinase A R1alpha Regulatory Subunit Cause Familial Cardiac Myxomas and Carney Complex. J. Clin. Investig. 2000, 106, R31–R38. [Google Scholar] [CrossRef]
- Horvath, A.; Bertherat, J.; Groussin, L.; Guillaud-Bataille, M.; Tsang, K.; Cazabat, L.; Libé, R.; Remmers, E.; René-Corail, F.; Faucz, F.R.; et al. Mutations and Polymorphisms in the Gene Encoding Regulatory Subunit Type 1-Alpha of Protein Kinase A (PRKAR1A): An Update. Hum. Mutat. 2010, 31, 369–379. [Google Scholar] [CrossRef]
- Horvath, A.; Bossis, I.; Giatzakis, C.; Levine, E.; Weinberg, F.; Meoli, E.; Robinson-White, A.; Siegel, J.; Soni, P.; Groussin, L.; et al. Large Deletions of the PRKAR1A Gene in Carney Complex. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Groussin, L.; Kirschner, L.S.; Vincent-Dejean, C.; Perlemoine, K.; Jullian, E.; Delemer, B.; Zacharieva, S.; Pignatelli, D.; Carney, J.A.; Luton, J.P.; et al. Molecular Analysis of the Cyclic AMP-Dependent Protein Kinase A (PKA) Regulatory Subunit 1A (PRKAR1A) Gene in Patients with Carney Complex and Primary Pigmented Nodular Adrenocortical Disease (PPNAD) Reveals Novel Mutations and Clues for Pathophysiology: Augmented PKA Signaling Is Associated with Adrenal Tumorigenesis in PPNAD. Am. J. Hum. Genet. 2002, 71, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Patronas, Y.; Horvath, A.; Greene, E.; Tsang, K.; Bimpaki, E.; Haran, M.; Nesterova, M.; Stratakis, C.A. In Vitro Studies of Novel PRKAR1A Mutants That Extend the Predicted RIα Protein Sequence into the 3′-Untranslated Open Reading Frame: Proteasomal Degradation Leads to RIα Haploinsufficiency and Carney Complex. J. Clin. Endocrinol. Metab. 2012, 97, E496–E502. [Google Scholar] [CrossRef] [PubMed]
- Amieux, P.S.; Howe, D.G.; Knickerbocker, H.; Lee, D.C.; Su, T.; Laszlo, G.S.; Idzerda, R.L.; McKnight, G.S. Increased Basal CAMP-Dependent Protein Kinase Activity Inhibits the Formation of Mesoderm-Derived Structures in the Developing Mouse Embryo. J. Biol. Chem. 2002, 277, 27294–27304. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Kusewitt, D.F.; Matyakhina, L.; Towns, W.H., 2nd; Carney, J.A.; Westphal, H.; Stratakis, C.A. A Mouse Model for the Carney Complex Tumor Syndrome Develops Neoplasia in Cyclic AMP-Responsive Tissues. Cancer Res. 2005, 65, 4506–4514. [Google Scholar] [CrossRef] [PubMed]
- Griffin, K.J.; Kirschner, L.S.; Matyakhina, L.; Stergiopoulos, S.; Robinson-White, A.; Lenherr, S.; Weinberg, F.D.; Claflin, E.; Meoli, E.; Cho-Chung, Y.S.; et al. Down-Regulation of Regulatory Subunit Type 1A of Protein Kinase A Leads to Endocrine and Other Tumors. Cancer Res. 2004, 64, 8811–8815. [Google Scholar] [CrossRef]
- Sahut-Barnola, I.; de Joussineau, C.; Val, P.; Lambert-Langlais, S.; Damon, C.; Lefrançois-Martinez, A.-M.; Pointud, J.-C.; Marceau, G.; Sapin, V.; Tissier, F.; et al. Cushing’s Syndrome and Fetal Features Resurgence in Adrenal Cortex-Specific Prkar1a Knockout Mice. PLoS Genet. 2010, 6, e1000980. [Google Scholar] [CrossRef]
- Dumontet, T.; Sahut-Barnola, I.; Septier, A.; Montanier, N.; Plotton, I.; Roucher-Boulez, F.; Ducros, V.; Lefrançois-Martinez, A.-M.; Pointud, J.-C.; Zubair, M.; et al. PKA Signaling Drives Reticularis Differentiation and Sexually Dimorphic Adrenal Cortex Renewal. JCI Insight 2018, 3, 98394. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, M.; Lippincott-Schwartz, J.; Stratakis, C.A.; Bossis, I. Depletion of Type IA Regulatory Subunit (RIalpha) of Protein Kinase A (PKA) in Mammalian Cells and Tissues Activates MTOR and Causes Autophagic Deficiency. Hum. Mol. Genet. 2006, 15, 2962–2971. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive Activation of PKA Catalytic Subunit in Adrenal Cushing’s Syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Lodish, M.B.; Yuan, B.; Levy, I.; Braunstein, G.D.; Lyssikatos, C.; Salpea, P.; Szarek, E.; Karageorgiadis, A.S.; Belyavskaya, E.; Raygada, M.; et al. Germline PRKACA Amplification Causes Variable Phenotypes That May Depend on the Extent of the Genomic Defect: Molecular Mechanisms and Clinical Presentations. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2015, 172, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Swords, F.M.; Baig, A.; Malchoff, D.M.; Malchoff, C.D.; Thorner, M.O.; King, P.J.; Hunyady, L.; Clark, A.J.L. Impaired Desensitization of a Mutant Adrenocorticotropin Receptor Associated with Apparent Constitutive Activity. Mol. Endocrinol. Baltim. Md 2002, 16, 2746–2753. [Google Scholar] [CrossRef][Green Version]
- Swords, F.M.; Noon, L.A.; King, P.J.; Clark, A.J.L. Constitutive Activation of the Human ACTH Receptor Resulting from a Synergistic Interaction between Two Naturally Occurring Missense Mutations in the MC2R Gene. Mol. Cell. Endocrinol. 2004, 213, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Libé, R.; Fratticci, A.; Coste, J.; Tissier, F.; Horvath, A.; Ragazzon, B.; Rene-Corail, F.; Groussin, L.; Bertagna, X.; Raffin-Sanson, M.L.; et al. Phosphodiesterase 11A (PDE11A) and Genetic Predisposition to Adrenocortical Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 4016–4024. [Google Scholar] [CrossRef]
- Vezzosi, D.; Libé, R.; Baudry, C.; Rizk-Rabin, M.; Horvath, A.; Levy, I.; René-Corail, F.; Ragazzon, B.; Stratakis, C.A.; Vandecasteele, G.; et al. Phosphodiesterase 11A (PDE11A) Gene Defects in Patients with Acth-Independent Macronodular Adrenal Hyperplasia (AIMAH): Functional Variants May Contribute to Genetic Susceptibility of Bilateral Adrenal Tumors. J. Clin. Endocrinol. Metab. 2012, 97, E2063–E2069. [Google Scholar] [CrossRef]
- Horvath, A.; Boikos, S.; Giatzakis, C.; Robinson-White, A.; Groussin, L.; Griffin, K.J.; Stein, E.; Levine, E.; Delimpasi, G.; Hsiao, H.P.; et al. A Genome-Wide Scan Identifies Mutations in the Gene Encoding Phosphodiesterase 11A4 (PDE11A) in Individuals with Adrenocortical Hyperplasia. Nat. Genet. 2006, 38, 794–800. [Google Scholar] [CrossRef]
- Horvath, A.; Mericq, V.; Stratakis, C.A. Mutation in PDE8B, a Cyclic AMP-Specific Phosphodiesterase in Adrenal Hyperplasia. N. Engl. J. Med. 2008, 358, 750–752. [Google Scholar] [CrossRef]
- Libé, R.; Horvath, A.; Vezzosi, D.; Fratticci, A.; Coste, J.; Perlemoine, K.; Ragazzon, B.; Guillaud-Bataille, M.; Groussin, L.; Clauser, E.; et al. Frequent Phosphodiesterase 11A Gene (PDE11A) Defects in Patients with Carney Complex (CNC) Caused by PRKAR1A Mutations: PDE11A May Contribute to Adrenal and Testicular Tumors in CNC as a Modifier of the Phenotype. J. Clin. Endocrinol. Metab. 2011, 96, E208–E214. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating Mutations of the Stimulatory G Protein in the McCune-Albright Syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A.; Young, W.F.; Stratakis, C.A. Primary Bimorphic Adrenocortical Disease: Cause of Hypercortisolism in McCune-Albright Syndrome. Am. J. Surg. Pathol. 2011, 35, 1311–1326. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Azevedo, M.F.; Xekouki, P.; Bimpaki, E.I.; Horvath, A.; Collins, M.T.; Karaviti, L.P.; Jeha, G.S.; Bhattacharyya, N.; Cheadle, C.; et al. Activation of Cyclic AMP Signaling Leads to Different Pathway Alterations in Lesions of the Adrenal Cortex Caused by Germline PRKAR1A Defects versus Those Due to Somatic GNAS Mutations. J. Clin. Endocrinol. Metab. 2012, 97, E687–E693. [Google Scholar] [CrossRef]
- Lacroix, A.; Bolté, E.; Tremblay, J.; Dupré, J.; Poitras, P.; Fournier, H.; Garon, J.; Garrel, D.; Bayard, F.; Taillefer, R. Gastric Inhibitory Polypeptide-Dependent Cortisol Hypersecretion--a New Cause of Cushing’s Syndrome. N. Engl. J. Med. 1992, 327, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Reznik, Y.; Allali-Zerah, V.; Chayvialle, J.A.; Leroyer, R.; Leymarie, P.; Travert, G.; Lebrethon, M.C.; Budi, I.; Balliere, A.M.; Mahoudeau, J. Food-Dependent Cushing’s Syndrome Mediated by Aberrant Adrenal Sensitivity to Gastric Inhibitory Polypeptide. N. Engl. J. Med. 1992, 327, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Bourdeau, I.; Lampron, A.; Mazzuco, T.L.; Tremblay, J.; Hamet, P. Aberrant G-Protein Coupled Receptor Expression in Relation to Adrenocortical Overfunction. Clin. Endocrinol. (Oxf.) 2010, 73, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Ndiaye, N.; Tremblay, J.; Hamet, P. Ectopic and Abnormal Hormone Receptors in Adrenal Cushing’s Syndrome. Endocr. Rev. 2001, 22, 75–110. [Google Scholar] [PubMed]
- Libé, R.; Coste, J.; Guignat, L.; Tissier, F.; Lefebvre, H.; Barrande, G.; Ajzenberg, C.; Tauveron, I.; Clauser, E.; Dousset, B.; et al. Aberrant Cortisol Regulations in Bilateral Macronodular Adrenal Hyperplasia: A Frequent Finding in a Prospective Study of 32 Patients with Overt or Subclinical Cushing’s Syndrome. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2010, 163, 129–138. [Google Scholar] [CrossRef] [PubMed]
- El Ghorayeb, N.; Bourdeau, I.; Lacroix, A. Multiple Aberrant Hormone Receptors in Cushing’s Syndrome. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2015, 173, M45–M60. [Google Scholar] [CrossRef]
- Groussin, L.; Perlemoine, K.; Contesse, V.; Lefebvre, H.; Tabarin, A.; Thieblot, P.; Schlienger, J.L.; Luton, J.P.; Bertagna, X.; Bertherat, J. The Ectopic Expression of the Gastric Inhibitory Polypeptide Receptor Is Frequent in Adrenocorticotropin-Independent Bilateral Macronodular Adrenal Hyperplasia, but Rare in Unilateral Tumors. J. Clin. Endocrinol. Metab. 2002, 87, 1980–1985. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Assie, G.; Louiset, E.; Sturm, N.; René-Corail, F.; Groussin, L.; Bertherat, J.; Thomas, M.; Lefebvre, H.; Feige, J.-J.; Clauser, E.; et al. Systematic Analysis of G Protein-Coupled Receptor Gene Expression in Adrenocorticotropin-Independent Macronodular Adrenocortical Hyperplasia Identifies Novel Targets for Pharmacological Control of Adrenal Cushing’s Syndrome. J. Clin. Endocrinol. Metab. 2010, 95, E253–E262. [Google Scholar] [CrossRef]
- Lampron, A.; Bourdeau, I.; Hamet, P.; Tremblay, J.; Lacroix, A. Whole Genome Expression Profiling of Glucose-Dependent Insulinotropic Peptide (GIP)- and Adrenocorticotropin-Dependent Adrenal Hyperplasias Reveals Novel Targets for the Study of GIP-Dependent Cushing’s Syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 3611–3618. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bertherat, J.; Contesse, V.; Louiset, E.; Barrande, G.; Duparc, C.; Groussin, L.; Emy, P.; Bertagna, X.; Kuhn, J.-M.; Vaudry, H.; et al. In Vivo and in Vitro Screening for Illegitimate Receptors in Adrenocorticotropin-Independent Macronodular Adrenal Hyperplasia Causing Cushing’s Syndrome: Identification of Two Cases of Gonadotropin/Gastric Inhibitory Polypeptide-Dependent Hypercortisolism. J. Clin. Endocrinol. Metab. 2005, 90, 1302–1310. [Google Scholar] [CrossRef]
- Louiset, E.; Contesse, V.; Groussin, L.; Cartier, D.; Duparc, C.; Barrande, G.; Bertherat, J.; Vaudry, H.; Lefebvre, H. Expression of Serotonin7 Receptor and Coupling of Ectopic Receptors to Protein Kinase A and Ionic Currents in Adrenocorticotropin-Independent Macronodular Adrenal Hyperplasia Causing Cushing’s Syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 4578–4586. [Google Scholar] [CrossRef] [PubMed]
- Hofland, J.; Hofland, L.J.; van Koetsveld, P.M.; Steenbergen, J.; de Herder, W.W.; van Eijck, C.H.; de Krijger, R.R.; van Nederveen, F.H.; van Aken, M.O.; de Groot, J.W.; et al. ACTH-Independent Macronodular Adrenocortical Hyperplasia Reveals Prevalent Aberrant in Vivo and in Vitro Responses to Hormonal Stimuli and Coupling of Arginine-Vasopressin Type 1a Receptor to 11β-Hydroxylase. Orphanet J. Rare Dis. 2013, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Ferruzzi, P.; Luciani, P.; Crescioli, C.; Buci, L.; Corona, G.; Serio, M.; Peri, A. Cushing’s Syndrome in a Patient with Bilateral Macronodular Adrenal Hyperplasia Responding to Cisapride: An in Vivo and in Vitro Study. J. Clin. Endocrinol. Metab. 2003, 88, 4616–4622. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazzuco, T.L.; Chabre, O.; Feige, J.-J.; Thomas, M. Aberrant Expression of Human Luteinizing Hormone Receptor by Adrenocortical Cells Is Sufficient to Provoke Both Hyperplasia and Cushing’s Syndrome Features. J. Clin. Endocrinol. Metab. 2006, 91, 196–203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazzuco, T.L.; Chabre, O.; Sturm, N.; Feige, J.-J.; Thomas, M. Ectopic Expression of the Gastric Inhibitory Polypeptide Receptor Gene Is a Sufficient Genetic Event to Induce Benign Adrenocortical Tumor in a Xenotransplantation Model. Endocrinology 2006, 147, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, C.; Ballarè, E.; Mantovani, G.; Ambrosi, B.; Spada, A.; Barbetta, L.; Colombo, P.; Travaglini, P.; Loli, P.; Beck-Peccoz, P. Assessing the Presence of Abnormal Regulation of Cortisol Secretion by Membrane Hormone Receptors: In Vivo and in Vitro Studies in Patients with Functioning and Non-Functioning Adrenal Adenoma. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Métabolisme 2004, 36, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Berthon, A.; Faucz, F.R.; Espiard, S.; Drougat, L.; Bertherat, J.; Stratakis, C.A. Age-Dependent Effects of Armc5 Haploinsufficiency on Adrenocortical Function. Hum. Mol. Genet. 2017, 26, 3495–3507. [Google Scholar] [CrossRef]
- Lecoq, A.-L.; Stratakis, C.A.; Viengchareun, S.; Chaligné, R.; Tosca, L.; Deméocq, V.; Hage, M.; Berthon, A.; Faucz, F.R.; Hanna, P.; et al. Adrenal GIPR Expression and Chromosome 19q13 Microduplications in GIP-Dependent Cushing’s Syndrome. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Vaczlavik, A.; Bouys, L.; Letouze, E.; Perlemoine, K.; Guignat, L.; Letourneur, F.; Sibony, M.; Bonnet, F.; Heurtier, V.; Espiard, S.; et al. Integrated Genomics Reveals Different Subgroups of Primary Bilateral Macronodular Adrenal Hyperplasia (PBMAH). In Endocrine Abstracts; Bioscientifica: Bristol, UK, 21 August 2020; Volume 70. [Google Scholar]
- Assié, G.; Libé, R.; Espiard, S.; Rizk-Rabin, M.; Guimier, A.; Luscap, W.; Barreau, O.; Lefèvre, L.; Sibony, M.; Guignat, L.; et al. ARMC5 Mutations in Macronodular Adrenal Hyperplasia with Cushing’s Syndrome. N. Engl. J. Med. 2013, 369, 2105–2114. [Google Scholar] [CrossRef]
- Faucz, F.R.; Zilbermint, M.; Lodish, M.B.; Szarek, E.; Trivellin, G.; Sinaii, N.; Berthon, A.; Libé, R.; Assié, G.; Espiard, S.; et al. Macronodular Adrenal Hyperplasia Due to Mutations in an Armadillo Repeat Containing 5 (ARMC5) Gene: A Clinical and Genetic Investigation. J. Clin. Endocrinol. Metab. 2014, jc20134280. [Google Scholar] [CrossRef]
- Albiger, N.M.; Regazzo, D.; Rubin, B.; Ferrara, A.M.; Rizzati, S.; Taschin, E.; Ceccato, F.; Arnaldi, G.; Pecori Giraldi, F.; Stigliano, A.; et al. A Multicenter Experience on the Prevalence of ARMC5 Mutations in Patients with Primary Bilateral Macronodular Adrenal Hyperplasia: From Genetic Characterization to Clinical Phenotype. Endocrine 2016. [Google Scholar] [CrossRef] [PubMed]
- Alencar, G.A.; Lerario, A.M.; Nishi, M.Y.; Mariani, B.M.d.P.; Almeida, M.Q.; Tremblay, J.; Hamet, P.; Bourdeau, I.; Zerbini, M.C.N.; Pereira, M.A.A.; et al. ARMC5 Mutations Are a Frequent Cause of Primary Macronodular Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2014, jc20134237. [Google Scholar] [CrossRef]
- Kyo, C.; Usui, T.; Kosugi, R.; Torii, M.; Yonemoto, T.; Ogawa, T.; Kotani, M.; Tamura, N.; Yamamoto, Y.; Katabami, T.; et al. ARMC5 Alterations in Primary Macronodular Adrenal Hyperplasia (PMAH) and the Clinical State of Variant Carriers. J. Endocr. Soc. 2019, 3, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Zilbermint, M.; Xekouki, P.; Faucz, F.R.; Berthon, A.; Gkourogianni, A.; Helene Schernthaner-Reiter, M.; Batsis, M.; Sinaii, N.; Quezado, M.M.; Merino, M.; et al. Primary Aldosteronism and ARMC5 Variants. J. Clin. Endocrinol. Metab. 2015, jc20144167. [Google Scholar] [CrossRef]
- Joseph, J.J.; Zhou, X.; Zilbermint, M.; Stratakis, C.A.; Faucz, F.R.; Lodish, M.B.; Berthon, A.; Wilson, J.G.; Hsueh, W.A.; Golden, S.H.; et al. The Association of ARMC5 with the Renin-Angiotensin-Aldosterone System, Blood Pressure, and Glycemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, dgaa290. [Google Scholar] [CrossRef]
- Bourdeau, I.; Oble, S.; Magne, F.; Lévesque, I.; Cáceres-Gorriti, K.Y.; Nolet, S.; Awadalla, P.; Tremblay, J.; Hamet, P.; Fragoso, M.C.B.V.; et al. ARMC5 Mutations in a Large French-Canadian Family with Cortisol-Secreting β-Adrenergic/Vasopressin Responsive Bilateral Macronodular Adrenal Hyperplasia. Eur. J. Endocrinol. 2016, 174, 85–96. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gagliardi, L.; Schreiber, A.W.; Hahn, C.N.; Feng, J.; Cranston, T.; Boon, H.; Hotu, C.; Oftedal, B.E.; Cutfield, R.; Adelson, D.L.; et al. ARMC5 Mutations Are Common in Familial Bilateral Macronodular Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2014, 99, E1784–E1792. [Google Scholar] [CrossRef] [PubMed]
- Elbelt, U.; Trovato, A.; Kloth, M.; Gentz, E.; Finke, R.; Spranger, J.; Galas, D.; Weber, S.; Wolf, C.; König, K.; et al. Molecular and Clinical Evidence for an ARMC5 Tumor Syndrome: Concurrent Inactivating Germline and Somatic Mutations Are Associated with Both Primary Macronodular Adrenal Hyperplasia and Meningioma. J. Clin. Endocrinol. Metab. 2014, jc20142648. [Google Scholar] [CrossRef]
- Jojima, T.; Kogai, T.; Iijima, T.; Kato, K.; Sagara, M.; Kezuka, A.; Kase, M.; Sakurai, S.; Akimoto, K.; Sakumoto, J.; et al. Genetic Alteration of ARMC5 in a Patient Diagnosed with Meningioma and Primary Macronodular Adrenal Hyperplasia: A Case Report. Eur. J. Endocrinol. 2020, 183, K7–K12. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Zilbermint, M.; Berthon, A.; Espiard, S.; Batsis, M.; Papadakis, G.Z.; Xekouki, P.; Lodish, M.B.; Bertherat, J.; Faucz, F.R.; et al. The ARMC5 Gene Shows Extensive Genetic Variance in Primary Macronodular Adrenocortical Hyperplasia. Eur. J. Endocrinol. 2015, 173, 435–440. [Google Scholar] [CrossRef]
- Drougat, L.; Espiard, S.; Bertherat, J. Genetics of Primary Bilateral Macronodular Adrenal Hyperplasia: A Model for Early Diagnosis of Cushing’s Syndrome? Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2015, 173, M121–M131. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tatsuno, I.; Oohara, E.; Nakayama, A.; Komai, E.; Shiga, A.; Kono, T.; Takiguchi, T.; Higuchi, S.; Sakuma, I.; et al. GERMLINE DELETION OF ARMC5 IN FAMILIAL PRIMARY MACRONODULAR ADRENAL HYPERPLASIA. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2015, 21, 1152–1160. [Google Scholar] [CrossRef]
- Berthon, A.; Faucz, F.; Bertherat, J.; Stratakis, C.A. Analysis of ARMC5 Expression in Human Tissues. Mol. Cell. Endocrinol. 2017, 441, 140–145. [Google Scholar] [CrossRef]
- Cavalcante, I.P.; Nishi, M.; Zerbini, M.C.N.; Almeida, M.Q.; Brondani, V.B.; Botelho, M.L.A.d.A.; Tanno, F.Y.; Srougi, V.; Chambo, J.L.; Mendonca, B.B.; et al. The Role of ARMC5 in Human Cell Cultures from Nodules of Primary Macronodular Adrenocortical Hyperplasia (PMAH). Mol. Cell. Endocrinol. 2018, 460, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, I.; Antonini, S.R.; Lacroix, A.; Kirschner, L.S.; Matyakhina, L.; Lorang, D.; Libutti, S.K.; Stratakis, C.A. Gene Array Analysis of Macronodular Adrenal Hyperplasia Confirms Clinical Heterogeneity and Identifies Several Candidate Genes as Molecular Mediators. Oncogene 2004, 23, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Wurth, R.; Tirosh, A.; Kamilaris, C.D.C.; Camacho, J.; Faucz, F.R.; Maria, A.G.; Berthon, A.; Papadakis, G.Z.; Nilubol, N.; Hamimi, A.; et al. Volumetric Modeling of Adrenal Gland Size in Primary Bilateral Macronodular Adrenocortical Hyperplasia. J. Endocr. Soc. 2021, 5, bvaa162. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lao, L.; Mao, J.; Jin, W.; Luo, H.; Charpentier, T.; Qi, S.; Peng, J.; Hu, B.; Marcinkiewicz, M.M.; et al. Armc5 Deletion Causes Developmental Defects and Compromises T-Cell Immune Responses. Nat. Commun. 2017, 8, 13834. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Harran, M.; Bimpaki, E.I.; Hsiao, H.-P.; Horvath, A.; Cheadle, C.; Watkins, T.; Nesterova, M.; Stratakis, C.A. Integrated Genomic Analysis of Nodular Tissue in Macronodular Adrenocortical Hyperplasia: Progression of Tumorigenesis in a Disorder Associated with Multiple Benign Lesions. J. Clin. Endocrinol. Metab. 2011, 96, E728–E738. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, I.P.; Vaczlavik, A.; Drougat, L.; Lotfi, C.F.P.; Perlemoine, K.; Ribes, C.; Rizk-Rabin, M.; Clauser, E.; Fragoso, M.C.B.V.; Bertherat, J.; et al. Cullin 3 Targets the Tumor Suppressor Gene ARMC5 for Ubiquitination and Degradation. Endocr. Relat. Cancer 2020, 27, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart-Bornstein, M.; Hinson, J.P.; Bornstein, S.R.; Scherbaum, W.A.; Vinson, G.P. Intraadrenal Interactions in the Regulation of Adrenocortical Steroidogenesis. Endocr. Rev. 1998, 19, 101–143. [Google Scholar] [CrossRef]
- Perraudin, V.; Delarue, C.; Lefebvre, H.; Contesse, V.; Kuhn, J.M.; Vaudry, H. Vasopressin Stimulates Cortisol Secretion from Human Adrenocortical Tissue through Activation of V1 Receptors. J. Clin. Endocrinol. Metab. 1993, 76, 1522–1528. [Google Scholar] [CrossRef]
- Lefebvre, H.; Duparc, C.; Prévost, G.; Bertherat, J.; Louiset, E. Cell-to-Cell Communication in Bilateral Macronodular Adrenal Hyperplasia Causing Hypercortisolism. Front. Endocrinol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Tomori, N.; Tozawa, F.; Demura, H.; Shizume, K.; Mouri, T.; Miura, Y.; Sasano, N. Immunoreactive Corticotropin and Corticotropin-Releasing Factor in Human Hypothalamus, Adrenal, Lung Cancer, and Pheochromocytoma. J. Clin. Endocrinol. Metab. 1984, 58, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Louiset, E.; Duparc, C.; Young, J.; Renouf, S.; Tetsi Nomigni, M.; Boutelet, I.; Libé, R.; Bram, Z.; Groussin, L.; Caron, P.; et al. Intraadrenal Corticotropin in Bilateral Macronodular Adrenal Hyperplasia. N. Engl. J. Med. 2013, 369, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A. Heredity and Cortisol Regulation in Bilateral Macronodular Adrenal Hyperplasia. N. Engl. J. Med. 2013, 369, 2147–2149. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, H.; Contesse, V.; Delarue, C.; Feuilloley, M.; Hery, F.; Grise, P.; Raynaud, G.; Verhofstad, A.A.; Wolf, L.M.; Vaudry, H. Serotonin-Induced Stimulation of Cortisol Secretion from Human Adrenocortical Tissue Is Mediated through Activation of a Serotonin4 Receptor Subtype. Neuroscience 1992, 47, 999–1007. [Google Scholar] [CrossRef]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Murad, M.H.; Newell-Price, J.; Savage, M.O.; Tabarin, A. Endocrine Society Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2015, 100, 2807–2831. [Google Scholar] [CrossRef] [PubMed]
- Ngaosuwan, K.; Johnston, D.G.; Godsland, I.F.; Cox, J.; Majeed, A.; Quint, J.K.; Oliver, N.; Robinson, S. Cardiovascular Disease in Patients With Primary and Secondary Adrenal Insufficiency and the Role of Comorbidities. J. Clin. Endocrinol. Metab. 2021, 106, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Tresoldi, A.S.; Sumilo, D.; Perrins, M.; Toulis, K.A.; Prete, A.; Reddy, N.; Wass, J.A.H.; Arlt, W.; Nirantharakumar, K. Increased Infection Risk in Addison’s Disease and Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2020, 105, dgz006. [Google Scholar] [CrossRef]
- Johannsson, G.; Falorni, A.; Skrtic, S.; Lennernäs, H.; Quinkler, M.; Monson, J.P.; Stewart, P.M. Adrenal Insufficiency: Review of Clinical Outcomes with Current Glucocorticoid Replacement Therapy. Clin. Endocrinol. (Oxf.) 2015, 82, 2–11. [Google Scholar] [CrossRef]
- Nieman, L.K. Update on Subclinical Cushing’s Syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 180–184. [Google Scholar] [CrossRef]
- Bancos, I.; Alahdab, F.; Crowley, R.K.; Chortis, V.; Delivanis, D.A.; Erickson, D.; Natt, N.; Terzolo, M.; Arlt, W.; Young, W.F.J.; et al. THERAPY OF ENDOCRINE DISEASE: Improvement of Cardiovascular Risk Factors after Adrenalectomy in Patients with Adrenal Tumors and Subclinical Cushing’s Syndrome: A Systematic Review and Meta-Analysis. Eur. J. Endocrinol. 2016, 175, R283–R295. [Google Scholar] [CrossRef]
- Powell, A.C.; Stratakis, C.A.; Patronas, N.J.; Steinberg, S.M.; Batista, D.; Alexander, H.R.; Pingpank, J.F.; Keil, M.; Bartlett, D.L.; Libutti, S.K. Operative Management of Cushing Syndrome Secondary to Micronodular Adrenal Hyperplasia. Surgery 2008, 143, 750–758. [Google Scholar] [CrossRef]
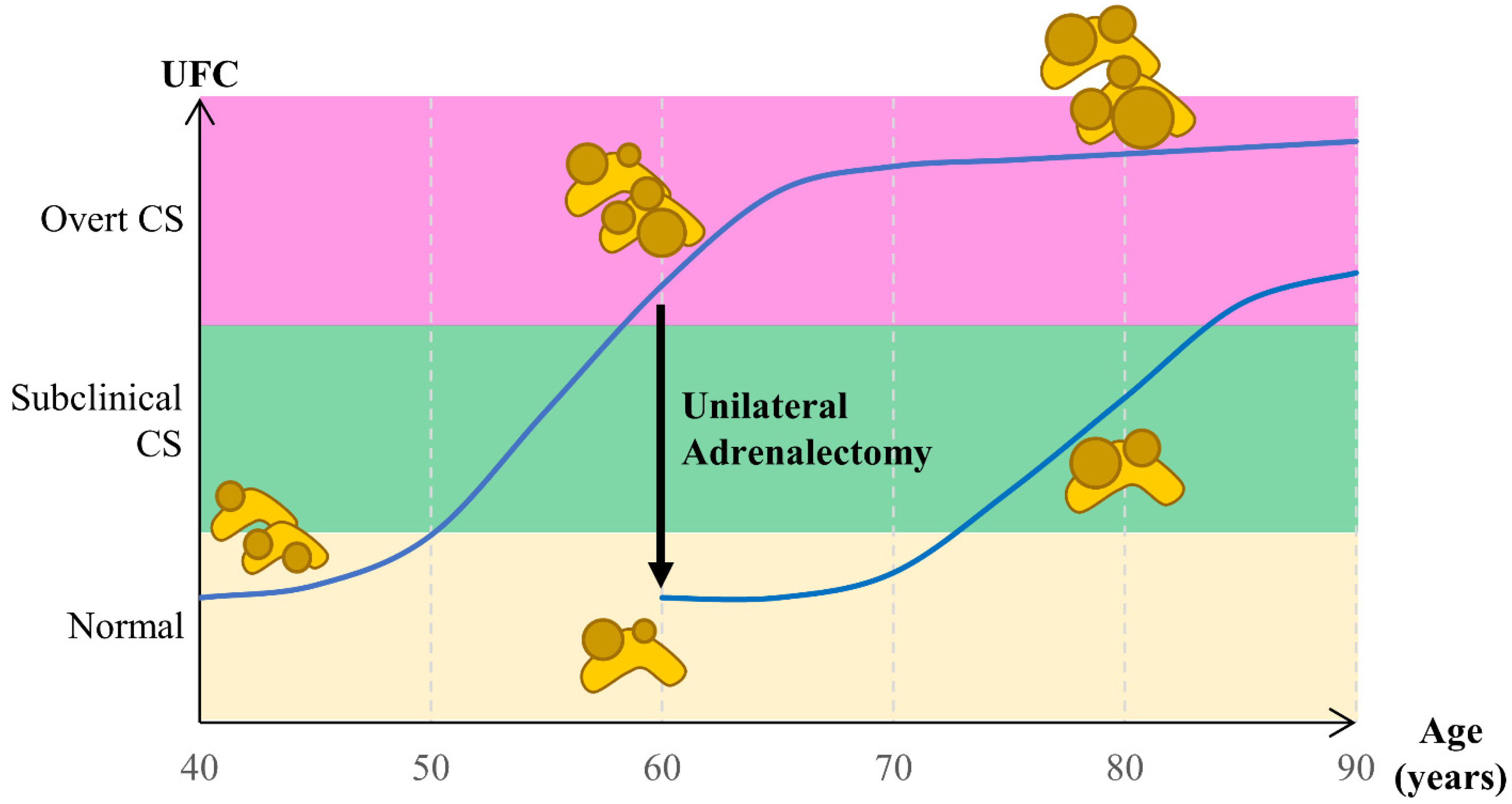
- Meloche-Dumas, L.; Mercier, F.; Lacroix, A. Role of Unilateral Adrenalectomy in Bilateral Adrenal Hyperplasias with Cushing’s Syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101486. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H. Classification and Surgical Treatment for 180 Cases of Adrenocortical Hyperplastic Disease. Int. J. Clin. Exp. Med. 2015, 8, 19311–19317. [Google Scholar]
- Carney, J.A.; Young, W.F., Jr. Primary Pigmented Nodular Adrenocortical Disease and Its Associated Conditions. The Endocrinologist 1992, 2. [Google Scholar] [CrossRef]
- Tadjine, M.; Lampron, A.; Ouadi, L.; Horvath, A.; Stratakis, C.A.; Bourdeau, I. Detection of Somatic Beta-Catenin Mutations in Primary Pigmented Nodular Adrenocortical Disease (PPNAD). Clin. Endocrinol. (Oxf.) 2008, 69, 367–373. [Google Scholar] [CrossRef]
- Sarlis, N.J.; Chrousos, G.P.; Doppman, J.L.; Carney, J.A.; Stratakis, C.A. Primary Pigmented Nodular Adrenocortical Disease: Reevaluation of a Patient with Carney Complex 27 Years after Unilateral Adrenalectomy. J. Clin. Endocrinol. Metab. 1997, 82, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M. The New Bone Biology: Pathologic, Molecular, and Clinical Correlates. Am. J. Med. Genet. A 2006, 140, 2646–2706. [Google Scholar] [CrossRef] [PubMed]
- Guanà, R.; Gesmundo, R.; Morino, M.; Matarazzo, P.; Pucci, A.; Pasini, B.; Lala, R.; Fiore, L.; Repici, M.; Canavese, F. Laparoscopic Unilateral Adrenalectomy in Children for Isolated Primary Pigmented Nodular Adrenocortical Disease (PPNAD): Case Report and Literature Review. Eur. J. Pediatr. Surg. Off. J. Austrian Assoc. Pediatr. Surg. Al Z. Kinderchir. 2010, 20, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Rui, W.; Qi, Y.; Zhang, C.; Zhao, J.; Wang, X.; Wu, Y.; Zhu, Q.; Shen, Z.; Ning, G.; et al. The Role of Unilateral Adrenalectomy in Corticotropin-Independent Bilateral Adrenocortical Hyperplasias. World J. Surg. 2013, 37, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Kyrilli, A.; Lytrivi, M.; Bouquegneau, M.S.; Demetter, P.; Lucidi, V.; Garcia, C.; Moreno-Reyes, R.; Tabarin, A.; Corvilain, B.; Driessens, N. Unilateral Adrenalectomy Could Be a Valid Option for Primary Nodular Adrenal Disease: Evidence From Twins. J. Endocr. Soc. 2019, 3, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kumorowicz-Czoch, M.; Dolezal-Oltarzewska, K.; Roztoczynska, D.; Chrupek, M.; Prokurat, A.I.; Drabik, G.; Starzyk, J. Causes and Consequences of Abandoning One-Stage Bilateral Adrenalectomy Recommended in Primary Pigmented Nodular Adrenocortical Disease--Case Presentation. J. Pediatr. Endocrinol. Metab. JPEM 2011, 24, 565–567. [Google Scholar] [CrossRef]
- Debillon, E.; Velayoudom-Cephise, F.-L.; Salenave, S.; Caron, P.; Chaffanjon, P.; Wagner, T.; Massoutier, M.; Lambert, B.; Benoit, M.; Young, J.; et al. Unilateral Adrenalectomy as a First-Line Treatment of Cushing’s Syndrome in Patients With Primary Bilateral Macronodular Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2015, 100, 4417–4424. [Google Scholar] [CrossRef]
- Acharya, R.; Dhir, M.; Bandi, R.; Yip, L.; Challinor, S. Outcomes of Adrenal Venous Sampling in Patients with Bilateral Adrenal Masses and ACTH-Independent Cushing’s Syndrome. World J. Surg. 2019, 43, 527–533. [Google Scholar] [CrossRef]
- Ueland, G.Å.; Methlie, P.; Jøssang, D.E.; Sagen, J.V.; Viste, K.; Thordarson, H.B.; Heie, A.; Grytaas, M.; Løvås, K.; Biermann, M.; et al. Adrenal Venous Sampling for Assessment of Autonomous Cortisol Secretion. J. Clin. Endocrinol. Metab. 2018, 103, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Osswald, A.; Quinkler, M.; Di Dalmazi, G.; Deutschbein, T.; Rubinstein, G.; Ritzel, K.; Zopp, S.; Bertherat, J.; Beuschlein, F.; Reincke, M. Long-Term Outcome of Primary Bilateral Macronodular Adrenocortical Hyperplasia After Unilateral Adrenalectomy. J. Clin. Endocrinol. Metab. 2019, 104, 2985–2993. [Google Scholar] [CrossRef]
- Yoshiaki Tanno, F.; Srougi, V.; Almeida, M.Q.; Ide Yamauchi, F.; Morbeck Almeida Coelho, F.; Nishi, M.Y.; Claudia Nogueira Zerbini, M.; Silvia Correa Soares, I.; Adelaide Albergaria Pereira, M.; Laiz Silva Charchar, H.; et al. A New Insight into the Surgical Treatment of Primary Macronodular Adrenal Hyperplasia. J. Endocr. Soc. 2020, 4, bvaa083. [Google Scholar] [CrossRef]
- Albiger, N.M.; Ceccato, F.; Zilio, M.; Barbot, M.; Occhi, G.; Rizzati, S.; Fassina, A.; Mantero, F.; Boscaro, M.; Iacobone, M.; et al. An Analysis of Different Therapeutic Options in Patients with Cushing’s Syndrome Due to Bilateral Macronodular Adrenal Hyperplasia: A Single-Centre Experience. Clin. Endocrinol. (Oxf.) 2015, 82, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Mazzuco, T.L.; Chaffanjon, P.; Martinie, M.; Sturm, N.; Chabre, O. Adrenal Cushing’s Syndrome Due to Bilateral Macronodular Adrenal Hyperplasia: Prediction of the Efficacy of Beta-Blockade Therapy and Interest of Unilateral Adrenalectomy. Endocr. J. 2009, 56, 867–877. [Google Scholar] [CrossRef]
- Oki, K.; Yamane, K.; Nakanishi, S.; Nakashima, R.; Jitsuiki, K.; Kohno, N. Improvement of Hypercortisolism by β-Blocker Therapy in Subclinical Cushing’s Syndrome Associated with ACTH-Independent Macronodular Adrenocortical Hyperplasia. Endocrine 2009, 36, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Preumont, V.; Mermejo, L.M.; Damoiseaux, P.; Lacroix, A.; Maiter, D. Transient Efficacy of Octreotide and Pasireotide (SOM230) Treatment in GIP-Dependent Cushing’s Syndrome. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Métabolisme 2011, 43, 287–291. [Google Scholar] [CrossRef]
- Karapanou, O.; Vlassopoulou, B.; Tzanela, M.; Stratigou, T.; Tsatlidis, V.; Tsirona, S.; Tsagarakis, S. Adrenocorticotropic Hormone Independent Macronodular Adrenal Hyperplasia Due to Aberrant Receptor Expression: Is Medical Treatment Always an Option? Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2013, 19, e77–e82. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Narita, I.; Omori, K.; Komura, S.; Arakawa, M. Adrenocorticotropic Hormone-Independent Bilateral Adrenocortical Macronodular Hyperplasia Treated with Mitotane. Intern. Med. Tokyo Jpn. 1999, 38, 969–973. [Google Scholar] [CrossRef][Green Version]
- Comte-Perret, S.; Zanchi, A.; Gomez, F. Long-Term Low-Dose Ketoconazole Treatment in Bilateral Macronodular Adrenal Hyperplasia. Endocrinol. Diabetes Metab. Case Rep. 2014, 2014, 140083. [Google Scholar] [CrossRef]
- Campo, M.R.; Lamacchia, O.; Farese, A.; Conserva, A.; Picca, G.; Grilli, G.; Cignarelli, M. Mitotane and Carney Complex: Ten Years Follow-up of a Low-Dose Mitotane Regimen Inducing a Sustained Correction of Hypercortisolism. Horm. Athens Greece 2015, 14, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Debono, M.; Harrison, R.F.; Chadarevian, R.; Gueroult, C.; Abitbol, J.-L.; Newell-Price, J. Resetting the Abnormal Circadian Cortisol Rhythm in Adrenal Incidentaloma Patients With Mild Autonomous Cortisol Secretion. J. Clin. Endocrinol. Metab. 2017, 102, 3461–3469. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Genetic | Function | Phenotype | Frequency of the Adrenal Hyperplasia in Case of Mutations of the Gene |
|---|---|---|---|---|
| PRKAR1A | Unique inactivating mutations spread along the gene. 3 hotspots (c.709(−7–2)del6, c.491–492delTG, c82C > T). Large deletions described | Regulatory subunit R1α of the PKA. Inhibition of PKA pathway | Isolated PPNAD (~12%) Carney complex: cardiac, skin and breast myxomas, lentigines, pituitary adenoma or hyperplasia (GH +/− PRL), LCCST, osteochondromyxoma, schwannomas | 26% to 60% [1,2,3] |
| PRKACA | Amplification of the gene | Catalytic subunit Cα of the PKA. Activation of PKA pathway | PBMAH Macroglossia? | NA 1 |
| GNAS | Post-zygotic activating mutations Two hotspots (p.R201H and p.C174Y) | G protein subunit alpha stimulating. Activation of PKA pathway | Macronodular adrenal hyperplasia Mc Cune Albright syndrome: precocious puberty, Café-au-lait spot, polyostotic fibrous dysplasia, somatotroph adenoma or prolactinoma, multinodular goiter, hyperthyroidism | Near 5% [4,5] |
| PED8B PDE11A | Unique inactivating mutations | Phosphodiesterase type 8B and 11A. Inactivation of PKA pathway | iMAD | NA 1 |
| MC2R | Unique activating mutations | ACTH receptor. Activation of the PKA pathway. | PBMAH | NA 1 |
| ARMC5 | Unique inactivating mutations spread along the gene. | Potentially control apoptosis and cell cycle. Interaction with PKA pathway and steroidogenesis? | PBMAH Meningioma (several cases described) | ND, High penetrance described in families |
| MEN1 | Unique inactivating mutations spread along the gene. Large deletions | Scaffold protein controlling gene transcription and many other cellular functions, such as proliferation | PBMAH Pituitary adenoma Primary hyperparathyroidism Neuroendocrine tumors | Case reports |
| FH | Unique inactivating mutations spread along the gene. | Krebs cycle | HLRCC: leiomyomatosis, renal cell cancer | Estimated at 0.8% [6] |
| APC | Unique inactivating mutations spread along the gene. | Inhibition of Wnt/β-catenin pathway | PBMAH Familial adenomatous polyposis | Case reports |
| Clinical Features | Frequency (%) [1,2,3] | Age at Diagnosis (Years) [2,3] |
|---|---|---|
| PPNAD | 45–68 | Median: 25 Bimodal age distribution: in the first 3 years of life or in the 2nd and 3rd decades |
| Skin lesion | ||
| Lentigines | 56–70 | From birth or appear progressively, fade after the 4th decade |
| Blue naevi | 17–50 | May appear in early childhood years |
| Cutaneous myxoma | 20–45 | May appear within the first 10 years of life |
| Cardiac myxoma | 23–53 | Median: 29 Described in the first years of life |
| Hypersomatotropism | 10–19 | Median: 35 |
| Thyroid tumors | 5–25 | May appear within the first 10 years of life |
| Psammomatous melanotic schwannoma | 8–10 | Median: 35 |
| Osteochondromyxoma | 2–6 | Described in the first years of life but also in adults |
| Breast lesions | 19–42 | Breast myxomas may appear in childhood |
| LCCSCT | 33–41 | Median: 28 Described from the first years of life |
| Receptor | Ligand | Diagnostic Tests |
|---|---|---|
| Ectopic receptors | ||
| GIP receptor | GIP | Standard mixed meal, IV GIP infusion |
| V2R/V3 receptor | AVP/Anti-diuretic hormone | Supine-to-upright posture test, AVP/IM/SC desmopressin infusion (terlipressin) |
| β-adrenergic receptor | β-epinephrine | Insulin hypoglycemia IV isoproterenol infusion |
| AT1 receptor | Angiotensin 2 | Supine-to-upright posture test, IV angiotensin 2 infusion |
| 5-HT7 receptor | Serotonin | Metoclopramide administration |
| Glucagon receptor | Glucagon | IV glucagon infusion |
| Eutopic receptors | ||
| V1R receptor | AVP/Anti-diuretic hormone | Supine-to-upright posture test IM desmopressin infusion (terlipressin) |
| 5-HT4 receptor | Serotonin | Metoclopramide administration |
| LH/HCG receptor | LH/HCG | IV GnRH infusion IM LH or HCG infusion |
| PRL receptor | Prolactin | Chlorpromazine administration IV TRH infusion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chevalier, B.; Vantyghem, M.-C.; Espiard, S. Bilateral Adrenal Hyperplasia: Pathogenesis and Treatment. Biomedicines 2021, 9, 1397. https://doi.org/10.3390/biomedicines9101397
Chevalier B, Vantyghem M-C, Espiard S. Bilateral Adrenal Hyperplasia: Pathogenesis and Treatment. Biomedicines. 2021; 9(10):1397. https://doi.org/10.3390/biomedicines9101397
Chicago/Turabian StyleChevalier, Benjamin, Marie-Christine Vantyghem, and Stéphanie Espiard. 2021. "Bilateral Adrenal Hyperplasia: Pathogenesis and Treatment" Biomedicines 9, no. 10: 1397. https://doi.org/10.3390/biomedicines9101397
APA StyleChevalier, B., Vantyghem, M.-C., & Espiard, S. (2021). Bilateral Adrenal Hyperplasia: Pathogenesis and Treatment. Biomedicines, 9(10), 1397. https://doi.org/10.3390/biomedicines9101397