
Possible Role of Cytochrome P450 1B1 in the Mechanism of Gemcitabine Resistance in Pancreatic Cancer

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tumor Tissues for Transplantation
2.3. Establishment of Xenografts
2.4. Gemcitabine (GEM) Administration
2.5. Immunohistochemistry
2.6. RNA Extraction and High-Throughput Sequencing
2.7. Gene Set Enrichment Analysis (GSEA)
2.8. Cell Culture
2.9. Quantitative Real-Time PCR (RT-qPCR)
2.10. Statistical Analysis
3. Results
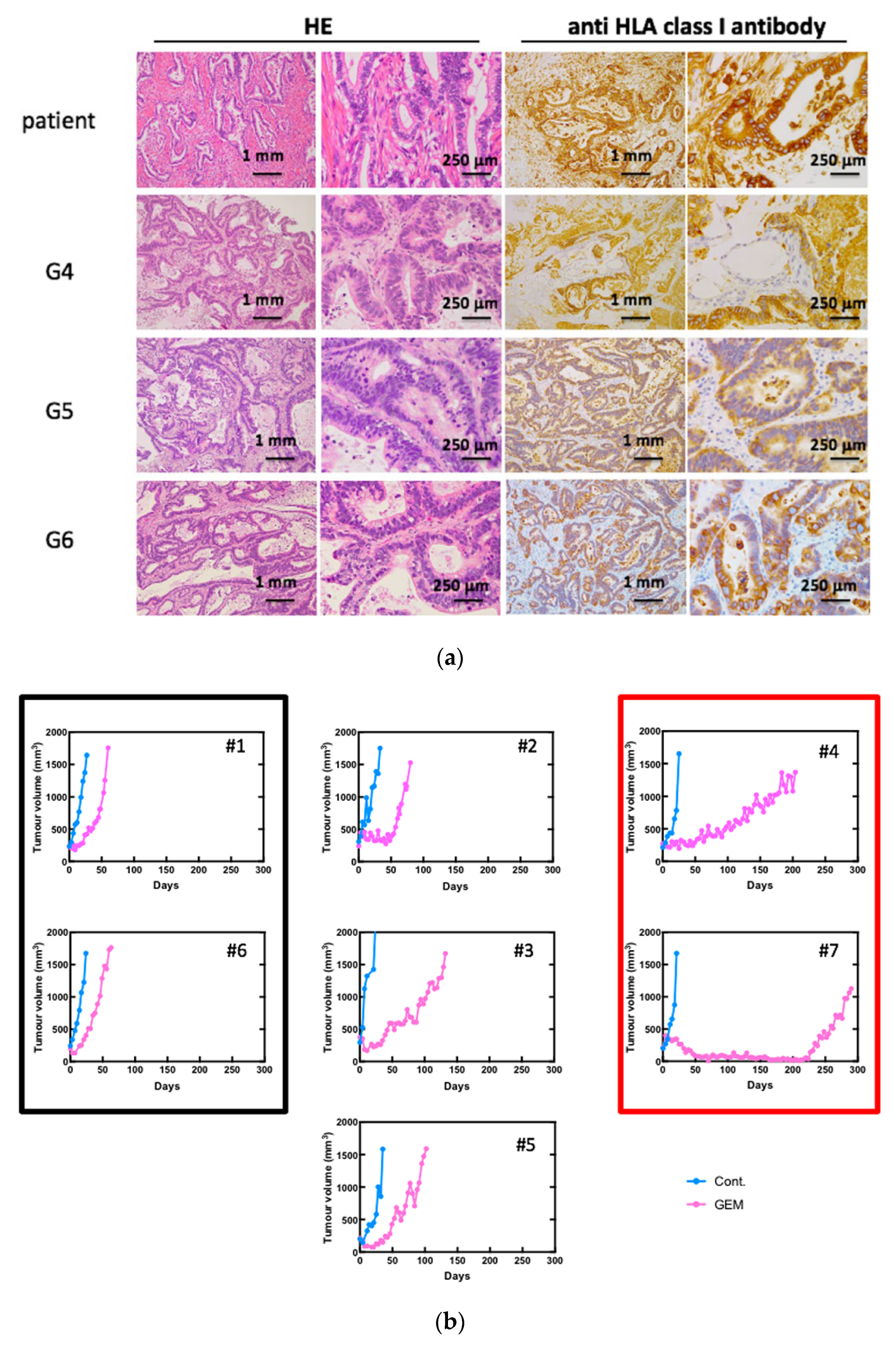
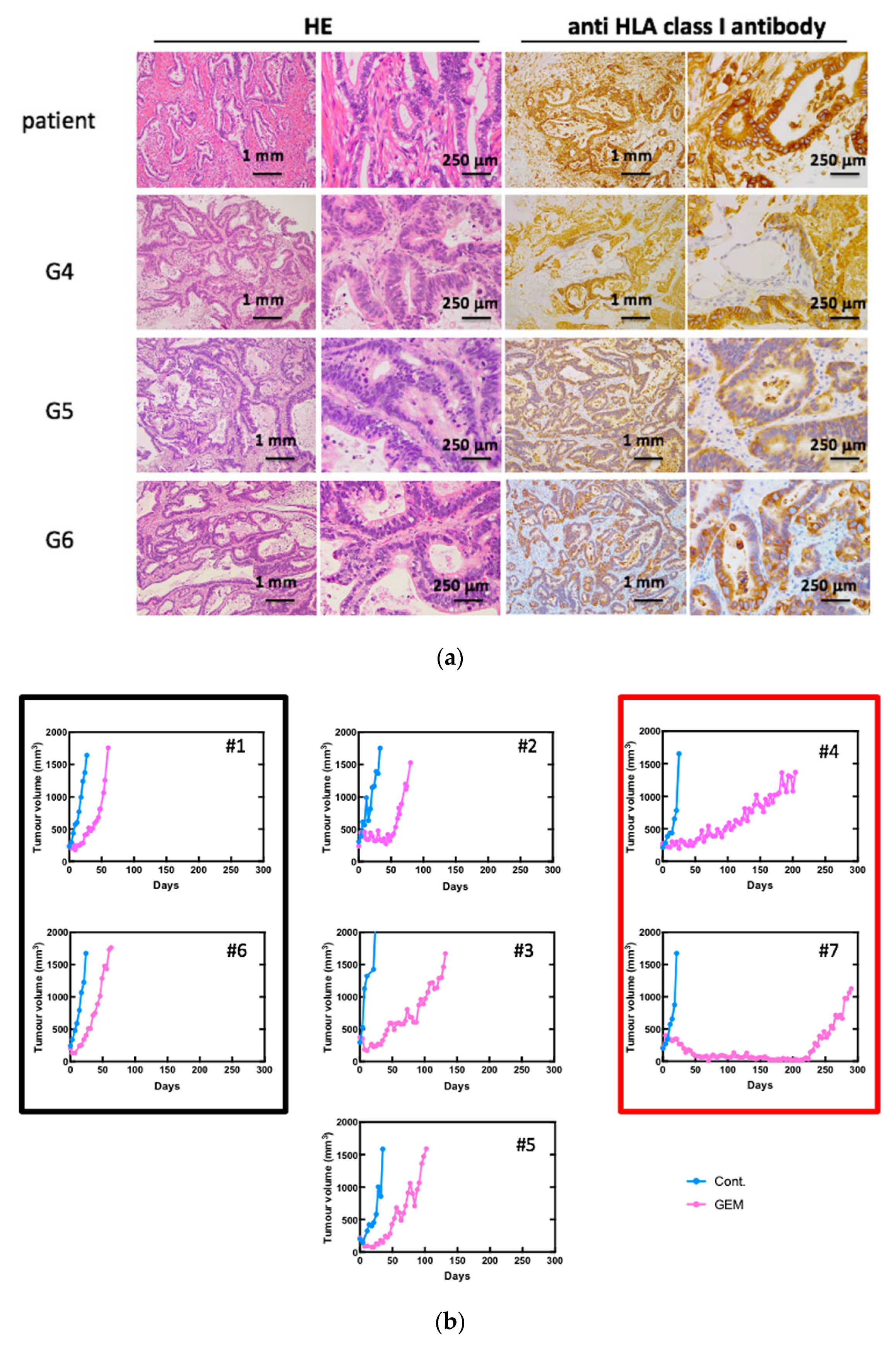
3.1. Establishment of Pancreatic Cancer PDXs and the Anti-Tumor Effects of GEM Treatment
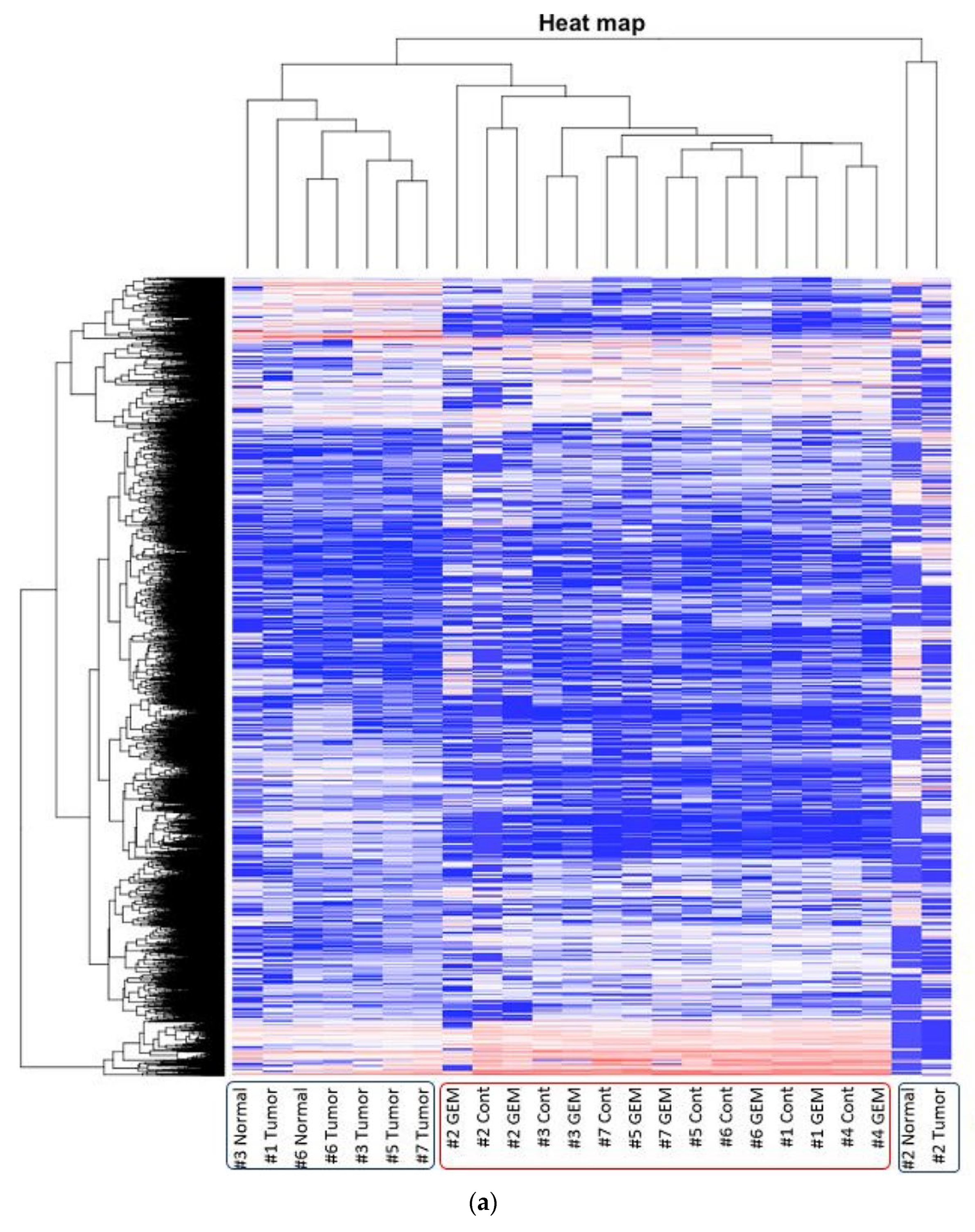
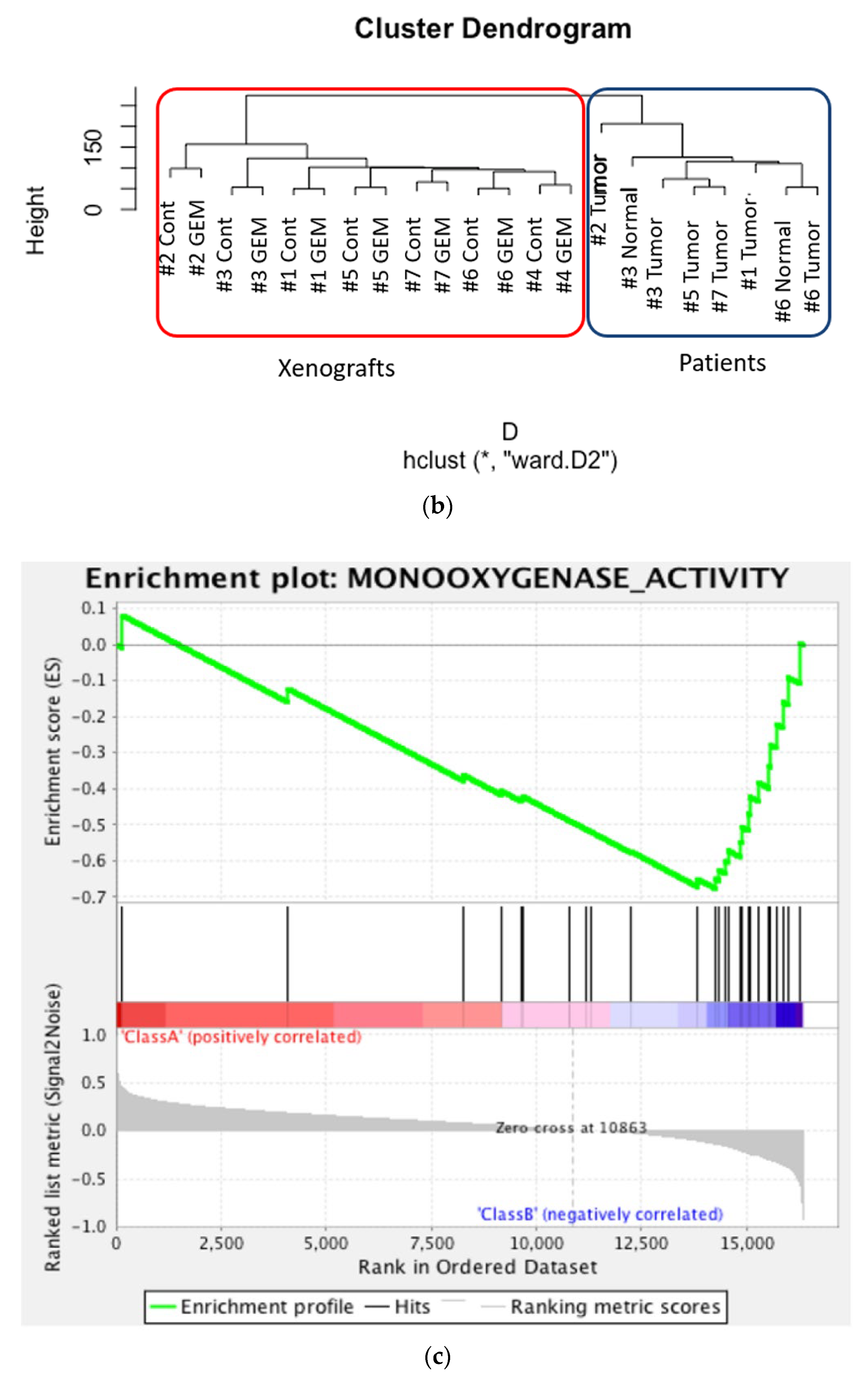
3.2. RNA Sequencing and Gene Pathway Analysis
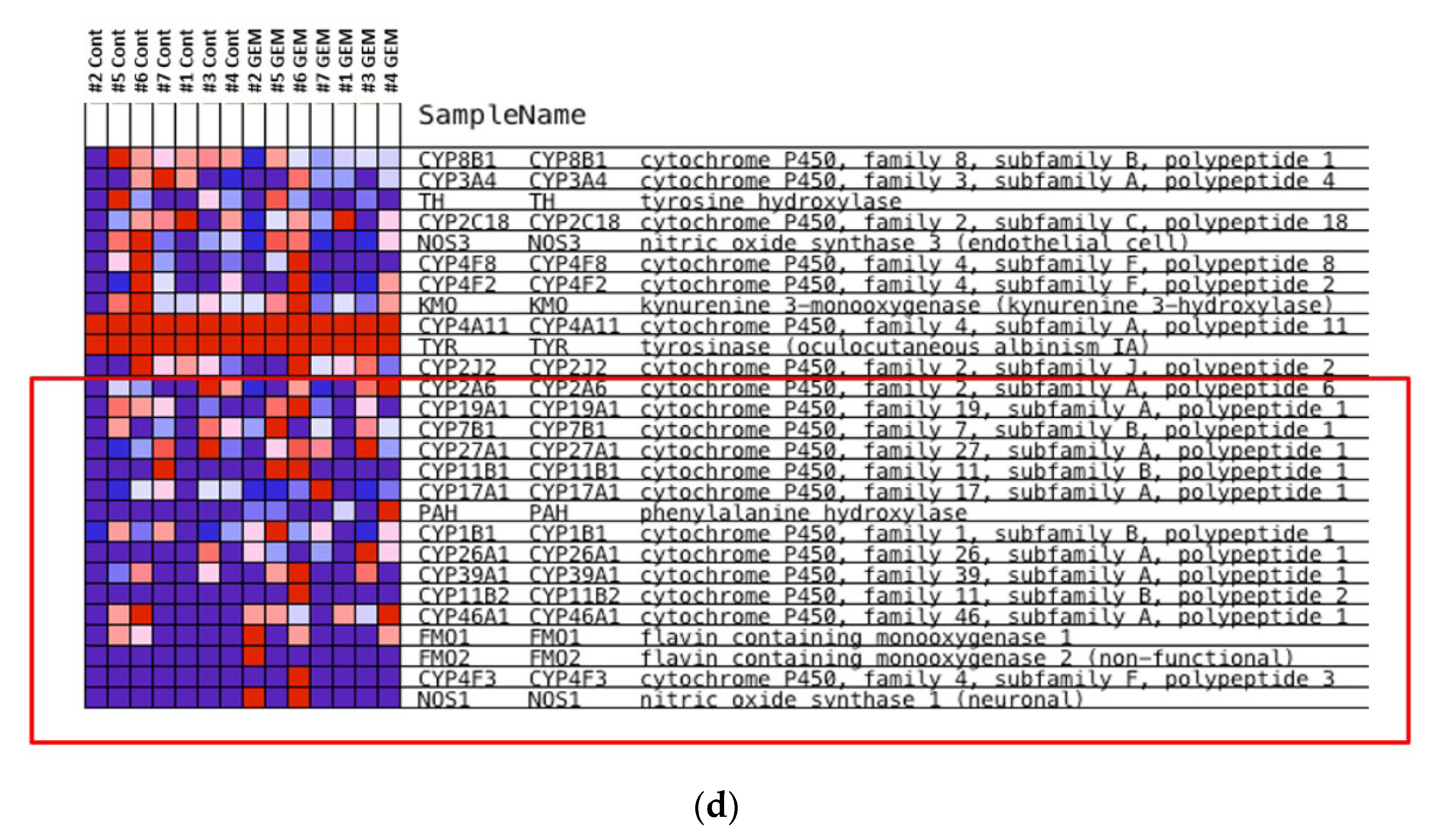
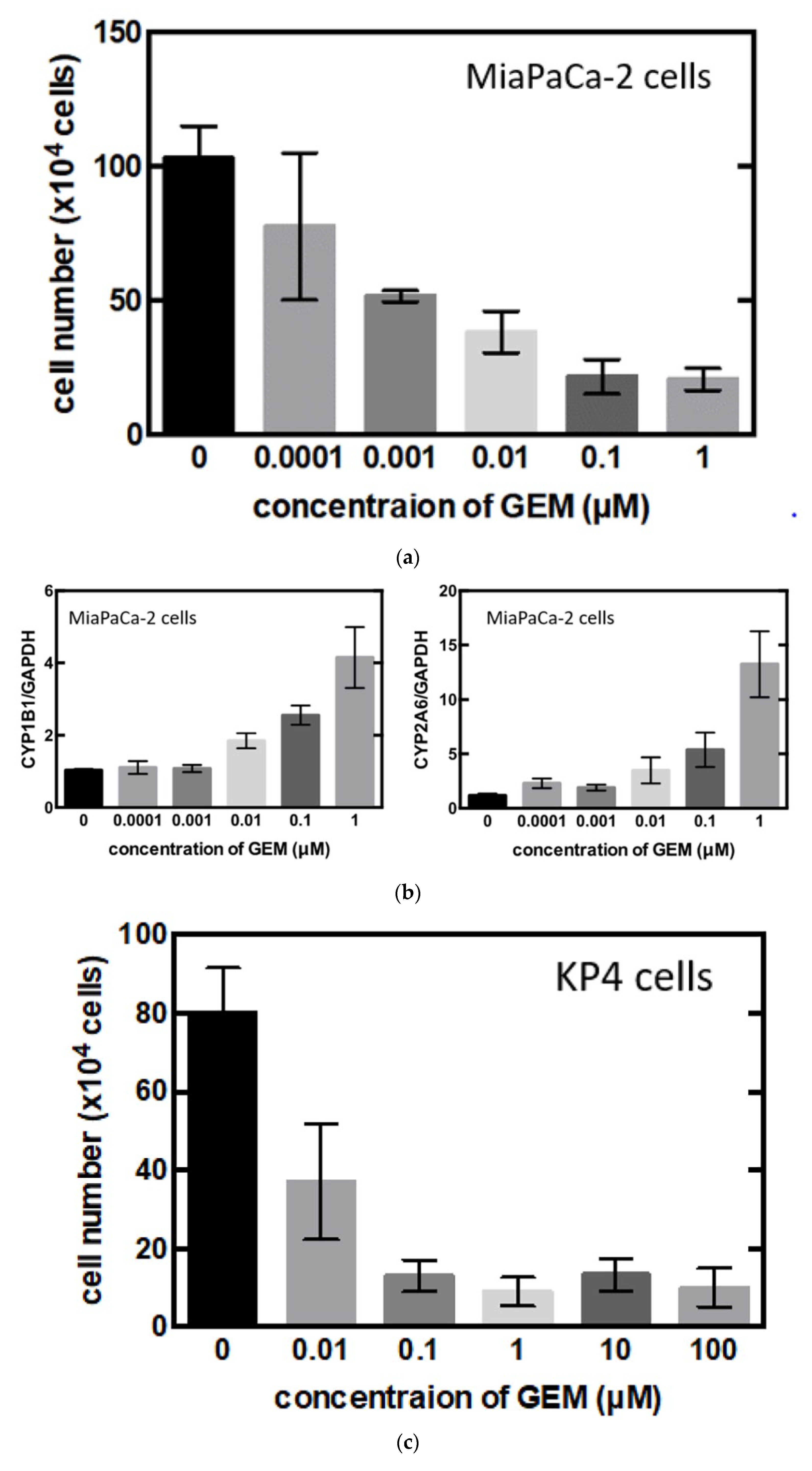
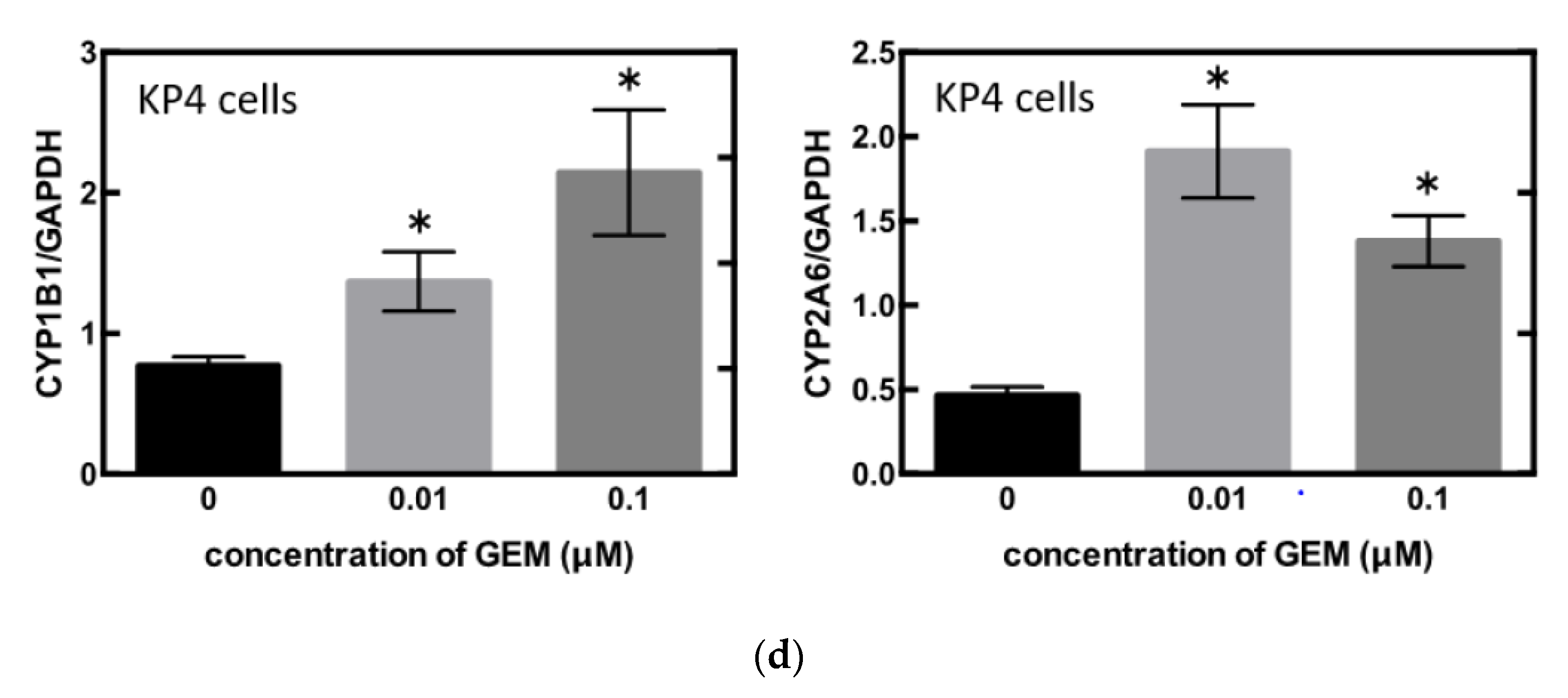
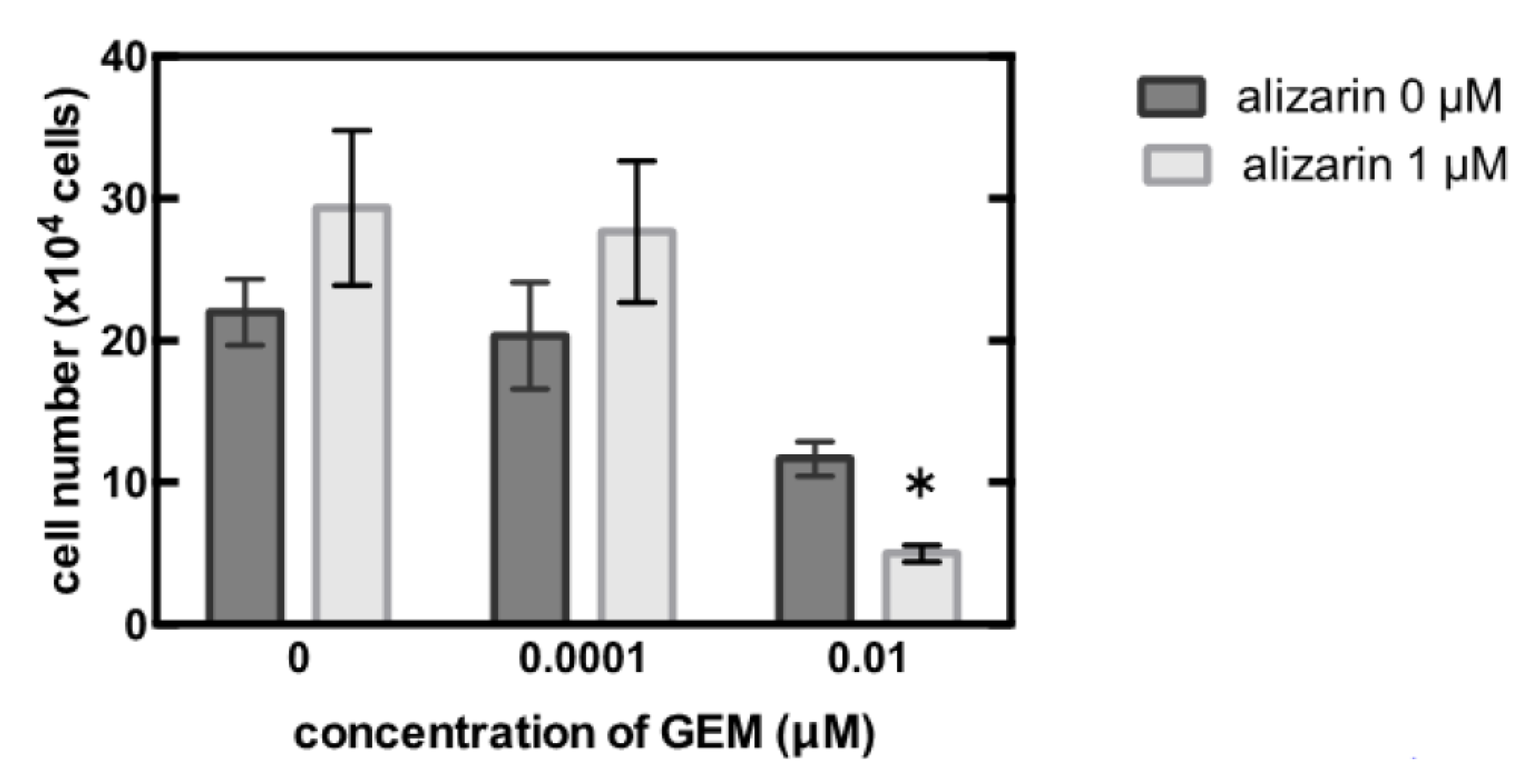
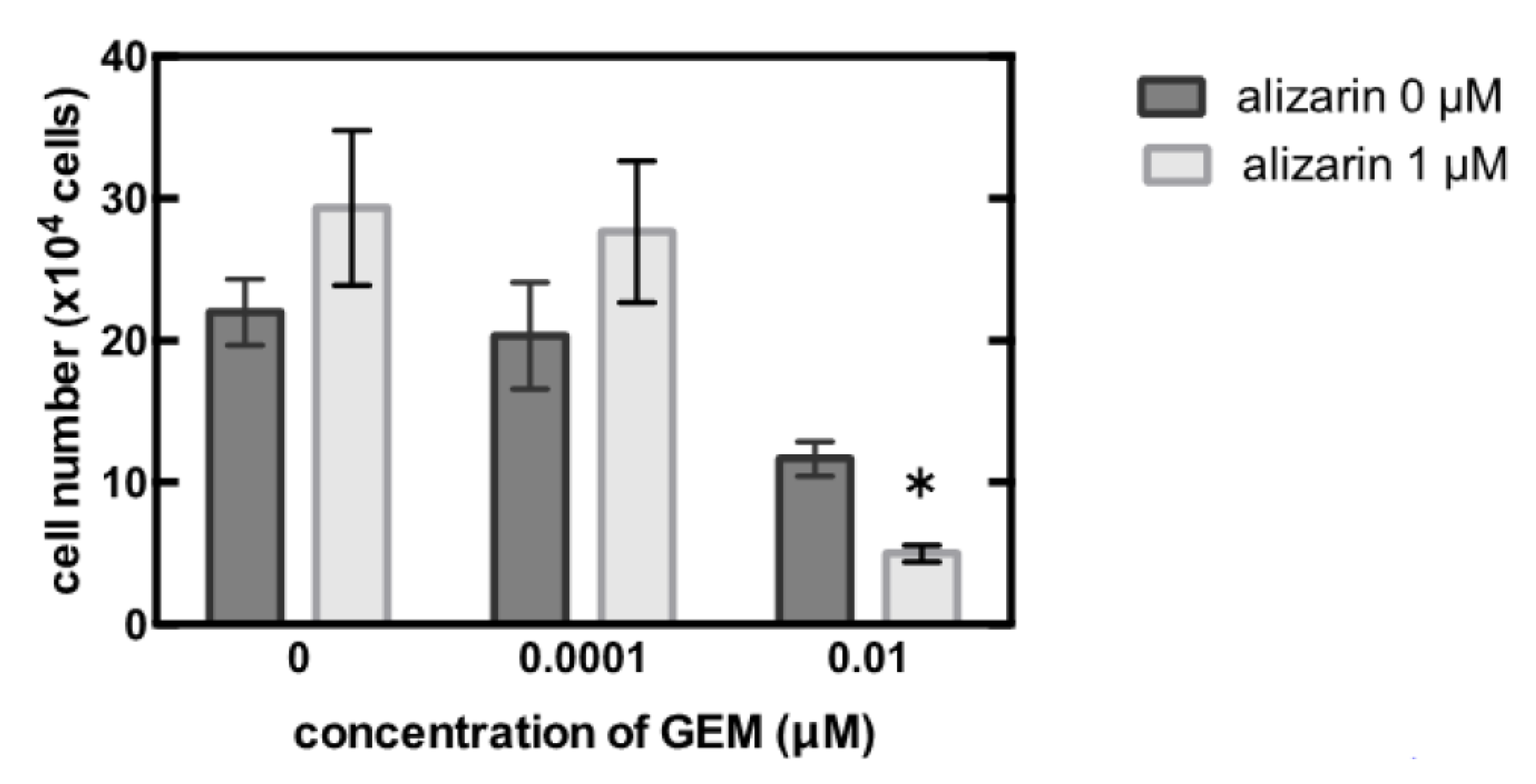
3.3. Identification of the Key Molecule of GEM Resistance in Pancreatic Cancer
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sausville, E.A.; Burger, A.M. Contributions of Human Tumor Xenografts to Anticancer Drug Development. Cancer Res. 2006, 66, 3351–3354; discussion 3354. [Google Scholar] [CrossRef] [Green Version]
- Yada, E.; Wada, S.; Yoshida, S.; Sasada, T. Use of Patient-Derived Xenograft Mouse Models in Cancer Research and Treatment. Future Sci. OA 2018, 4, FSO271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: A Randomized Trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1 Alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in Determining Chemo-Resistance in Pancreatic Cancer: Neighborhood Matters. Pancreatology 2017, 17, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Chijiwa, T.; Kawai, K.; Noguchi, A.; Sato, H.; Hayashi, A.; Cho, H.; Shiozawa, M.; Kishida, T.; Morinaga, S.; Yokose, T.; et al. Establishment of Patient-Derived Cancer Xenografts in Immunodeficient NOG Mice. Int. J. Oncol. 2015, 47, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of Orthotopic and Heterotopic Human Pancreatic Cancer Xenografts in Immunodeficient Mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G. Kingsford C. Salmon: Accurate, Versatile and Ultrafast Quantification from RNA-Seq Data Using Lightweight-Alignment. bioRxiv 2015, 9, 021592. [Google Scholar]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome-A Tool for Classifying Reads from Xenograft Samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Ohkuma, R.; Yada, E.; Ishikawa, S.; Komura, D.; Ishizaki, H.; Tamada, K.; Kubota, Y.; Hamada, K.; Ishida, H.; Hirasawa, Y.; et al. High Expression of olfactomedin-4 Is Correlated with Chemoresistance and Poor Prognosis in Pancreatic Cancer. PLoS ONE 2020, 15, e0226707. [Google Scholar] [CrossRef] [PubMed]
- Barton-Burke, M. Gemcitabine: A pharmacologic and clinical overview. Cancer Nurs. 1999, 22, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar] [PubMed]
- Gandhi, V.; Plunkett, W. Modulatory activity of 2′,2′-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990, 50, 3675–3680. [Google Scholar] [PubMed]
- Hertel, L.W.; Boder, G.B.; Kroin, J.S.; Rinzel, S.M.; Poore, G.A.; Todd, G.C.; Grindey, G.B. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [Google Scholar] [PubMed]
- Elfaki, I.; Mir, R.; Almutairi, F.M.; Duhier, F.M.A. Cytochrome P450: Polymorphisms and Roles in Cancer, Diabetes and Atherosclerosis. Asian Pac. J. Cancer Prev. 2018, 19, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical Importance of the Cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Porter, T.D.; Coon, M.J. Cytochrome P-450 Multiplicity of Isoforms, Substrates, and Catalytic and Regulatory Mechanisms. J. Biol. Chem. 1991, 266, 13469–13472. [Google Scholar] [CrossRef]
- Thelen, K.; Dressman, J.B. Cytochrome P450-Mediated Metabolism in the Human Gut Wall. J. Pharm. Pharmacol. 2009, 61, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Renaud, H.J.; Cui, J.Y.; Khan, M.; Klaassen, C.D. Tissue Distribution and Gender-Divergent Expression of 78 Cytochrome P450 mRNAs in Mice. Toxicol. Sci. 2011, 124, 261–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.W.; Kam, P.C. The Physiological and Pharmacological Roles of Cytochrome P450 Isoenzymes. Anaesthesia 1999, 54, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Sutter, T.R.; Tang, Y.M.; Hayes, C.L.; Wo, Y.Y.; Jabs, E.W.; Li, X.; Yin, H.; Cody, C.W.; Greenlee, W.F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [Google Scholar] [CrossRef]
- Murray, G.I.; Melvin, W.T.; Greenlee, W.F.; Burke, M.D. Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Kyo, S.; Kanaya, T.; Inoue, M.; Yokoi, T. Human CYP1B1 is regulated by estradiol via estrogen receptor. Cancer Res. 2004, 64, 3119–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zordoky, B.N.; El-Kadi, A.O. Role of NF-kappaB in the regulation of cytochrome P450 enzymes. Curr. Drug Metab. 2009, 10, 164–178. [Google Scholar] [CrossRef]
- Theken, K.N.; Deng, Y.; Kannon, M.A.; Miller, T.M.; Poloyac, S.M.; Lee, C.R. Activation of the acute inflammatory response alters cytochrome P450 expression and eicosanoid metabolism. Drug Metab. Dispos. 2011, 39, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Smerdová, L.; Šmerdová, J.; Kabátková, M.; Kohoutek, J.; Blažek, D.; Machala, M.; Vondráček, J. Upregulation of CYP1B1 expression by inflammatory cytokines is mediated by the p38 MAP kinase signal transduction pathway. Carcinogenesis 2014, 35, 2534–3543. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, N.; Awwad, K.; Fisslthaler, B.; Reis, M.; Devraj, K.; Corada, M.; Minardi, S.P.; Dejana, E.; Plate, K.H.; Fleming, I.; et al. β-Catenin Is Required for Endothelial Cyp1b1 Regulation Influencing Metabolic Barrier Function. J. Neurosci. 2016, 36, 8921–8935. [Google Scholar] [CrossRef] [PubMed]
- Muskhelishvili, L.; Thompson, P.A.; Kusewitt, D.F.; Wang, C.; Kadlubar, F.F. In situ hybridization and immunohistochemical analysis of cytochrome P450 1B1 expression in human normal tissues. J. Histochem. Cytochem. 2001, 49, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.I.; Taylor, M.C.; McFadyen, M.C.; McKay, J.A.; Greenlee, W.F.; Burke, M.D.; Melvin, W.T. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997, 57, 3026–3031. [Google Scholar]
- Takahashi, E.; Fujita, K.; Kamataki, T.; Arimoto-Kobayashi, S.; Okamoto, K.; Negishi, T. Inhibition of Human Cytochrome P450 1B1, 1A1 and 1A2 by Antigenotoxic Compounds, Purpurin and Alizarin. Mutat. Res. 2002, 508, 147–156. [Google Scholar] [CrossRef]
- Chun, Y.J.; Kim, S.; Kim, D.; Lee, S.K.; Guengerich, F.P. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001, 61, 8164–8170. [Google Scholar] [PubMed]
- Zhu, Z.; Mu, Y.; Qi, C.; Wang, J.; Xi, G.; Guo, J.; Mi, R.; Zhao, F. CYP1B1 Enhances the Resistance of Epithelial Ovarian Cancer Cells to Paclitaxel In Vivo and In Vitro. Int. J. Mol. Med. 2015, 35, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadyen, M.C.; McLeod, H.L.; Jackson, F.C.; Melvin, W.T.; Doehmer, J.; Murray, G.I. Cytochrome P450 CYP1B1 Protein Expression: A Novel Mechanism of Anticancer Drug Resistance. Biochem. Pharmacol. 2001, 62, 207–212. [Google Scholar] [CrossRef]
- Martinez, V.G.; O’Connor, R.; Liang, Y.; Clynes, M. CYP1B1 expression is induced by docetaxel: Effect on cell viability and drug resistance. Br. J. Cancer 2008, 98, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Tu, Z.; Xiong, J.; Kang, G.; Zhao, L.; Hu, W.; Tan, H.; Tembo, K.M.; Ding, Q.; Deng, X.; et al. CXCR4 Promotes Cisplatin-Resistance of Non-Small Cell Lung Cancer in a CYP1B1-Dependent Manner. Oncol. Rep. 2017, 37, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikstacka, R.; Dutkiewicz, Z. New Perspectives of CYP1B1 Inhibitors in the Licht of Molecular Studies. Processes 2021, 9, 817. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Size | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value | Rank at Max | Leading Edge |
|---|---|---|---|---|---|---|---|---|
| ANION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 56 | −0.4869358 | −2.101442 | 0 | 0.033455465 | 0.051 | 2391 | tags = 39%, list = 15%, signal = 46% |
| EXTRACELLULAR_MATRIX_STRUCTURAL_CONSTITUENT | 26 | −0.58796436 | −2.0886946 | 0 | 0.024774604 | 0.057 | 2216 | tags = 46%, list = 14%, signal = 53% |
| COLLAGEN | 23 | −0.603812 | −2.0409071 | 0 | 0.028811615 | 0.085 | 1935 | tags = 43%, list = 12%, signal = 49% |
| INORGANIC_ANION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 19 | −0.54800725 | −1.8485612 | 0.006849315 | 0.13804246 | 0.414 | 1759 | tags = 37%, list = 11%, signal = 41% |
| PEPTIDE_RECEPTOR_ACTIVITY | 44 | −0.4471173 | −1.7894889 | 0.003921569 | 0.19005722 | 0.582 | 1946 | tags = 36%, list = 12%, signal = 41% |
| ANION_CATION_SYMPORTER_ACTIVITY | 16 | −0.56837684 | −1.7396722 | 0.021341464 | 0.24313329 | 0.713 | 1378 | tags = 38%, list = 8%, signal = 41% |
| SULFOTRANSFERASE_ACTIVITY | 22 | −0.49270993 | −1.701363 | 0.009646302 | 0.27869517 | 0.805 | 1937 | tags = 36%, list = 12%, signal = 41% |
| PROTEOGLYCAN_METABOLIC_PROCESS | 17 | −0.53180295 | −1.6986526 | 0.01618123 | 0.25181675 | 0.811 | 1768 | tags = 29%, list = 11%, signal = 33% |
| LIGAND_GATED_CHANNEL_ACTIVITY | 35 | −0.44609684 | −1.6825303 | 0 | 0.25340274 | 0.833 | 1930 | tags = 34%, list = 12%, signal = 39% |
| ION_HOMEOSTASIS | 115 | −0.34891573 | −1.6815822 | 0 | 0.23189601 | 0.834 | 2002 | tags = 28%, list = 12%, signal = 31% |
| HORMONE_METABOLIC_PROCESS | 29 | −0.4490808 | −1.6701156 | 0.015444015 | 0.23098724 | 0.863 | 1011 | tags = 24%, list = 6%, signal = 26% |
| CORTICAL_CYTOSKELETON | 18 | −0.5224792 | −1.6676438 | 0.015625 | 0.21772708 | 0.869 | 2712 | tags = 44%, list = 17%, signal = 53% |
| HEPARIN_BINDING | 20 | −0.5068244 | −1.6632581 | 0.014492754 | 0.20953959 | 0.877 | 1576 | tags = 40%, list = 10%, signal = 44% |
| AMINO_ACID_TRANSPORT | 25 | −0.47122175 | −1.658062 | 0.013793103 | 0.20178199 | 0.885 | 1581 | tags = 28%, list = 10%, signal = 31% |
| TRANSFERASE_ACTIVITY_TRANSFERRING_SULFUR_CONTAINING_GROUPS | 26 | −0.46915933 | −1.6546559 | 0.010830325 | 0.19338286 | 0.891 | 1937 | tags = 31%, list = 12%, signal = 35% |
| EXTRACELLULAR_MATRIX_PART | 52 | −0.39951557 | −1.6543405 | 0.004149378 | 0.18208581 | 0.891 | 1935 | tags = 31%, list = 12%, signal = 35% |
| ION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 263 | −0.30467132 | −1.6533009 | 0 | 0.1726972 | 0.892 | 2264 | tags = 26%, list = 14%, signal = 30% |
| AMINE_TRANSPORT | 34 | −0.42308894 | −1.6444278 | 0.01171875 | 0.17343472 | 0.902 | 1581 | tags = 29%, list = 10%, signal = 32% |
| POLYSACCHARIDE_BINDING | 29 | −0.45253384 | −1.633435 | 0.014981274 | 0.17863564 | 0.921 | 1576 | tags = 34%, list = 10%, signal = 38% |
| CHEMICAL_HOMEOSTASIS | 138 | −0.33011207 | −1.6284368 | 0 | 0.17518602 | 0.931 | 2002 | tags = 28%, list = 12%, signal = 31% |
| SYNAPTOGENESIS | 18 | −0.5083024 | −1.6254768 | 0.013745705 | 0.17086776 | 0.932 | 2646 | tags = 33%, list = 16%, signal = 40% |
| EXTRACELLULAR_MATRIX | 91 | −0.34340963 | −1.6218148 | 0 | 0.1676017 | 0.933 | 2251 | tags = 32%, list = 14%, signal = 37% |
| PROTEINACEOUS_EXTRACELLULAR_MATRIX | 90 | −0.3495134 | −1.6129957 | 0 | 0.17004865 | 0.946 | 2251 | tags = 32%, list = 14%, signal = 37% |
| NEUROTRANSMITTER_BINDING | 49 | −0.3874927 | −1.6098273 | 0.015444015 | 0.16672221 | 0.95 | 1913 | tags = 31%, list = 12%, signal = 35% |
| STEROID_BIOSYNTHETIC_PROCESS | 22 | −0.4757495 | −1.6048489 | 0.032467533 | 0.16537002 | 0.956 | 2297 | tags = 45%, list = 14%, signal = 53% |
| G_PROTEIN_COUPLED_RECEPTOR_ACTIVITY | 170 | −0.3093993 | −1.5869783 | 0 | 0.18080884 | 0.975 | 2000 | tags = 25%, list = 12%, signal = 29% |
| PATTERN_BINDING | 35 | −0.40934092 | −1.5851388 | 0.010830325 | 0.17625666 | 0.977 | 2241 | tags = 37%, list = 14%, signal = 43% |
| REGULATION_OF_BODY_FLUID_LEVELS | 55 | −0.36799893 | −1.5495309 | 0.00952381 | 0.22113751 | 0.993 | 2331 | tags = 31%, list = 14%, signal = 36% |
| GLYCOSAMINOGLYCAN_BINDING | 27 | −0.42942247 | −1.5400821 | 0.02734375 | 0.22773191 | 0.994 | 1576 | tags = 33%, list = 10%, signal = 37% |
| NEUROTRANSMITTER_RECEPTOR_ACTIVITY | 46 | −0.38586268 | −1.5308675 | 0.023809524 | 0.234069 | 0.996 | 1913 | tags = 30%, list = 12%, signal = 34% |
| GLUTAMATE_RECEPTOR_ACTIVITY | 18 | −0.47083804 | −1.527544 | 0.038709678 | 0.2313607 | 0.997 | 705 | tags = 22%, list = 4%, signal = 23% |
| CATION_HOMEOSTASIS | 98 | −0.31595644 | −1.5160195 | 0.011428571 | 0.24062736 | 0.997 | 2002 | tags = 27%, list = 12%, signal = 30% |
| ACETYLCHOLINE_BINDING | 17 | −0.47908843 | −1.5119975 | 0.04452055 | 0.23989336 | 0.997 | 2446 | tags = 41%, list = 15%, signal = 48% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yada, E.; Kasajima, R.; Niida, A.; Imoto, S.; Miyano, S.; Miyagi, Y.; Sasada, T.; Wada, S. Possible Role of Cytochrome P450 1B1 in the Mechanism of Gemcitabine Resistance in Pancreatic Cancer. Biomedicines 2021, 9, 1396. https://doi.org/10.3390/biomedicines9101396
Yada E, Kasajima R, Niida A, Imoto S, Miyano S, Miyagi Y, Sasada T, Wada S. Possible Role of Cytochrome P450 1B1 in the Mechanism of Gemcitabine Resistance in Pancreatic Cancer. Biomedicines. 2021; 9(10):1396. https://doi.org/10.3390/biomedicines9101396
Chicago/Turabian StyleYada, Erica, Rika Kasajima, Atsushi Niida, Seiya Imoto, Satoru Miyano, Yohei Miyagi, Tetsuro Sasada, and Satoshi Wada. 2021. "Possible Role of Cytochrome P450 1B1 in the Mechanism of Gemcitabine Resistance in Pancreatic Cancer" Biomedicines 9, no. 10: 1396. https://doi.org/10.3390/biomedicines9101396
APA StyleYada, E., Kasajima, R., Niida, A., Imoto, S., Miyano, S., Miyagi, Y., Sasada, T., & Wada, S. (2021). Possible Role of Cytochrome P450 1B1 in the Mechanism of Gemcitabine Resistance in Pancreatic Cancer. Biomedicines, 9(10), 1396. https://doi.org/10.3390/biomedicines9101396