Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals

 , , , ,
, , , ,
Abstract
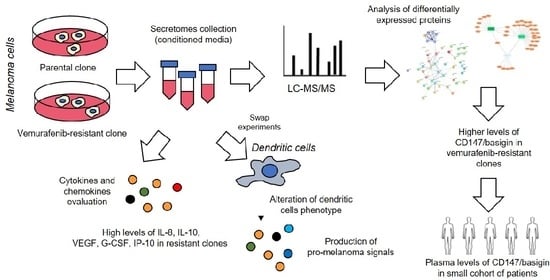
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Generation of Vemurafenib-Resistant Cell Lines
2.3. Expression Levels of Cytokines/Chemokines in Conditioned Medium
2.4. Generation of Dendritic Cells and Swap Experiments
2.5. FACS Analysis
2.6. Collection of Cell Line Secretome, Mass Spectrometry, and Protein Identification
2.7. Bioinformatics Analysis
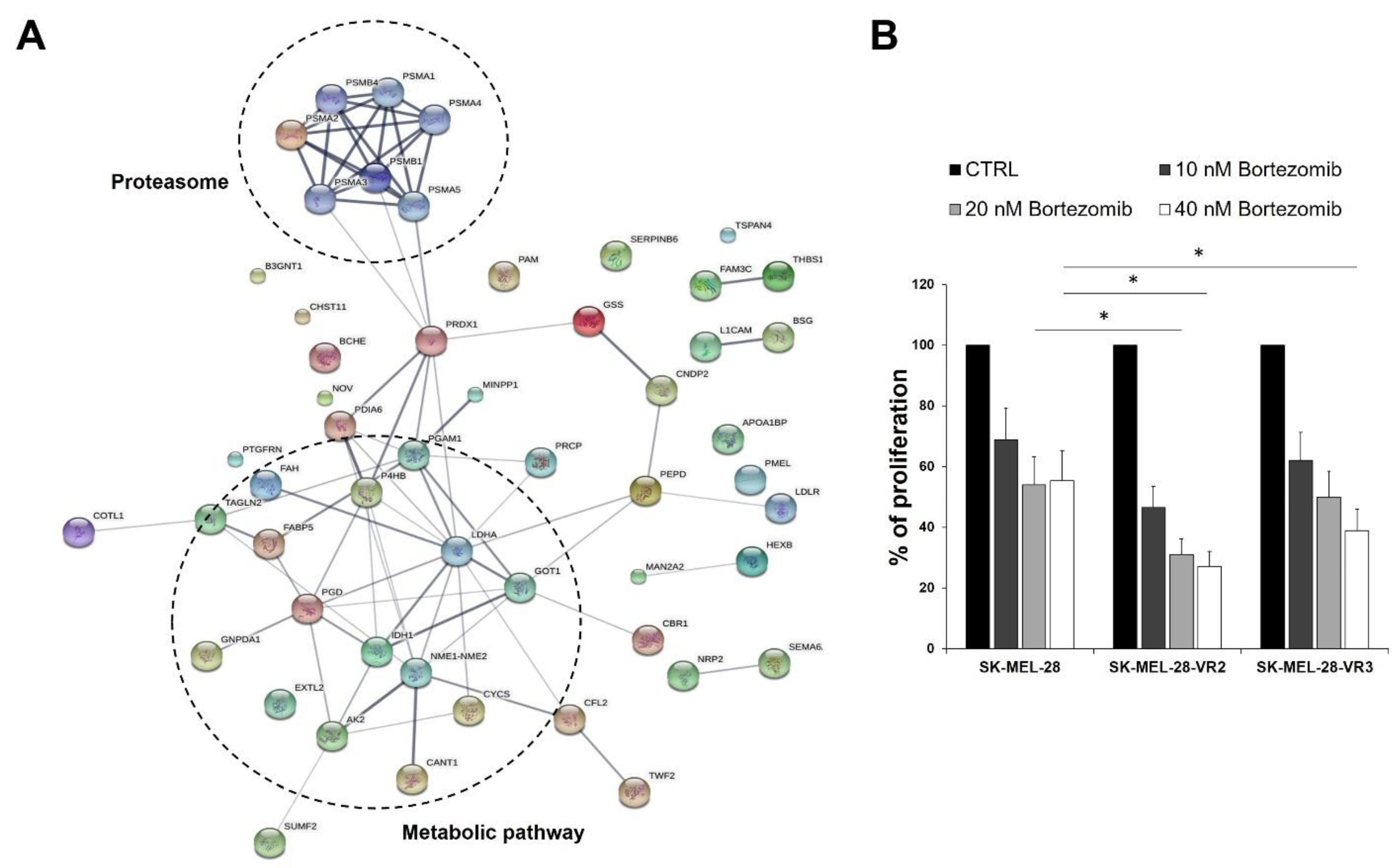
2.8. Evaluation of Melanoma Cell Sensitivity to Bortezomib, Adhesion Assay, and Determination of CD147/Basigin and MMP-2
2.9. Patients
2.10. Plasma Preparation
2.11. Statistical Analysis
3. Results
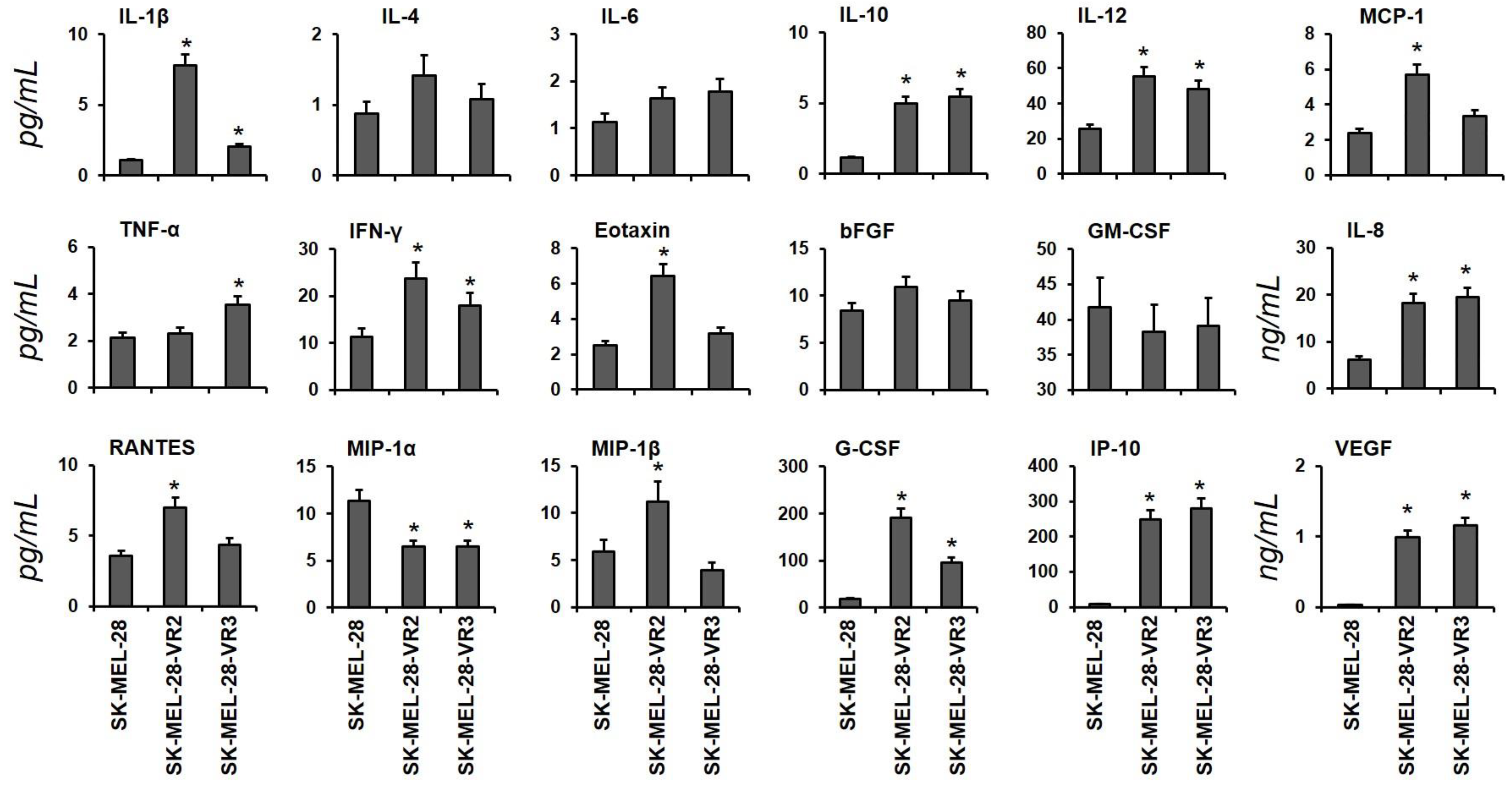
3.1. Identification of Cytokines in Conditioned Media from Vemurafenib-Resistant Cells
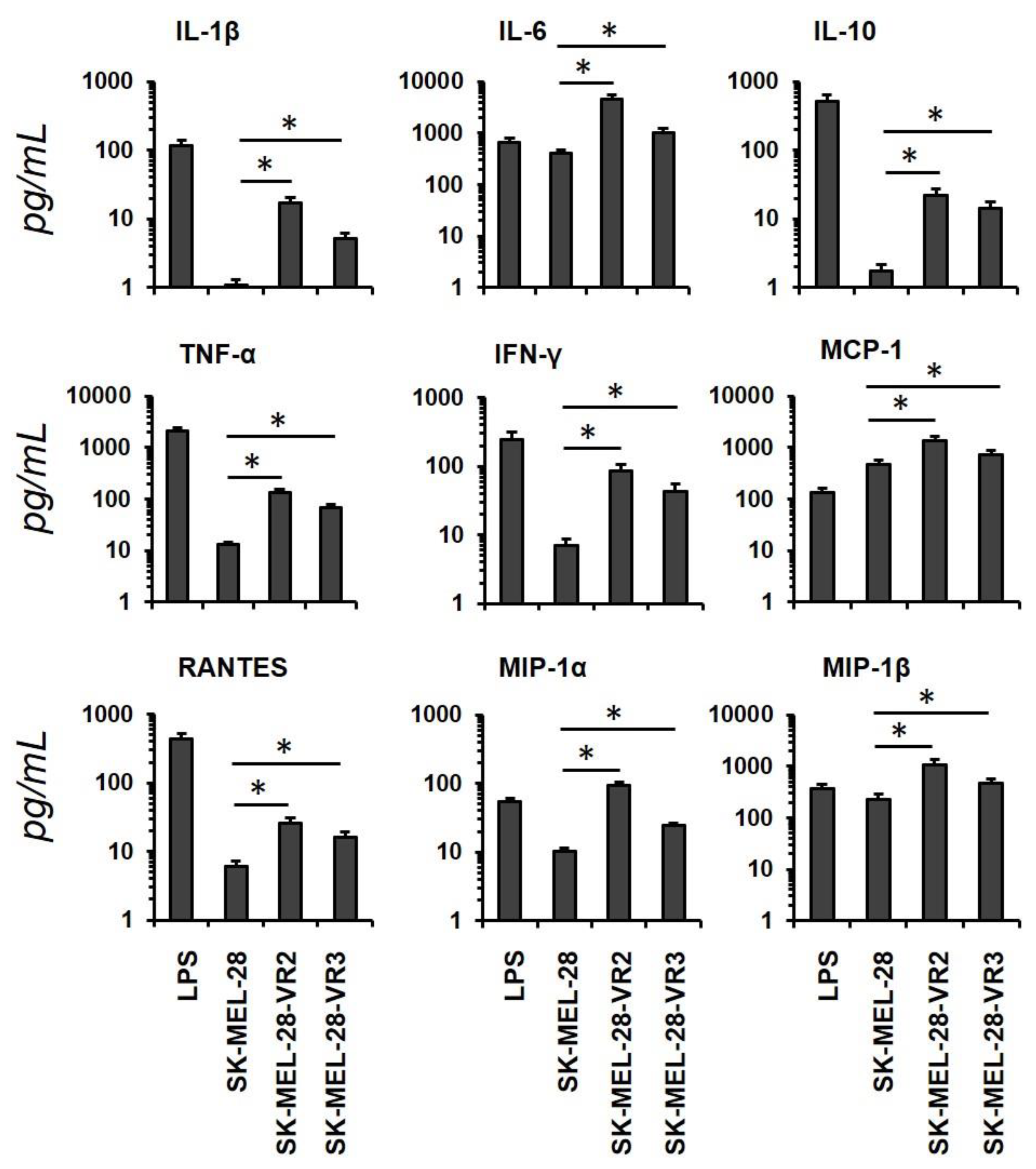
3.2. Conditioned Media from Resistant Cells Alter Dendritic Cells Phenotype and Cytokines/Chemokine Secretion Pattern
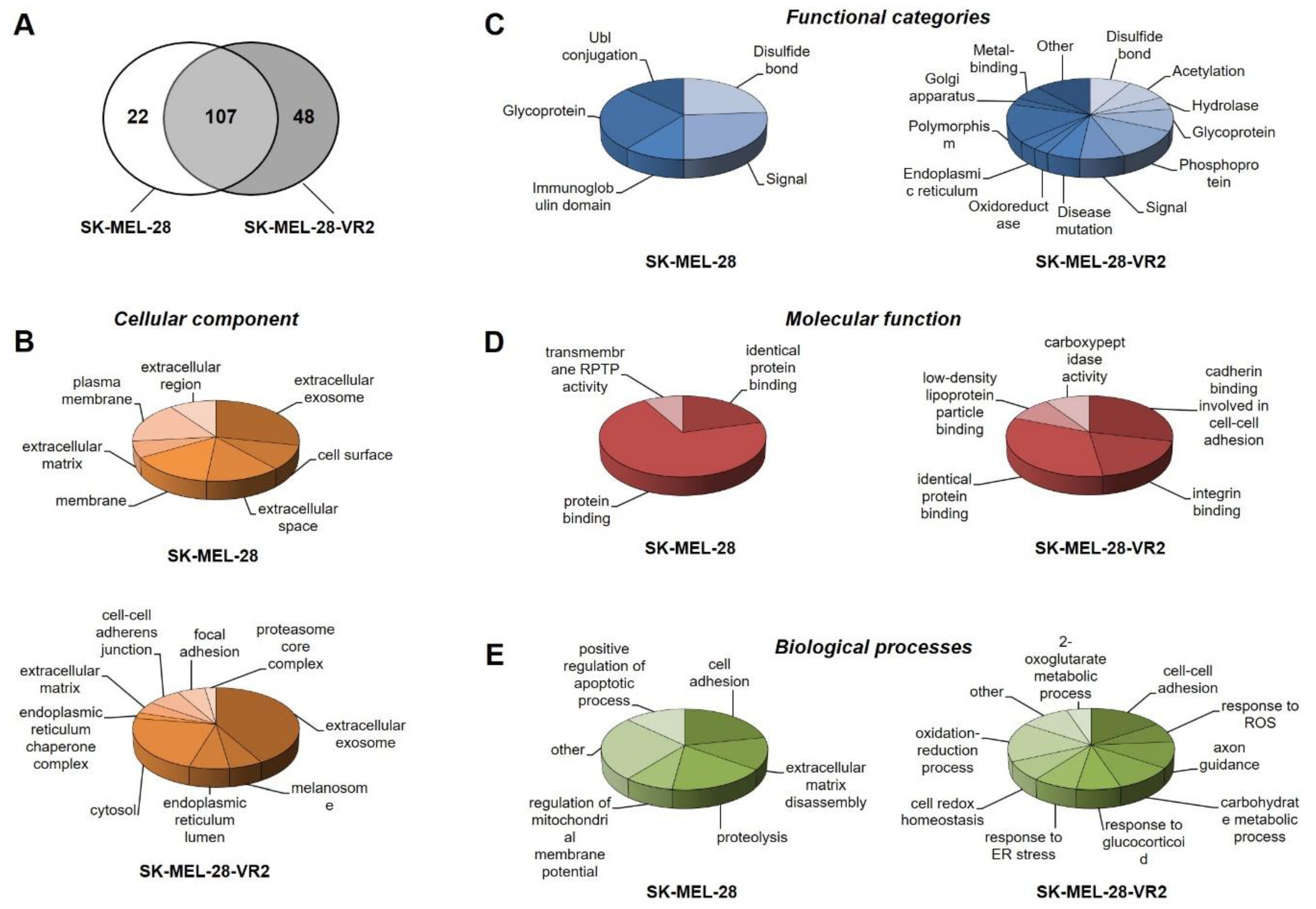
3.3. Secretome Profiling of Vemurafenib-Resistant Cells
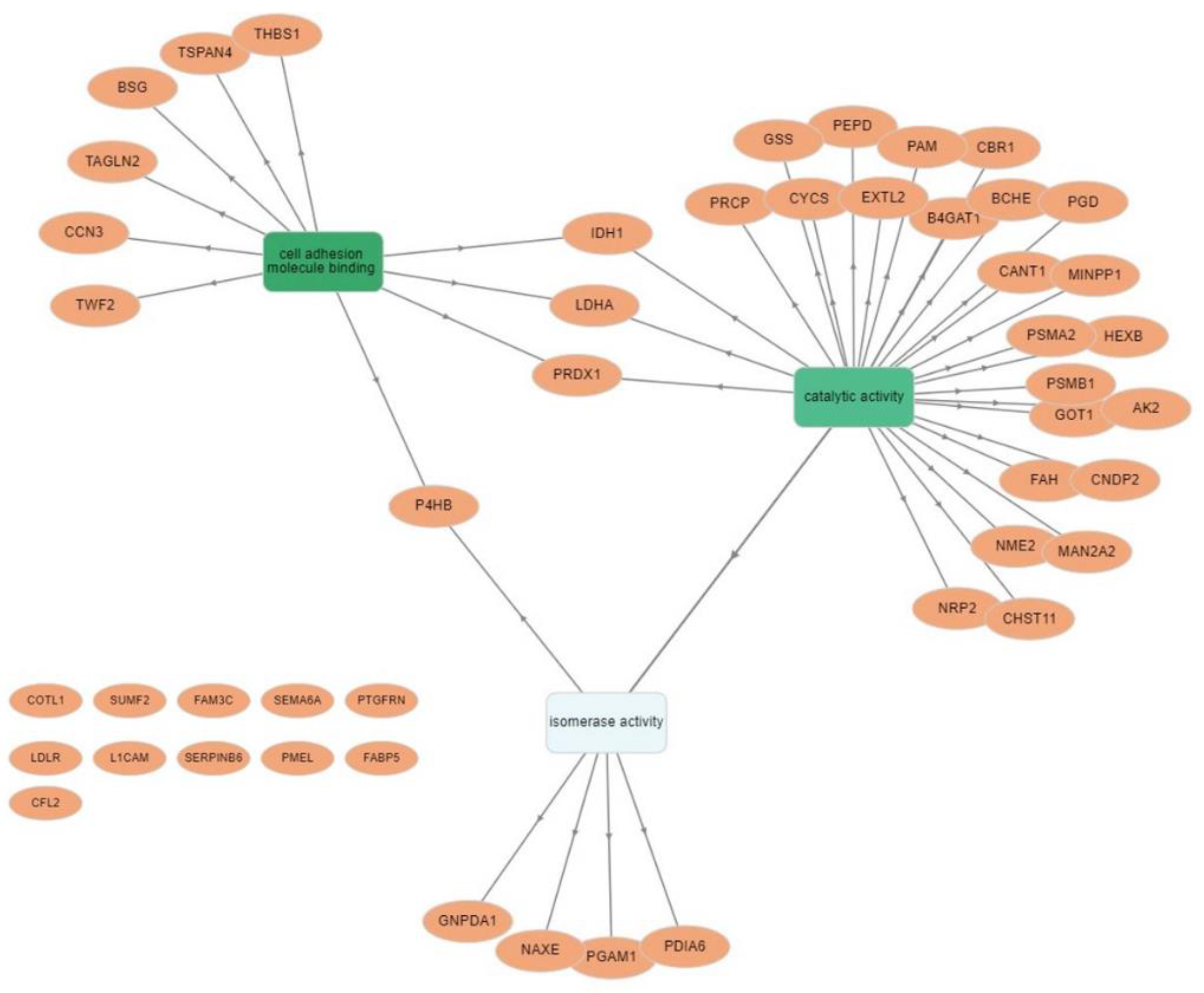
3.4. Protein–Protein Interaction Network Analysis
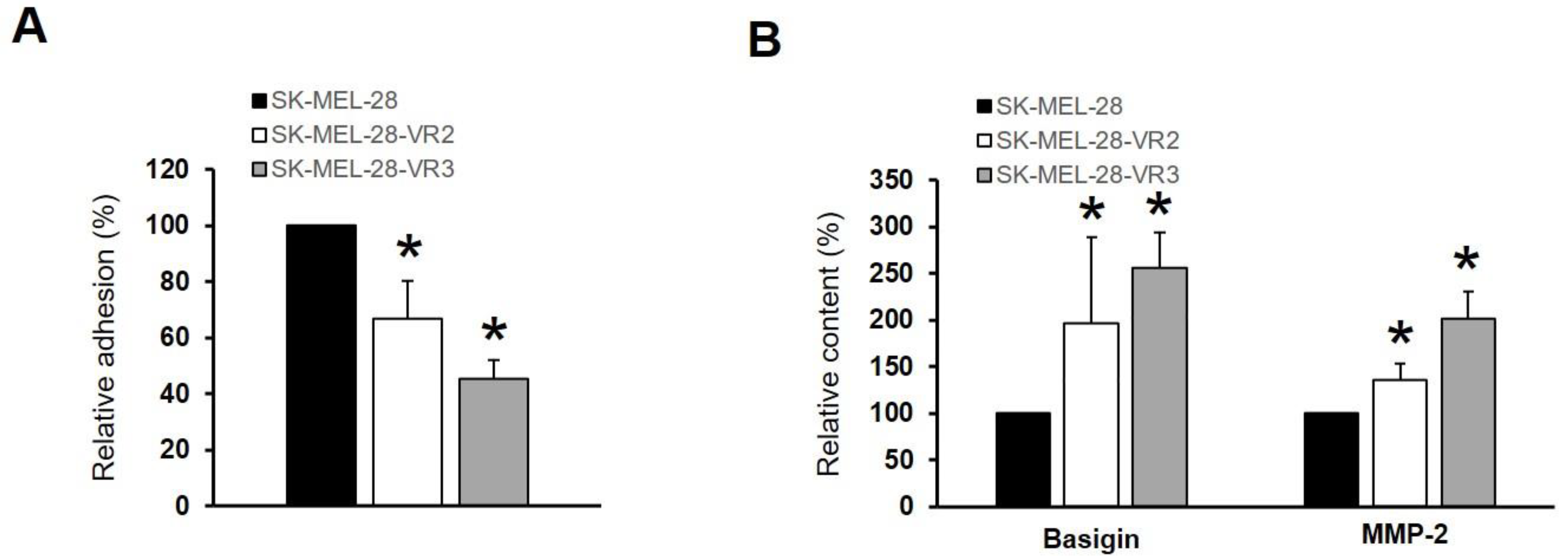
3.5. CD147/Basigin Expression in Vemurafenib-Resistant Cells
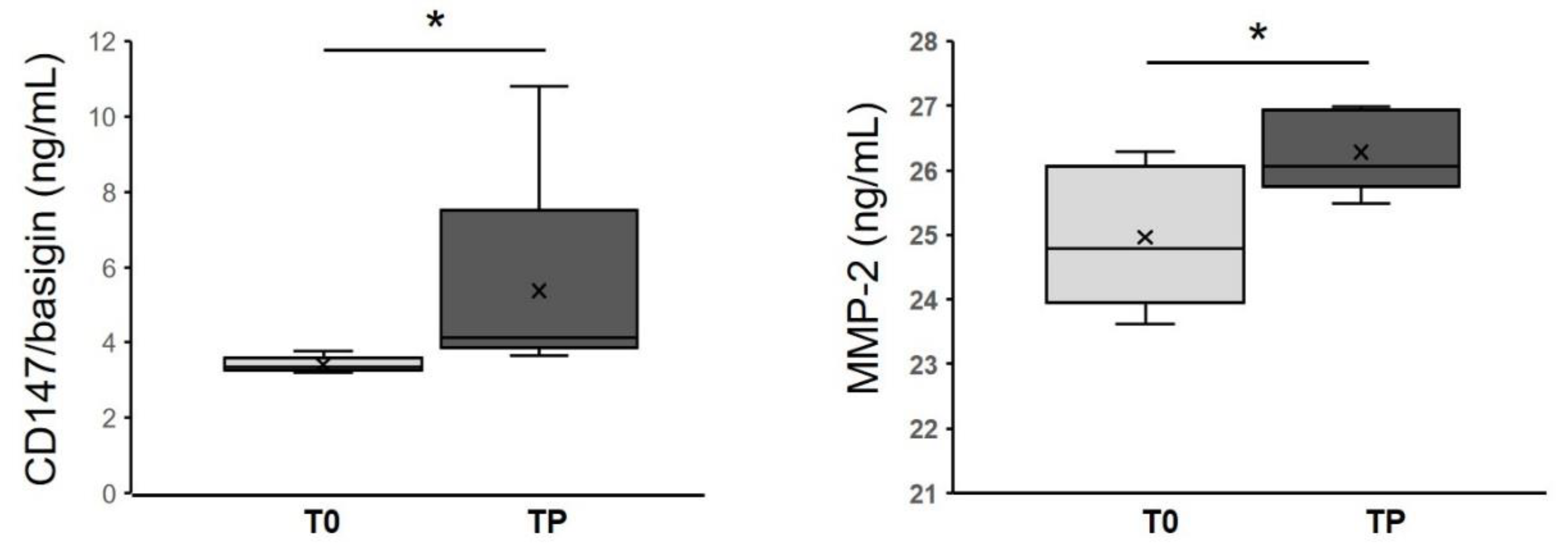
3.6. CD147/Basigin and MMP-2 Levels in Melanoma Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motofei, I.G. Malignant Melanoma: Autoimmunity and Supracellular Messaging as New Therapeutic Approaches. Curr. Treat. Options Oncol. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Alqathama, A. BRAF in malignant melanoma progression and metastasis: Potentials and challenges. Am. J. Cancer Res. 2020, 10, 1103–1114. [Google Scholar]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Re-sistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Redmond, K.L.; Papafili, A.; Lawler, M.; Van Schaeybroeck, S. Overcoming Resistance to Targeted Therapies in Cancer. Semin. Oncol. 2015, 42, 896–908. [Google Scholar] [CrossRef]
- Zhang, W. BRAF inhibitors: The current and the future. Curr. Opin. Pharmacol. 2015, 23, 68–73. [Google Scholar] [CrossRef]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug resistance of BRAF-mutant melano-ma: Review of up-to-date mechanisms of action and promising targeted agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Immunogenic, cellular, and angiogenic drivers of tumor dormancy-a melanoma view. Pigment. Cell Melanoma Res. 2015, 29, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Janco, J.M.T.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Stucci, S.; Passarelli, A.; Giudice, G.; Dammacco, F.; Silvestris, F. The immune escape in melanoma: Role of the impaired dendritic cell function. Expert Rev. Clin. Immunol. 2014, 10, 1395–1404. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef]
- Pal, H.C.; Sharma, S.; Strickland, L.R.; Katiyar, S.K.; Ballestas, M.E.; Athar, M.; Elmets, C.A.; Afaq, F. Fisetin inhibits hu-man melanoma cell invasion through promotion of mesenchymal to epithelial transition and by targeting MAPK and NFκB signaling pathways. PLoS ONE 2014, 9, e86338. [Google Scholar] [CrossRef]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef]
- Houghton, P.; Fang, R.; Techatanawat, I.; Steventon, G.; Hylands, P.J.; Lee, C.C. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 2007, 42, 377–387. [Google Scholar] [CrossRef]
- Mariotti, S.; Pardini, M.; Gagliardi, M.C.; Teloni, R.; Giannoni, F.; Fraziano, M.; Lozupone, F.; Meschini, S.; Nisini, R. Dormant Mycobacterium tuberculosis Fails to Block Phagosome Maturation and Shows Unexpected Capacity To Stimulate Specific Human T Lymphocytes. J. Immunol. 2013, 191, 274–282. [Google Scholar] [CrossRef]
- Verdoliva, V.; Senatore, C.; Polci, M.L.; Rossi, S.; Cordella, M.; Carlucci, G.; Marchetti, P.; Antonini-Cappellini, G.; Facchiano, A.; D’Arcangelo, D.; et al. Differential Denaturation of Serum Proteome Reveals a Significant Amount of Hidden Information in Complex Mixtures of Proteins. PLoS ONE 2013, 8, e57104. [Google Scholar] [CrossRef]
- Cordella, M.; Tabolacci, C.; Senatore, C.; Rossi, S.; Mueller, S.; Lintas, C.; Eramo, A.; D’Arcangelo, D.; Valitutti, S.; Fac-chiano, A.; et al. Theophylline induces differentiation and modulates cytoskeleton dynamics and cytokines secre-tion in human melanoma-initiating cells. Life Sci. 2019, 230, 121–131. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioin-formatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Pomaznoy, M.; Ha, B.; Peters, B. GOnet: A tool for interactive Gene Ontology analysis. BMC Bioinform. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Mohan, M.; Matin, A.; Davies, F.E. Update on the optimal use of bortezomib in the treatment of multiple myeloma. Cancer Manag. Res. 2017, 9, 51–63. [Google Scholar] [CrossRef]
- Shahshahan, M.A.; Beckley, M.N.; Jazirehi, A.R. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: Basic and clinical aspects. Am. J. Cancer Res. 2011, 1, 913–924. [Google Scholar]
- Postovit, L.M.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Influence of the microenvironment on melanoma cell fate determi-nation and phenotype. Cancer Res. 2006, 66, 7833–7836. [Google Scholar] [CrossRef]
- Kanekura, T.; Chen, X. CD147/basigin promotes progression of malignant melanoma and other cancers. J. Dermatol. Sci. 2010, 57, 149–154. [Google Scholar] [CrossRef]
- Fukumura, D.; Jai, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell Biochem. 2006, 101, 937–949. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.Y.; Dummer, R.; Geertsen, R.; Hofbauer, G.; Laine, E.; Manolio, S.; Burg, G. Interleukin-10 is a growth factor for human melanoma cells and down-regulates HLA class I, HLA class II and ICAM-1 molecules. Int. J. Cancer 1997, 71, 630–637. [Google Scholar] [CrossRef]
- Inozume, T.; Tsunoda, T.; Morisaki, T.; Harada, K.; Shirasawa, S.; Kawamura, T. Acquisition of resistance to vemurafenib leads to interleukin-10 production through an aberrant activation of Akt in a melanoma cell line. J. Dermatol. 2018, 45, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Antonini Cappellini, G.C.; D’Atri, S. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of mel-anoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflam-masome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleu-kin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.-Y.; Chen, L.-P.; Chen, Z.-S.; Fu, L.; Huang, R.-P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; Vazquez, V.D.L.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]
- Luca, M.; Huang, S.; Gershenwald, J.E.; Singh, R.K.; Reich, R.; Bar-Eli, M. Expression of interleukin-8 by human melanoma cells up-regulates MMP-2 activity and increases tumor growth and metastasis. Am. J. Pathol. 1997, 151, 1105–1113. [Google Scholar]
- Hartman, M.L.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Czyz, M. Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells 2020, 9, 142. [Google Scholar] [CrossRef]
- Smalley, I.; Kim, E.; Li, J.; Spence, P.; Wyatt, C.J.; Eroglu, Z.; Sondak, V.K.; Babacan, N.A.; Maria-Engler, S.S.; De Armas, L.; et al. Leveraging transcriptional dynamics to improve BRAF inhibitor responses in melanoma. EBioMedicine 2019, 48, 178–190. [Google Scholar] [CrossRef]
- Bosco, M.C.; Puppo, M.; Blengio, F.; Fraone, T.; Cappello, P.; Giovarelli, M.; Varesio, L. Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 2008, 213, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metasta-sis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2015, 7, 4428–4441. [Google Scholar] [CrossRef]
- Zou, G.M.; Tam, Y.K. Cytokines in the generation and maturation of dendritic cells: Recent advances. Eur. Cytokine Netw. 2002, 13, 186–199. [Google Scholar]
- Hoejberg, L.; Bastholt, L.; Schmidt, H. Interleukin-6 and melanoma. Melanoma Res. 2012, 22, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Huang, R.R.; Wen, D.R.; Paul, E.; Wünsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells corre-lates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Laxmanan, S.; Robertson, S.W.; Wang, E.; Lau, J.S.; Briscoe, D.M.; Mukhopadhyay, D. Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways. Biochem. Biophys. Res. Commun. 2005, 334, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Bishop, J.D.; Brandt, J.P.; Hand, Z.C.; Ararso, Y.T.; Forrest, O.A. Melanoma-derived factors alter the maturation and activation of differentiated tissue-resident dendritic cells. Immunol. Cell Biol. 2015, 94, 24–38. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, M.; Wang, J.; Wang, Q.; Xia, D.; Sun, W.; Zhang, L.; Yu, H.; Liu, Y.; Cao, X. Interferon-gamma is an autocrine mediator for dendritic cell maturation. Immunol. Lett. 2004, 94, 141–151. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Kenkel, J.A.; Tseng, W.W.; Davidson, M.G.; Tolentino, L.L.; Choi, O.; Bhattacharya, N.; Seeley, E.S.; Winer, D.A.; Retick-er-Flynn, N.E.; Engleman, E.G. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Pro-motes Metastasis in Pancreatic Cancer. Cancer Res. 2017, 77, 4158–4170. [Google Scholar] [PubMed]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Ba-layssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [PubMed]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires met-abolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [PubMed]
- Brown, K.J.; Formolo, C.A.; Seol, H.; Marathi, R.L.; Duguez, S.; An, E.; Pillai, D.; Nazarian, J.; Rood, B.R.; Hathout, Y. Advances in the proteomic investigation of the cell secretome. Expert Rev. Proteom. 2012, 9, 337–345. [Google Scholar] [CrossRef]
- Sixt, S.U.; Dahlmann, B. Extracellular, circulating proteasomes and ubiquitin—Incidence and relevance. Biochim. Biophys. Acta 2008, 1782, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, F.; Jones, R.J.; Singh, R.K.; Zou, J.; Kuiatse, I.; Berkova, Z.; Wang, H.; Lee, H.C.; Hong, S.; Dick, L.; et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplas-mic reticulum stress in multiple myeloma. Proc. Natl. Acad. Sci. USA 2020, 117, 20004–20014. [Google Scholar] [PubMed]
- Zecchin, D.; Boscaro, V.; Medico, E.; Barault, L.; Martini, M.; Arena, S.; Cancelliere, C.; Bartolini, A.; Crowley, E.H.; Bardelli, A.; et al. BRAF V600E Is a Determinant of Sensitivity to Proteasome Inhibitors. Mol. Cancer Ther. 2013, 12, 2950–2961. [Google Scholar] [CrossRef]
- Tsumagari, K.; Elmageed, Z.Y.A.; Sholl, A.B.; Green, E.A.; Sobti, S.; Khan, A.R.; Kandil, A.; Murad, F.; Friedlander, P.; Boulares, A.H.; et al. Bortezomib sensitizes thyroid cancer to BRAF inhibitor in vitro and in vivo. Endocr. Relat. Cancer 2018, 25, 99–109. [Google Scholar] [CrossRef]
- Pellom, S.T., Jr.; Dudimah, D.F.; Thounaojam, M.C.; Sayers, T.J.; Shanker, A. Modulatory effects of bortezomib on host im-mune cell functions. Immunotherapy 2015, 7, 1011–1022. [Google Scholar]
- Jayachandran, A.; Anaka, M.; Prithviraj, P.; Hudson, C.; McKeown, S.J.; Lo, P.-H.; Vella, L.J.; Goding, C.R.; Cebon, J.; Behren, A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014, 5, 5782–5797. [Google Scholar] [CrossRef]
- Waerner, T.; Alacakaptan, M.; Tamir, I.; Oberauer, R.; Gal, A.; Brabletz, T.; Schreiber, M.; Jechlinger, M.; Beug, H. ILEI: A cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006, 10, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Tuck, J.; Kim, O.-J.; Panova, I.; Symanowski, J.; Mahalingam, M.; Riker, A.; Alani, R.; Ryu, B. Neuropilin-2 gene expression correlates with malignant progression in cutaneous melanoma. Br. J. Dermatol. 2014, 171, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Zeiderman, M.R.; Egger, M.E.; Kimbrough, C.W.; England, C.G.; Dupre, T.; McMasters, K.M.; McNally, L.R. Targeting of BRAF resistant melanoma via extracellular matrix metalloproteinase inducer receptor. J. Surg. Res. 2014, 190, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Koshikawa, N.; Tomari, T.; Nabeshima, K.; Isobe, T.; Seiki, M. Membrane Type 1 Matrix Metalloproteinase (MT1-MMP/MMP-14) Cleaves and Releases a 22-kDa Extracellular Matrix Metalloproteinase Inducer (EMMPRIN) Fragment from Tumor Cells. J. Biol. Chem. 2006, 281, 37576–37585. [Google Scholar] [CrossRef]
- Wu, J.; Hao, Z.W.; Zhao, Y.X.; Yang, X.M.; Tang, H.; Zhang, X.; Song, F.; Sun, X.X.; Wang, B.; Nan, G.; et al. Full-length soluble CD147 promotes MMP-2 expression and is a potential serological marker in detection of hepatocel-lular carcinoma. J. Transl. Med. 2014, 12, 190. [Google Scholar] [CrossRef]
- Hu, X.; Su, J.; Zhou, Y.; Xie, X.; Peng, C.; Yuan, Z.; Chen, X. Repressing CD147 is a novel therapeutic strategy for malig-nant melanoma. Oncotarget 2017, 8, 25806–25813. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, L.; Kuang, Y.; Su, J.; Luo, Z.; Wang, Y.; Li, J.; Zhang, J.; Chen, W.; Li, F.; et al. Downregulation of CD147 induces malignant melanoma cell apoptosis via the regulation of IGFBP2 expression. Int. J. Oncol. 2018, 53, 2397–2408. [Google Scholar] [CrossRef]
- Chen, X.; Lin, J.; Kanekura, T.; Su, J.; Lin, W.; Xie, H.; Wu, Y.; Li, J.; Chen, M.; Chang, J. A Small Interfering CD147-Targeting RNA Inhibited the Proliferation, Invasiveness, and Metastatic Activity of Malignant Melanoma. Cancer Res. 2006, 66, 11323–11330. [Google Scholar] [CrossRef]
- Rossi, S.; Cordella, M.; Tabolacci, C.; Nassa, G.; D’Arcangelo, D.; Senatore, C.; Pagnotto, P.; Magliozzi, R.; Salvati, A.; Weisz, A.; et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J. Exp. Clin. Cancer Res. 2018, 37, 1–17. [Google Scholar] [CrossRef]
- Zheng, H.-C.; Gong, B.-C. CD147 expression was positively linked to aggressiveness and worse prognosis of gastric cancer: A meta and bioinformatics analysis. Oncotarget 2017, 8, 90358–90370. [Google Scholar] [CrossRef][Green Version]
- Luo, Z.; Zeng, W.; Tang, W.; Long, T.; Zhang, J.; Xie, X.; Kuang, Y.; Chen, M.; Su, J.; Chen, X. CD147 interacts with NDUFS6 in regulating mitochondrial complex I activity and the mitochondrial apoptotic pathway in human malignant melanoma cells. Curr. Mol. Med. 2014, 14, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.G.; Wang, Y.; Wang, S.; Shao, D.; Tian, L.; Li, Y.X.; Jiang, J.L.; Chen, Z.N.; Wen, N. Synthesis and Evaluation of a Novel Small-molecule Compound as an Anticancer Inhibitor of CD147. Biomed. Environ. Sci 2019, 32, 673–686. [Google Scholar] [PubMed]
- Wang, M.; Zhang, S.; Sun, Q.; Yang, X.; Wang, Y.; Shang, R.; Zhu, Y.; Yao, H.; Li, Y. Dual effects of an anti-CD147 anti-body for Esophageal cancer therapy. Cancer Biol. Ther. 2019, 20, 1443–1452. [Google Scholar] [CrossRef]
- Paulitschke, V.; Haudek-Prinz, V.; Griss, J.; Berger, W.; Mohr, T.; Pehamberger, H.; Kunstfeld, R.; Gerner, C. Functional Classification of Cellular Proteome Profiles Support the Identification of Drug Resistance Signatures in Melanoma Cells. J. Proteome Res. 2013, 12, 3264–3276. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Accession | Gene Symbol | Description |
|---|---|---|---|
| SK-MEL-28 | P08195 | SLC3A2 | 4F2 cell-surface antigen heavy chain |
| SK-MEL-28 | Q9ULZ3 | PYCARD | Apoptosis-associated speck-like protein containing a CARD |
| SK-MEL-28 | P06865 | HEXA | Beta-hexosaminidase subunit alpha |
| SK-MEL-28 | P07711 | CTSL | Cathepsin L1 |
| SK-MEL-28 | O00299 | CLIC1 | Chloride intracellular channel protein 1 |
| SK-MEL-28 | P63241 | EIF5A | Eukaryotic translation initiation factor 5A-1 |
| SK-MEL-28 | P04439 | HLA-A | HLA class I histocompatibility antigen, A-3 alpha chain |
| SK-MEL-28 | P19013 | KRT4 | Keratin, type II cytoskeletal 4 |
| SK-MEL-28 | Q92859 | NEO1 | Neogenin |
| SK-MEL-28 | Q02818 | NUCB1 | Nucleobindin-1 |
| SK-MEL-28 | P19338 | NCL | Nucleolin |
| SK-MEL-28 | P10451 | SPP1 | Osteopontin |
| SK-MEL-28 | Q86UD1 | OAF | out at first protein homolog |
| SK-MEL-28 | Q6S8J3 | POTEE | POTE ankyrin domain family member E |
| SK-MEL-28 | Q15262 | PTPRK | Receptor-type tyrosine-protein phosphatase kappa |
| SK-MEL-28 | Q13332 | PTPRS | Receptor-type tyrosine-protein phosphatase S |
| SK-MEL-28 | Q12765 | SCRN1 | Secernin-1 |
| SK-MEL-28 | Q8N474 | SFRP1 | Secreted frizzled-related protein 1 |
| SK-MEL-28 | Q92743 | HTRA1 | Serine protease HTRA1 |
| SK-MEL-28 | P00441 | SOD1 | Superoxide dismutase [Cu-Zn] |
| SK-MEL-28 | P78324 | SIRPA | Tyrosine-protein phosphatase non-receptor type substrate 1 |
| SK-MEL-28 | P08670 | VIM | Vimentin |
| SK-MEL-28-VR2 | O14817 | TSPAN4 | Tetraspanin-4 |
| SK-MEL-28-VR2 | O43505 | B3GNT1 | Beta-1,4-glucuronyltransferase 1 |
| SK-MEL-28-VR2 | O60462 | NRP2 | Neuropilin-2 |
| SK-MEL-28-VR2 | O75874 | IDH1 | Isocitrate dehydrogenase |
| SK-MEL-28-VR2 | P00338 | LDHA | L-lactate dehydrogenase A chain |
| SK-MEL-28-VR2 | P01130 | LDLR | Low-density lipoprotein receptor |
| SK-MEL-28-VR2 | P06276 | BCHE | Cholinesterase |
| SK-MEL-28-VR2 | P07237 | P4HB | Protein disulfide-isomerase |
| SK-MEL-28-VR2 | P32004 | L1CAM | Neural cell adhesion molecule L1 |
| SK-MEL-28-VR2 | P35237 | SERPINB6 | Serpin B6 |
| SK-MEL-28-VR2 | P35613 | BSG | Basigin |
| SK-MEL-28-VR2 | P37802 | TAGLN2 | Transgelin-2 |
| SK-MEL-28-VR2 | P40967 | PMEL | Melanocyte protein PMEL |
| SK-MEL-28-VR2 | P42785 | PRCP | Lysosomal Pro-X carboxypeptidase |
| SK-MEL-28-VR2 | P46926 | GNPDA1 | Glucosamine-6-phosphate isomerase 1 |
| SK-MEL-28-VR2 | P48637 | GSS | Glutathione synthetase |
| SK-MEL-28-VR2 | P48745 | NOV | Protein NOV homolog |
| SK-MEL-28-VR2 | P49641 | MAN2A2 | Alpha-mannosidase 2x |
| SK-MEL-28-VR2 | P52209 | PGD | 6-phosphogluconate dehydrogenase, decarboxylating |
| SK-MEL-28-VR2 | P54819 | AK2 | Adenylate kinase 2, mitochondrial |
| SK-MEL-28-VR2 | P16152 | CBR1 | Carbonyl reductase |
| SK-MEL-28-VR2 | P16930 | FAH | Fumarylacetoacetase |
| SK-MEL-28-VR2 | P17174 | GOT1 | Aspartate aminotransferase, cytoplasmic |
| SK-MEL-28-VR2 | P18669 | PGAM1 | Phosphoglycerate mutase 1 |
| SK-MEL-28-VR2 | P19021 | PAM | Peptidyl-glycine alpha-amidating monooxygenase |
| SK-MEL-28-VR2 | P20618 | PSMB1 | Proteasome subunit beta type-1 |
| SK-MEL-28-VR2 | P22392 | NME1 | Nucleoside diphosphate kinase B |
| SK-MEL-28-VR2 | P25787 | PSMA2 | Proteasome subunit alpha type-2 |
| SK-MEL-28-VR2 | P12955 | PEPD | Xaa-Pro dipeptidase |
| SK-MEL-28-VR2 | P07686 | HEXB | Beta-hexosaminidase subunit beta |
| SK-MEL-28-VR2 | P07996 | THBS1 | Thrombospondin-1 |
| SK-MEL-28-VR2 | P99999 | CYCS | Cytochrome c |
| SK-MEL-28-VR2 | Q01469 | FABP5 | Fatty acid-binding protein, epidermal |
| SK-MEL-28-VR2 | Q96KP4 | CNDP2 | Cytosolic non-specific dipeptidase |
| SK-MEL-28-VR2 | Q9H2E6 | SEMA6A | Semaphorin-6A |
| SK-MEL-28-VR2 | Q9NPF2 | CHST11 | Carbohydrate sulfotransferase 11 |
| SK-MEL-28-VR2 | Q9P2B2 | PTGFRN | Prostaglandin F2 receptor negative regulator |
| SK-MEL-28-VR2 | Q9UBQ6 | EXTL2 | Exostosin-like 2 |
| SK-MEL-28-VR2 | Q9UNW1 | MINPP1 | Multiple inositol polyphosphate phosphatase 1 |
| SK-MEL-28-VR2 | Q9Y281 | CFL2 | Cofilin-2 |
| SK-MEL-28-VR2 | Q6IBS0 | TWF2 | Twinfilin-2 |
| SK-MEL-28-VR2 | Q8NBJ7 | SUMF2 | Sulfatase-modifying factor 2 |
| SK-MEL-28-VR2 | Q8NCW5 | APOA1BP | NAD(P)H-hydrate epimerase |
| SK-MEL-28-VR2 | Q8WVQ1 | CANT1 | Soluble calcium-activated nucleotidase 1 |
| SK-MEL-28-VR2 | Q92520 | FAM3C | Protein FAM3C |
| SK-MEL-28-VR2 | Q06830 | PRDX1 | Peroxiredoxin-1 |
| SK-MEL-28-VR2 | Q14019 | COTL1 | Coactosin-like protein |
| SK-MEL-28-VR2 | Q15084 | PDIA6 | Protein disulfide-isomerase A6 |
| Case | Sex | Age (Years) | Stage a | LDH b | Previous Therapy | Targeted Therapy c | BR d | TTF e (Days) |
|---|---|---|---|---|---|---|---|---|
| Pt#1 | M | 57 | M1c | H | None | VEMU | PR | 223 |
| Pt#2 | M | 49 | M1c | H | None | VEMU | PR | 147 |
| Pt#3 | F | 74 | M1b | N | None | VEMU | PR | 195 |
| Pt#4 | M | 50 | M1a | N | None | VEMU + COBI | PR | 362 |
| Pt#5 | M | 58 | M1a | H | None | VEMU + COBI | PR | 195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, C.; Cordella, M.; Mariotti, S.; Rossi, S.; Senatore, C.; Lintas, C.; Levati, L.; D’Arcangelo, D.; Facchiano, A.; D’Atri, S.; et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines 2021, 9, 79. https://doi.org/10.3390/biomedicines9010079
Tabolacci C, Cordella M, Mariotti S, Rossi S, Senatore C, Lintas C, Levati L, D’Arcangelo D, Facchiano A, D’Atri S, et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines. 2021; 9(1):79. https://doi.org/10.3390/biomedicines9010079
Chicago/Turabian StyleTabolacci, Claudio, Martina Cordella, Sabrina Mariotti, Stefania Rossi, Cinzia Senatore, Carla Lintas, Lauretta Levati, Daniela D’Arcangelo, Antonio Facchiano, Stefania D’Atri, and et al. 2021. "Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals" Biomedicines 9, no. 1: 79. https://doi.org/10.3390/biomedicines9010079
APA StyleTabolacci, C., Cordella, M., Mariotti, S., Rossi, S., Senatore, C., Lintas, C., Levati, L., D’Arcangelo, D., Facchiano, A., D’Atri, S., Nisini, R., & Facchiano, F. (2021). Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines, 9(1), 79. https://doi.org/10.3390/biomedicines9010079