Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence?

 , ,
, ,  ,
,  and
and 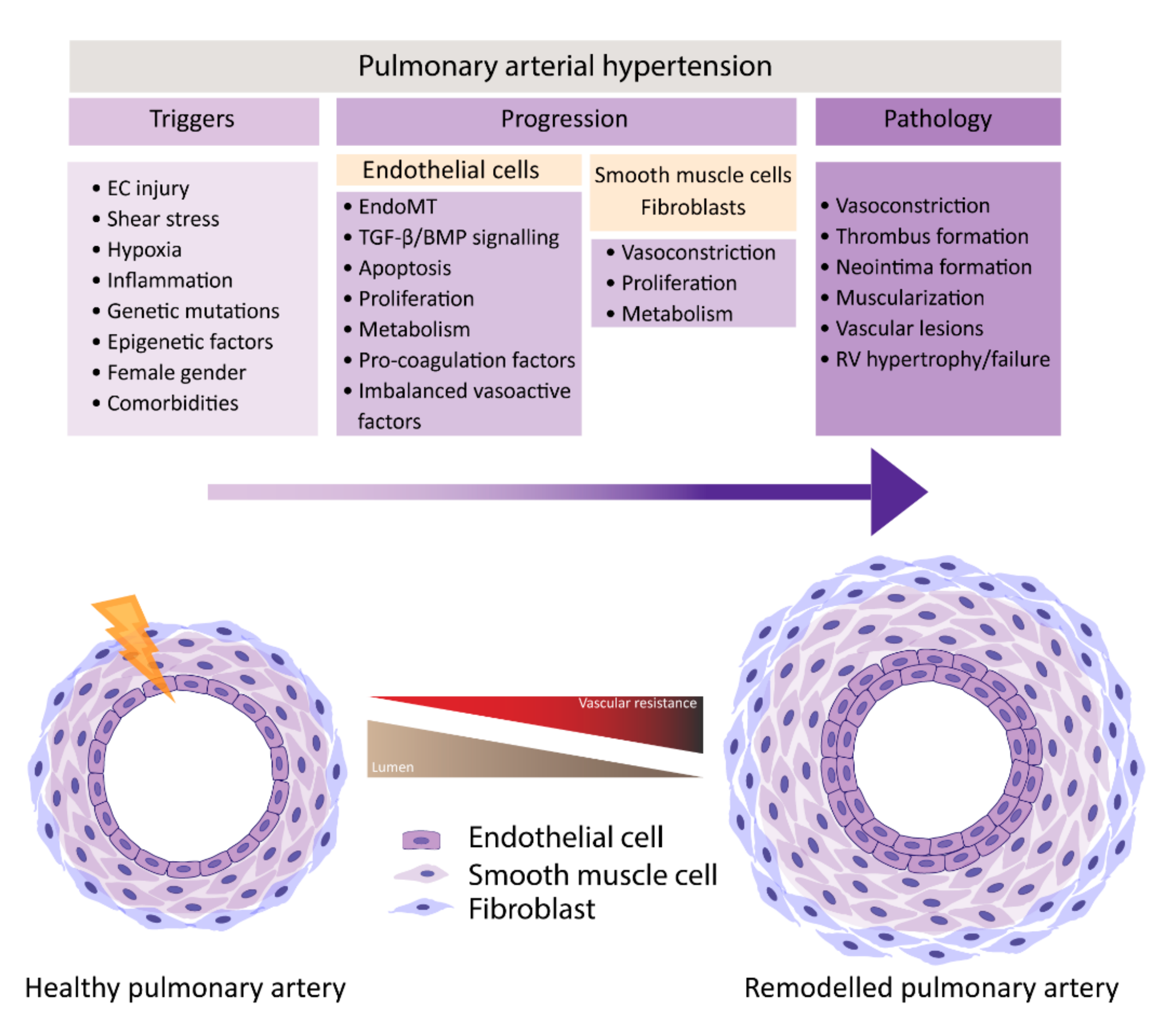{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Factors contributing to EC Dysfunction in PH
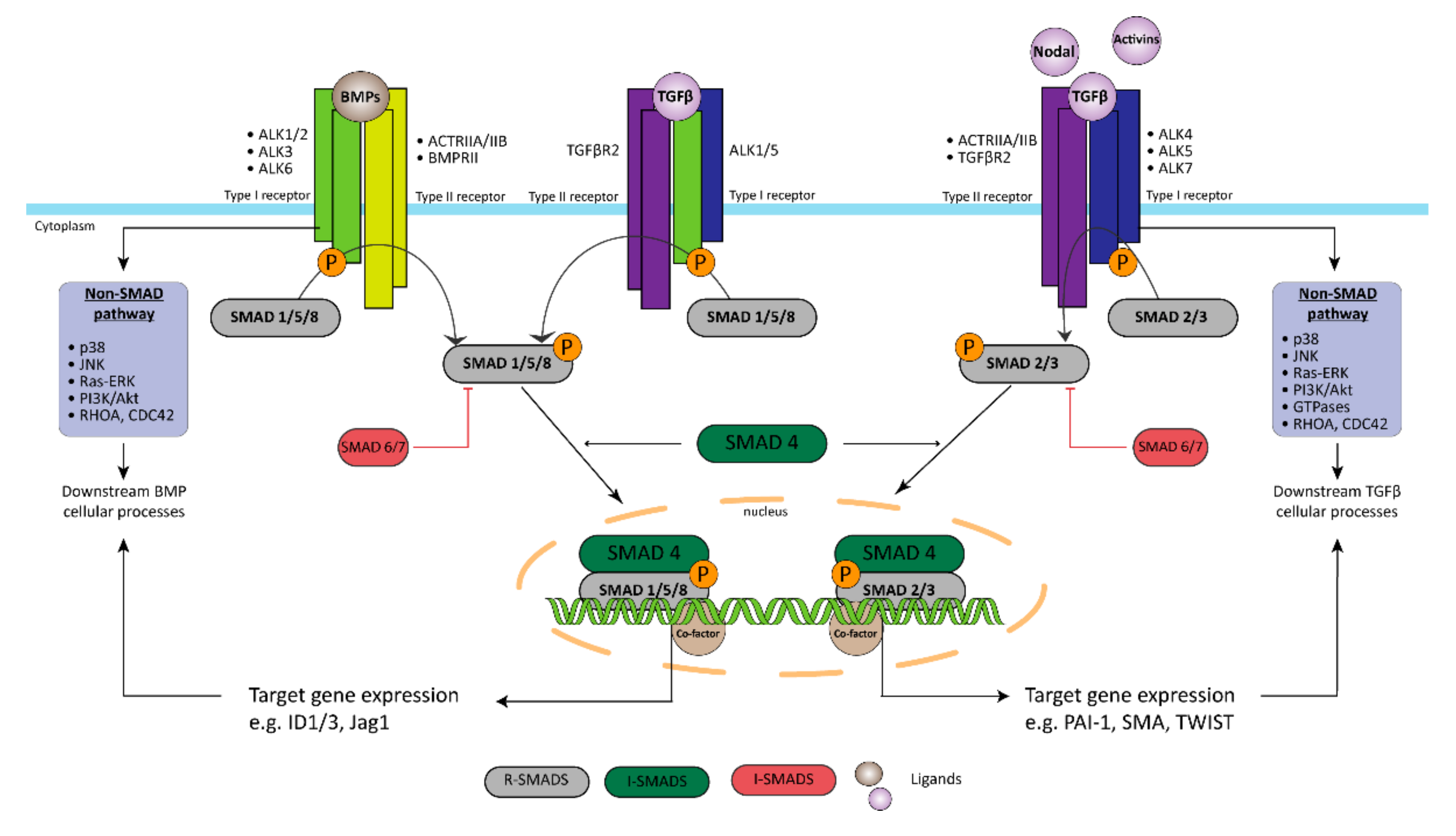
2.1. Bone Morphogenic Type 2 Receptor
2.2. Inflammation
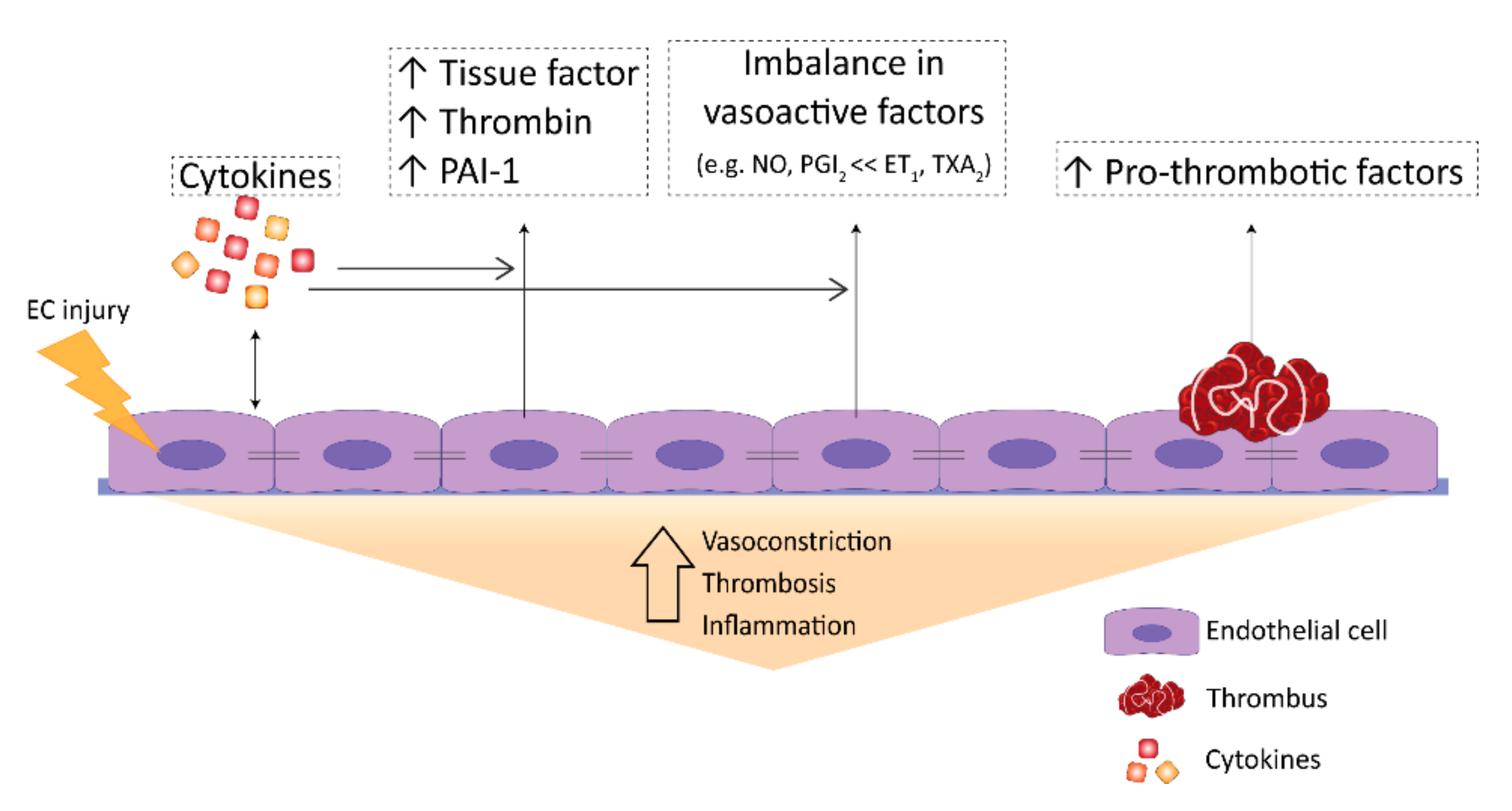
2.3. Thrombosis in PAH
2.4. Coagulation in PAH
2.5. EC Metabolism
2.6. Shear Stress
3. Features of EC Dysfunction
3.1. Perturbations in Vasoactivity
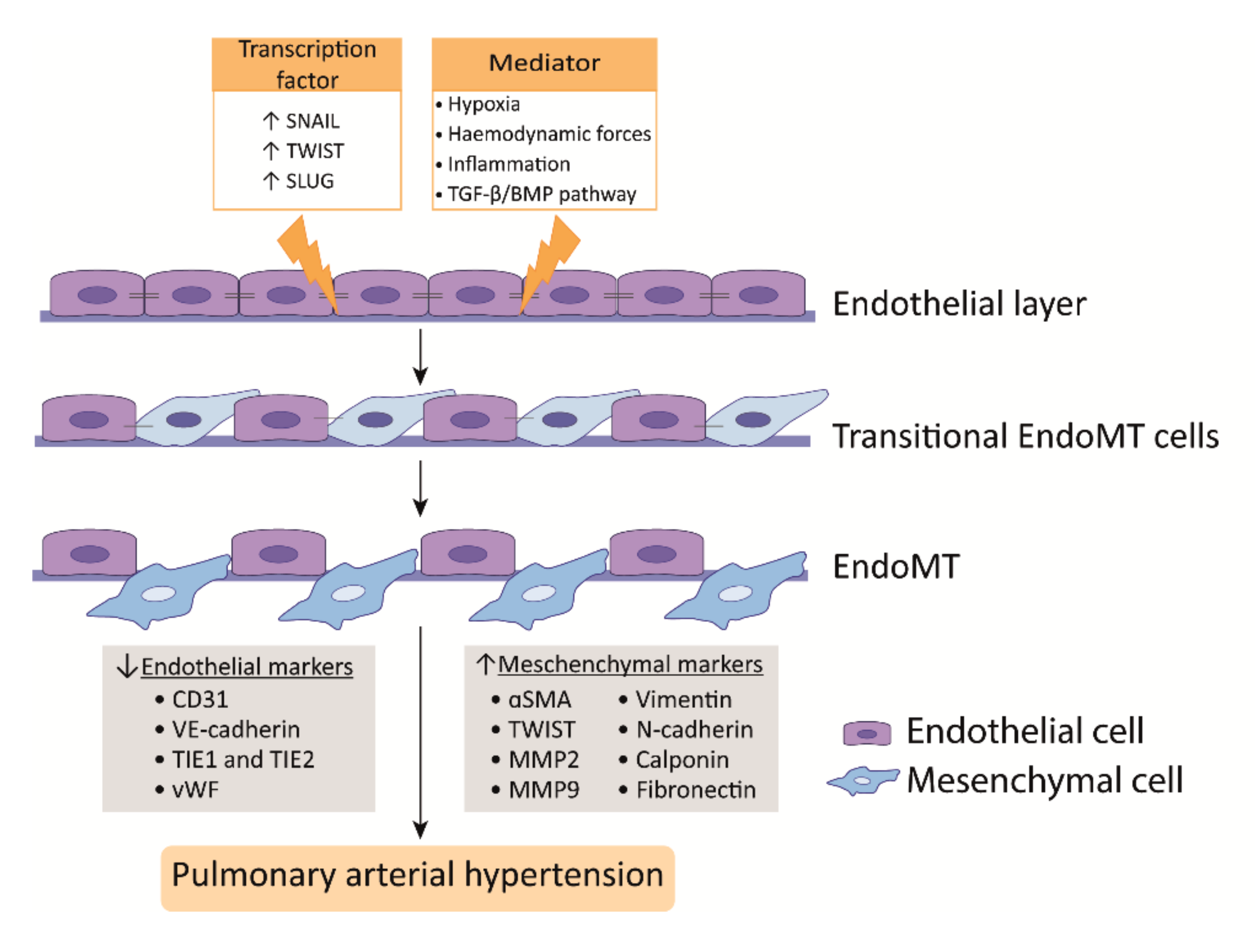
3.2. Endothelial to Mesenchymal Transition
3.3. Apoptosis
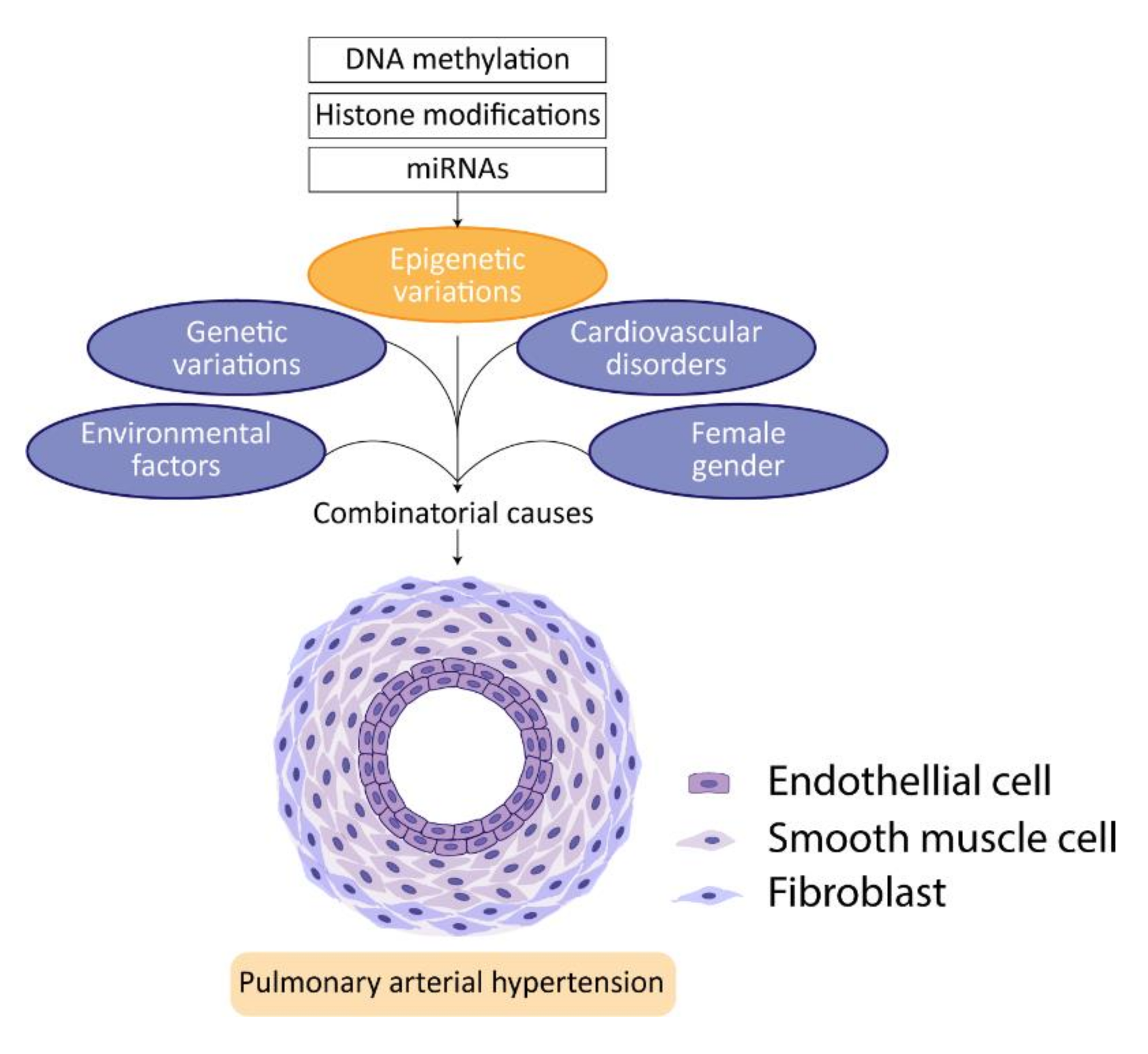
4. Epigenetics
5. EC Dysfunction in Other PH Groups
5.1. Group 2 PH
5.2. Group 3 PH
5.3. Group 4 PH
6. Current and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dumitrescu, D.; Hager, A.; Held, M.; Sinning, C.; Greiner, S.; Kruck, I.; Meyer, J.; Pabst, S.; Köhler, T.; Kovacs, G.; et al. Definition, clinical classification and initial diagnosis of pulmonary hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 11–19. [Google Scholar]
- Simonneau, G.; Hoeper, M.M. The revised definition of pulmonary hypertension: Exploring the impact on patient management. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2019, 21, K4–K8. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Vonk Noordegraaf, A.; Groeneveldt, J.A.; Bogaard, H.J. Pulmonary hypertension. Eur. Respir. Rev. 2016, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.C.; Gibbs, J.S.R. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial Dysfunction in Pulmonary Hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Humbert, M.; Montani, D.; Perros, F.; Dorfmüller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vasc. Pharmacol. 2008, 49, 113–118. [Google Scholar] [CrossRef]
- Nicod, L.P. The endothelium and genetics in pulmonary arterial hypertension. Swiss Med. Wkly. 2007, 137, 437–442. [Google Scholar] [PubMed]
- Dummer, A.; Rol, N.; Szulcek, R.; Kurakula, K.; Pan, X.; Visser, B.I.; Bogaard, H.J.; DeRuiter, M.C.; Goumans, M.J.; Hierck, B.P. Endothelial dysfunction in pulmonary arterial hypertension: Loss of cilia length regulation upon cytokine stimulation. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef]
- Tu, L.; Dewachter, L.; Gore, B.; Fadel, E.; Dartevelle, P.; Simonneau, G.; Humbert, M.; Eddahibi, S.; Guignabert, C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 311–322. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802. [Google Scholar] [CrossRef]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.J.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef]
- Orriols, M.; Gomez-Puerto, M.C.; Ten Dijke, P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell. Mol. Life Sci. 2017, 74, 2979–2995. [Google Scholar] [CrossRef]
- Newman, J.H.; Wheeler, L.; Lane, K.B.; Loyd, E.; Gaddipati, R.; Phillips, J.A., 3rd; Loyd, J.E. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N. Engl. J. Med. 2001, 345, 319–324. [Google Scholar] [CrossRef]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., 3rd; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ventura, F.; Doody, J.; Massagué, J. Human type II receptor for bone morphogenic proteins (BMPs): Extension of the two-kinase receptor model to the BMPs. Mol. Cell. Biol. 1995, 15, 3479–3486. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Goumans, M.J.; Ten Dijke, P. Regulatory RNAs controlling vascular (dys)function by affecting TGF-ß family signalling. EXCLI J. 2015, 14, 832–850. [Google Scholar] [PubMed]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef]
- Teichert-Kuliszewska, K.; Kutryk, M.J.B.; Kuliszewski, M.A.; Karoubi, G.; Courtman, D.W.; Zucco, L.; Granton, J.; Stewart, D.J. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 2006, 98, 209–217. [Google Scholar] [CrossRef]
- Zhang, S.; Fantozzi, I.; Tigno, D.D.; Yi, E.S.; Platoshyn, O.; Thistlethwaite, P.A.; Kriett, J.M.; Yung, G.; Rubin, L.J.; Yuan, J.X. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L740–L754. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Frump, A.; Prewitt, A.; de Caestecker, M.P. BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production: A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, J.; Kinzel, B.; Müeller, M.; Mao, X.; Valdez, R.; Liu, Y.; Li, E. Dosage-dependent requirement of BMP type II receptor for maintenance of vascular integrity. Blood 2007, 110, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; MacLean, M.R.; Jeffery, T.K.; Morecroft, I.; Yang, X.; Rudarakanchana, N.; Southwood, M.; James, V.; Trembath, R.C.; Morrell, N.W. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ. Res. 2006, 98, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef]
- Happé, C.; Kurakula, K.; Sun, X.Q.; da Silva Goncalves Bos, D.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. [Google Scholar] [CrossRef]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef]
- Majka, S.; Hagen, M.; Blackwell, T.; Harral, J.; Johnson, J.A.; Gendron, R.; Paradis, H.; Crona, D.; Loyd, J.E.; Nozik-Grayck, E.; et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir. Res. 2011, 12, 84. [Google Scholar] [CrossRef]
- Yang, X.; Long, L.; Reynolds, P.N.; Morrell, N.W. Expression of Mutant BMPR-II in Pulmonary Endothelial Cells Promotes Apoptosis and a Release of Factors that Stimulate Proliferation of Pulmonary Arterial Smooth Muscle Cells. Pulm. Circ. 2011, 1, 103–110. [Google Scholar] [CrossRef]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of GDF2 Mutations and Levels of BMP9 and BMP10 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Gräf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Vattulainen, S.; Aho, J.; Orcholski, M.; Rojas, V.; Yuan, K.; Helenius, M.; Taimen, P.; Myllykangas, S.; De Jesus Perez, V.; et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA Repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Im, H.; Cao, A.; Hennigs, J.K.; Wang, L.; Sa, S.; Chen, P.I.; Nickel, N.P.; Miyagawa, K.; Hopper, R.K.; et al. RNA Sequencing Analysis Detection of a Novel Pathway of Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 356–366. [Google Scholar] [CrossRef]
- Rol, N.; de Raaf, M.A.; Sun, X.Q.; Kuiper, V.P.; da Silva Gonçalves Bos, D.; Happé, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib improves cardiac fibrosis but leaves pulmonary vascular remodelling unaltered in experimental pulmonary hypertension. Cardiovasc. Res. 2019, 115, 432–439. [Google Scholar] [CrossRef]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Sung, Y.K.; Shuffle, E.; Wu, T.H.; Kao, P.N.; Tu, A.B.; Dorfmüller, P.; Cao, A.; Wang, L.; et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019, 140, 1409–1425. [Google Scholar] [CrossRef]
- Miyata, M.; Sakuma, F.; Yoshimura, A.; Ishikawa, H.; Nishimaki, T.; Kasukawa, R. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int. Arch. Allergy Immunol. 1995, 108, 287–291. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Perros, F.; Balabanian, K.; Humbert, M. Inflammation in pulmonary arterial hypertension. Eur. Respir. J. 2003, 22, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Jasiewicz, M.; Knapp, M.; Waszkiewicz, E.; Ptaszynska-Kopczynska, K.; Szpakowicz, A.; Sobkowicz, B.; Musial, W.J.; Kaminski, K.A. Enhanced IL-6 trans-signaling in pulmonary arterial hypertension and its potential role in disease-related systemic damage. Cytokine 2015, 76, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Sanchez-Duffhues, G.; Rol, N.; Pan, X.; Tsonaka, R.; Dickhoff, C.; Yung, L.M.; Manz, X.D.; Kurakula, K.; Kiełbasa, S.M.; et al. Exacerbated inflammatory signaling underlies aberrant response to BMP9 in pulmonary arterial hypertension lung endothelial cells. Angiogenesis 2020, 23, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Veyssier-Belot, C.; Cacoub, P. Role of endothelial and smooth muscle cells in the physiopathology and treatment management of pulmonary hypertension. Cardiovasc. Res. 1999, 44, 274–282. [Google Scholar] [CrossRef]
- Fuster, V.; Steele, P.M.; Edwards, W.D.; Gersh, B.J.; McGoon, M.D.; Frye, R.L. Primary pulmonary hypertension: Natural history and the importance of thrombosis. Circulation 1984, 70, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Granton, J.T.; Mehta, S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest 2006, 130, 545–552. [Google Scholar] [CrossRef]
- Berger, G.; Azzam, Z.S.; Hoffman, R.; Yigla, M. Coagulation and anticoagulation in pulmonary arterial hypertension. Isr. Med. Assoc. J. 2009, 11, 376–379. [Google Scholar]
- Kawut, S.M.; Horn, E.M.; Berekashvili, K.K.; Widlitz, A.C.; Rosenzweig, E.B.; Barst, R.J. Von Willebrand factor independently predicts long-term survival in patients with pulmonary arterial hypertension. Chest 2005, 128, 2355–2362. [Google Scholar] [CrossRef]
- Damås, J.K.; Otterdal, K.; Yndestad, A.; Aass, H.; Solum, N.O.; Frøland, S.S.; Simonsen, S.; Aukrust, P.; Andreassen, A.K. Soluble CD40 ligand in pulmonary arterial hypertension: Possible pathogenic role of the interaction between platelets and endothelial cells. Circulation 2004, 110, 999–1005. [Google Scholar] [CrossRef]
- Kroone, C.; Vos, M.; Rademakers, T.; Kuijpers, M.; Hoogenboezem, M.; van Buul, J.; Heemskerk, J.W.M.; Ruf, W.; van Hylckama Vlieg, A.; Versteeg, H.H.; et al. LIM-only protein FHL2 attenuates vascular tissue factor activity, inhibits thrombus formation in mice and FHL2 genetic variation associates with human venous thrombosis. Haematologica 2020, 105, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Koenis, D.S.; Herzik, M.A., Jr.; Liu, Y.; Craft, J.W., Jr.; van Loenen, P.B.; Vos, M.; Tran, M.K.; Versteeg, H.H.; Goumans, M.T.H.; et al. Structural and cellular mechanisms of peptidyl-prolyl isomerase Pin1-mediated enhancement of Tissue Factor gene expression, protein half-life, and pro-coagulant activity. Haematologica 2018, 103, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Meoli, D.F.; Swarthout, R.F.; Kallop, D.Y.; Galaria, I.I.; Harvey, J.L.; Miller, C.M.; Blaxall, B.C.; Hall, C.M.; Pierce, R.A.; et al. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L583–L590. [Google Scholar] [CrossRef] [PubMed]
- Bakouboula, B.; Morel, O.; Faure, A.; Zobairi, F.; Jesel, L.; Trinh, A.; Zupan, M.; Canuet, M.; Grunebaum, L.; Brunette, A.; et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Tournier, A.; Wahl, D.; Chaouat, A.; Max, J.P.; Regnault, V.; Lecompte, T.; Chabot, F. Calibrated automated thrombography demonstrates hypercoagulability in patients with idiopathic pulmonary arterial hypertension. Thromb. Res. 2010, 126, e418–e422. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Beckmann, R.; Frank, H.; Kneussl, M.; Mlczoch, J.; Binder, B.R. Fibrinogen, t-PA, and PAI-1 plasma levels in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1994, 150, 929–933. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Smolders, V.F.; Zodda, E.; Quax, P.H.A.; Carini, M.; Barberà, J.A.; Thomson, T.M.; Tura-Ceide, O.; Cascante, M. Metabolic Alterations in Cardiopulmonary Vascular Dysfunction. Front. Mol. Biosci. 2018, 5, 120. [Google Scholar] [CrossRef]
- Maron, B.A.; Leopold, J.A. Emerging Concepts in the Molecular Basis of Pulmonary Arterial Hypertension: Part II: Neurohormonal Signaling Contributes to the Pulmonary Vascular and Right Ventricular Pathophenotype of Pulmonary Arterial Hypertension. Circulation 2015, 131, 2079–2091. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Kheyfets, V.O.; Schroeder, J.D.; Dunning, J.; Shandas, R.; Buckner, J.K.; Browning, J.; Hertzberg, J.; Hunter, K.S.; Fenster, B.E. Main pulmonary arterial wall shear stress correlates with invasive hemodynamics and stiffness in pulmonary hypertension. Pulm. Circ. 2016, 6, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.P.; Schelegle, E.S.; Bennett, S.H. Diverse forms of pulmonary hypertension remodel the arterial tree to a high shear phenotype. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H405–H417. [Google Scholar] [CrossRef][Green Version]
- Gatzoulis, M.A.; Alonso-Gonzalez, R.; Beghetti, M. Pulmonary arterial hypertension in paediatric and adult patients with congenital heart disease. Eur. Respir. Rev. 2009, 18, 154–161. [Google Scholar] [CrossRef]
- Abe, K.; Shinoda, M.; Tanaka, M.; Kuwabara, Y.; Yoshida, K.; Hirooka, Y.; McMurtry, I.F.; Oka, M.; Sunagawa, K. Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc. Res. 2016, 111, 16–25. [Google Scholar] [CrossRef]
- van der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Hirata, M.; Ousaka, D.; Arai, S.; Okuyama, M.; Tarui, S.; Kobayashi, J.; Kasahara, S.; Sano, S. Novel Model of Pulmonary Artery Banding Leading to Right Heart Failure in Rats. BioMed Res. Int. 2015, 2015, 753210. [Google Scholar] [CrossRef]
- Szulcek, R.; Happé, C.M.; Rol, N.; Fontijn, R.D.; Dickhoff, C.; Hartemink, K.J.; Grünberg, K.; Tu, L.; Timens, W.; Nossent, G.D.; et al. Delayed Microvascular Shear Adaptation in Pulmonary Arterial Hypertension. Role of Platelet Endothelial Cell Adhesion Molecule-1 Cleavage. Am. J. Respir. Crit. Care Med. 2016, 193, 1410–1420. [Google Scholar] [CrossRef]
- Li, M.; Tan, Y.; Stenmark, K.R.; Tan, W. High Pulsatility Flow Induces Acute Endothelial Inflammation through Overpolarizing Cells to Activate NF-κB. Cardiovasc. Eng. Technol. 2013, 4, 26–38. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.A.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis In Vivo and endothelial cell growth and migration In Vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Babaei, S.; Teichert-Kuliszewska, K.; Monge, J.C.; Mohamed, F.; Bendeck, M.P.; Stewart, D.J. Role of nitric oxide in the angiogenic response In Vitro to basic fibroblast growth factor. Circ. Res. 1998, 82, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef]
- Quinlan, T.R.; Li, D.; Laubach, V.E.; Shesely, E.G.; Zhou, N.; Johns, R.A. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000. [Google Scholar] [CrossRef]
- Epstein, F.H.; Vane, J.R.; Änggård, E.E.; Botting, R.M. Regulatory Functions of the Vascular Endothelium. N. Engl. J. Med. 2010, 323, 27–36. [Google Scholar] [CrossRef]
- Chen, Y.F.; Oparil, S. Endothelial Dysfunction in the Pulmonary Vascular Bed. Am. J. Med. Sci. 2000, 320, 223–232. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O. Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Ahmetaj-Shala, B.; Kirkby, N.S.; Wright, W.R.; Mackenzie, L.S.; Reed, D.M.; Mohamed, N. Role of prostacyclin in pulmonary hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 382–393. [Google Scholar] [CrossRef]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999. [Google Scholar] [CrossRef] [PubMed]
- Geraci, M.W.; Gao, B.; Shepherd, D.C.; Moore, M.D.; Westcott, J.Y.; Fagan, K.A.; Alger, L.A.; Tuder, R.M.; Voelkel, N.F. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J. Clin. Investig. 1999, 103, 1509–1515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chester, A.H.; Yacoub, M.H. The role of endothelin-1 in pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Park, J.E.S.; Wort, S.J. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharm. Res. 2011, 63, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Kato, H.; Marumo, F.; Hirata, Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 1997, 30, 1198–1203. [Google Scholar] [CrossRef]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Li, H.B.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced Endothelin-1 and Endothelin Receptor Gene-Expression in Chronic Hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459. [Google Scholar] [CrossRef]
- Frasch, H.F.; Marshall, C.; Marshall, B.E. Endothelin-1 is elevated in monocrotaline pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L304–L310. [Google Scholar] [CrossRef]
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405. [Google Scholar] [CrossRef]
- Galié, N.; Manes, A.; Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 2004, 61, 227–237. [Google Scholar] [CrossRef]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Säleby, J.; Bouzina, H.; Ahmed, S.; Lundgren, J.; Rådegran, G. Plasma receptor tyrosine kinase RET in pulmonary arterial hypertension diagnosis and differentiation. ERJ Open Res. 2019, 5, 00037–02019. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α Contributes to Severe Pulmonary Hypertension by Inducing Endothelial-to-Mesenchymal Transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 314, L256–L275. [Google Scholar] [CrossRef]
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef]
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thomé, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faça, V.; Olsen Câmara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-β2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458. [Google Scholar] [CrossRef]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-β–Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207. [Google Scholar] [CrossRef]
- Hopper, R.K.; Moonen, J.R.A.J.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In pulmonary arterial hypertension, reduced bmpr2 promotes endothelial-to-Mesenchymal transition via hmga1 and its target slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; He, Y.; Shui, X.; Li, G.; Yan, G.; Zhang, Y.; Huang, S.; Chen, C.; Ding, Y. Expression and analyses of the HIF-1 pathway in the lungs of humans with pulmonary arterial hypertension. Mol. Med. Rep. 2016, 14, 4383–4390. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelial-mesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Zhang, X.; Guo, Y.; Wang, X. miR-181b-5p inhibits endothelial-mesenchymal transition in monocrotaline-induced pulmonary arterial hypertension by targeting endocan and TGFBR1. Toxicol. Appl. Pharmacol. 2020, 386, 114827. [Google Scholar] [CrossRef]
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef]
- Sakao, S.; Taraseviciene-Stewart, L.; Lee, J.D.; Wood, K.; Cool, C.D.; Voelkel, N.F. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Masri, F.A.; Xu, W.; Comhair, S.A.; Asosingh, K.; Koo, M.; Vasanji, A.; Drazba, J.; Anand-Apte, B.; Erzurum, S.C. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Dempsie, Y.; Caruso, P.; Wallace, E.; McDonald, R.A.; Stevens, H.; Hatley, M.E.; Van Rooij, E.; Morrell, N.W.; Maclean, M.R.; et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014, 64, 185–194. [Google Scholar] [CrossRef]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P.I.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S.; et al. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ. Res. 2019, 124, 211–224. [Google Scholar] [CrossRef]
- Hautefort, A.; Chesné, J.; Preussner, J.; Pullamsetti, S.S.; Tost, J.; Looso, M.; Antigny, F.; Girerd, B.; Riou, M.; Eddahibi, S.; et al. Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 2017, 8, 52995–53016. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Stenmark, K.R.; McKinsey, T.A. Emerging Roles for Histone Deacetylases in Pulmonary Hypertension and Right Ventricular Remodeling (2013 Grover Conference series). Pulm. Circ. 2015, 5, 63–72. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone Deacetylation Inhibition in Pulmonary Hypertension: Therapeutic of Valproic Acid and SuPotentialberoylanilide Hydroxamic Acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Chabot, S.; Boucherat, O.; Ruffenach, G.; Breuils-Bonnet, S.; Tremblay, E.; Provencher, S.; Bonnet, S. HDAC6-HSP90 interplay in pulmonary arterial hypertension. FASEB J. 2016, 30, 774.4. [Google Scholar]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 4546. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.M.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class i histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 2012, 110, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; El Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of Fibroblasts with a Proinflammatory Epigenetically Altered Phenotype in Severe Hypoxic Pulmonary Hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwangbo, C.; Hu, X.; Kang, Y.; Papangeli, I.; Mehrotra, D.; Park, H.; Ju, H.; McLean, D.L.; Comhair, S.A.; et al. Restoration of Impaired Endothelial MEF2 Function Rescues Pulmonary Arterial Hypertension. Circulation 2015, 131, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Feen, D.E.V.D.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L. Multicenter preclinical validation of BET inhibition for the treatment of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Gegonne, A.; Singer, D.S. Bromodomain 4: A cellular Swiss army knife. J. Leukoc. Biol. 2016, 100, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-Containing Protein 4. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef]
- Fernández, A.I.; Yotti, R.; González-Mansilla, A.; Mombiela, T.; Gutiérrez-Ibanes, E.; Pérez Del Villar, C.; Navas-Tejedor, P.; Chazo, C.; Martínez-Legazpi, P.; Fernández-Avilés, F.; et al. The Biological Bases of Group 2 Pulmonary Hypertension. Int. J. Mol. Sci. 2019, 20, 5884. [Google Scholar] [CrossRef]
- Ontkean, M.; Gay, R.; Greenberg, B. Diminished endothelium-derived relaxing factor activity in an experimental model of chronic heart failure. Circ. Res. 1991, 69, 1088–1096. [Google Scholar] [CrossRef]
- Givertz, M.M.; Colucci, W.S.; LeJemtel, T.H.; Gottlieb, S.S.; Hare, J.M.; Slawsky, M.T.; Leier, C.V.; Loh, E.; Nicklas, J.M.; Lewis, B.E. Acute endothelin A receptor blockade causes selective pulmonary vasodilation in patients with chronic heart failure. Circulation 2000, 101, 2922–2927. [Google Scholar] [CrossRef] [PubMed]
- Meoli, D.F.; Su, Y.R.; Brittain, E.L.; Robbins, I.M.; Hemnes, A.R.; Monahan, K. The transpulmonary ratio of endothelin 1 is elevated in patients with preserved left ventricular ejection fraction and combined pre- and post-capillary pulmonary hypertension. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.D.; Kansal, M.; Desai, A.A.; Riden, K.; Arwood, M.J.; Yacob, A.A.; Stamos, T.D.; Cavallari, L.H.; Zamanian, R.T.; Shah, S.J.; et al. Endothelial nitric oxide synthase genotype is associated with pulmonary hypertension severity in left heart failure patients. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Vachiéry, J.L.; Tedford, R.J.; Rosenkranz, S.; Palazzini, M.; Lang, I.; Guazzi, M.; Coghlan, G.; Chazova, I.; De Marco, T. Pulmonary hypertension due to left heart disease. Eur. Respir. J. 2019, 53, 1801897. [Google Scholar] [CrossRef]
- Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. Int. J. Mol. Sci. 2019, 20, 4329. [Google Scholar] [CrossRef]
- Barberà, J.A.; Peinado, V.I.; Santos, S.; Ramirez, J.; Roca, J.; Rodriguez-Roisin, R. Reduced Expression of Endothelial Nitric Oxide Synthase in Pulmonary Arteries of Smokers. Am. J. Respir. Crit. Care Med. 2001, 164, 709–713. [Google Scholar] [CrossRef]
- Nana-Sinkam, S.P.; Jong, D.L.; Sotto-Santiago, S.; Stearman, R.S.; Keith, R.L.; Choudhury, Q.; Cool, C.; Parr, J.; Moore, M.D.; Bull, T.M.; et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am. J. Respir. Crit. Care Med. 2007. [Google Scholar] [CrossRef]
- Santos, S.; Peinado, V.I.; Ramírez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barberà, J.A. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003. [Google Scholar] [CrossRef]
- Carratu, P.; Scoditti, C.; Maniscalco, M.; Seccia, T.; Di Gioia, G.; Gadaleta, F.; Cardone, R.; Dragonieri, S.; Pierucci, P.; Spanevello, A.; et al. Exhaled and arterial levels of endothelin-1 are increased and correlate with pulmonary systolic pressure in COPD with pulmonary hypertension. BMC Pulm. Med. 2008, 8, 20. [Google Scholar] [CrossRef]
- Xiong, P.Y.; Potus, F.; Chan, W.; Archer, S.L. Models and Molecular Mechanisms of World Health Organization Group 2 to 4 Pulmonary Hypertension. Hypertension 2018, 71, 34–55. [Google Scholar] [CrossRef]
- Reimann, S.; Fink, L.; Wilhelm, J.; Hoffmann, J.; Bednorz, M.; Seimetz, M.; Dessureault, I.; Troesser, R.; Ghanim, B.; Klepetko, W.; et al. Increased S100A4 expression in the vasculature of human COPD lungs and murine model of smoke-induced emphysema. Respir. Res. 2015, 16, 127. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, H.; Behr, J.; Bremer, H.; Claussen, M.; Douschan, P.; Halank, M.; Held, M.; Hoeper, M.M.; Holt, S.; Klose, H.; et al. Pulmonary hypertension due to lung diseases: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 63–68. [Google Scholar] [CrossRef]
- Simonneau, G.; Torbicki, A.; Dorfmüller, P.; Kim, N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 2017, 26, 160112. [Google Scholar] [CrossRef] [PubMed]
- Yaoita, N.; Shirakawa, R.; Fukumoto, Y.; Sugimura, K.; Miyata, S.; Miura, Y.; Nochioka, K.; Miura, M.; Tatebe, S.; Aoki, T.; et al. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M. Pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: Pathophysiology. Eur. Respir. Rev. 2010, 19, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Hao, H.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Tatsumi, K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: Crosstalk with myofibroblast-like cells. Respir. Res. 2011, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Mercier, O.; Arthur Ataam, J.; Langer, N.B.; Dorfmüller, P.; Lamrani, L.; Lecerf, F.; Decante, B.; Dartevelle, P.; Eddahibi, S.; Fadel, E. Abnormal pulmonary endothelial cells may underlie the enigmatic pathogenesis of chronic thromboembolic pulmonary hypertension. J. Heart Lung Transpl. 2017, 36, 305–314. [Google Scholar] [CrossRef]
- Tura-Ceide, O.; Aventín, N.; Piccari, L.; Morén, C.; Guitart-Mampel, M.; Garrabou, G.; García-Lucio, J.; Chamorro, N.; Blanco, I.; Peinado, V.; et al. Endothelial dysfunction in patients with chronic thromboembolic pulmonary hypertension (CTEPH). Eur. Respir. Soc. 2016, 48, PA3606. [Google Scholar]
- Naito, A.; Sakao, S.; Lang, I.M.; Voelkel, N.F.; Jujo, T.; Ishida, K.; Sugiura, T.; Matsumiya, G.; Yoshino, I.; Tanabe, N.; et al. Endothelial cells from pulmonary endarterectomy specimens possess a high angiogenic potential and express high levels of hepatocyte growth factor. BMC Pulm. Med. 2018, 18, 197. [Google Scholar] [CrossRef]
- Quarck, R.; Wynants, M.; Verbeken, E.; Meyns, B.; Delcroix, M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2015, 46, 431–443. [Google Scholar] [CrossRef]
- Arthur Ataam, J.; Mercier, O.; Lamrani, L.; Amsallem, M.; Ataam, J.A.; Ataam, S.A.; Guihaire, J.; Lecerf, F.; Capuano, V.; Ghigna, M.R.; et al. ICAM-1 promotes the abnormal endothelial cell phenotype in chronic thromboembolic pulmonary hypertension. J. Heart Lung Transplant. 2019, 38, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Smolders, V.; Rodríguez, C.; Morén, C.; Blanco, I.; Osorio, J.; Piccari, L.; Bonjoch, C.; Quax, P.H.A.; Peinado, V.I.; Castellà, M.; et al. Decreased Glycolysis as Metabolic Fingerprint of Endothelial Cells in Chronic Thromboembolic Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhong, Z.; Wu, D.; Chen, Y.; Lian, N.; Ding, H.; Zhang, Q.; Lin, Q.; Wu, S. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci. Rep. 2017, 7, 2210. [Google Scholar] [CrossRef] [PubMed]
- Newnham, M.; South, K.; Bleda, M.; Auger, W.R.; Barberà, J.A.; Bogaard, H.; Bunclark, K.; Cannon, J.E.; Delcroix, M.; Hadinnapola, C.; et al. The ADAMTS13-VWF axis is dysregulated in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801805. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Nagaraj, C.; Stacher, E.; Lang, I.M.; Nierlich, P.; Klepetko, W.; Heinemann, A.; Olschewski, H.; Bálint, Z.; Olschewski, A. Angiostatic factors in the pulmonary endarterectomy material from chronic thromboembolic pulmonary hypertension patients cause endothelial dysfunction. PLoS ONE 2012, 7, e43793. [Google Scholar] [CrossRef] [PubMed]
- Conole, D.; Scott, L.J. Riociguat: First global approval. Drugs 2013, 73, 1967–1975. [Google Scholar] [CrossRef]
- Prins, K.W.; Thenappan, T. WHO Group I Pulmonary Hypertension: Epidemiology and Pathophysiology. Cardiol. Clin. 2016, 34, 363–374. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Apitz, C.; Grünig, E.; Halank, M.; Ewert, R.; Kaemmerer, H.; Kabitz, H.J.; Kähler, C.; Klose, H.; Leuchte, H.; et al. Targeted therapy of pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272S, 37–45. [Google Scholar] [CrossRef]
- Lan, N.S.H.; Massam, B.D.; Kulkarni, S.S.; Lang, C.C. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases 2018, 6, 38. [Google Scholar] [CrossRef]
- Humbert, M.; Ghofrani, H.A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016, 71, 73–83. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Ibrahim, Y.F.; Shults, N.V. Apoptosis-based therapy to treat pulmonary arterial hypertension. J. Rare Dis. Res. Treat. 2016, 1, 17–24. [Google Scholar] [PubMed]
- Ibrahim, Y.F.; Wong, C.M.; Pavlickova, L.; Liu, L.; Trasar, L.; Bansal, G.; Suzuki, Y.J. Mechanism of the susceptibility of remodeled pulmonary vessels to drug-induced cell killing. J. Am. Heart Assoc. 2014, 3, e000520. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, J.H.; Huh, J.W.; Kim, H.J.; Park, M.K.; Ro, J.Y.; Oh, Y.M.; Lee, S.D.; Lee, Y.S. Bortezomib alleviates experimental pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2012, 47, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Russell, R.R.; Schwartz, R.G.; Panjrath, G.S.; Aronow, W. Cardiac Complications of Cancer Therapy: Pathophysiology, Identification, Prevention, Treatment, and Future Directions. Curr. Cardiol. Rep. 2017, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Quaife, R.A.; Leinwand, L.A.; Barst, R.J.; McGoon, M.D.; Meldrum, D.R.; Dupuis, J.; Long, C.S.; Rubin, L.J.; Smart, F.W.; et al. Right ventricular function and failure: Report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006, 114, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective Transforming Growth Factor-β Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef]
- Guo, Y.; Li, P.; Bledsoe, G.; Yang, Z.R.; Chao, L.; Chao, J. Kallistatin inhibits TGF-β-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp. Cell Res. 2015, 337, 103–110. [Google Scholar] [CrossRef]
- Marsh, L.M.; Jandl, K.; Grünig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef]
- Kumar, R.; Graham, B. How does inflammation contribute to pulmonary hypertension? Eur. Respir. J. 2018, 51, 1702403. [Google Scholar] [CrossRef]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Desroches-Castan, A.; Mallet, C.; Guyon, L.; Cumont, A.; Phan, C.; Robert, F.; Thuillet, R.; Bordenave, J.; Sekine, A.; et al. Selective BMP-9 Inhibition Partially Protects Against Experimental Pulmonary Hypertension. Circ. Res. 2019, 124, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Sanada, T.J.; Sun, X.-Q.; Happé, C.; Guignabert, C.; Tu, L.; Schalij, I.; Bogaard, H.-J.; Goumans, M.-J.; Kurakula, K. AlteredTGFβ/SMAD Signaling in Human and Rat Models of Pulmonary Hypertension: An Old Target Needs Attention. Cells 2021, 10, 84. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Quarck, R.; Perros, F. Rescuing BMPR2-driven endothelial dysfunction in PAH: A novel treatment strategy for the future? Stem Cell Investig. 2017, 4, 56. [Google Scholar] [CrossRef]
- Kurakula, K.; Sun, X.Q.; Happé, C.; da Silva Goncalves Bos, D.; Szulcek, R.; Schalij, I.; Wiesmeijer, K.C.; Lodder, K.; Tu, L.; Guignabert, C.; et al. 6-mercaptopurine, an agonist of Nur77, reduces progression of pulmonary hypertension by enhancing BMP signalling. Eur. Respir. J. 2019, 54, 1802400. [Google Scholar] [CrossRef]
- Botros, L.; Szulcek, R.; Jansen, S.M.; Kurakula, K.; Goumans, M.T.; van Kuilenburg, A.B.P.; Vonk Noordegraaf, A.; de Man, F.S.; Aman, J.; Bogaard, H.J. The Effects of Mercaptopurine on Pulmonary Vascular Resistance and BMPR2 Expression in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Dumont, F.; Ruellou, G.; Lambert, M.; Balliau, T.; Quatredeniers, M.; Girerd, B.; Cohen-Kaminsky, S.; Mercier, O.; Yen-Nicolaÿ, S.; et al. Proteomic Analysis of KCNK3 Loss of Expression Identified Dysregulated Pathways in Pulmonary Vascular Cells. Int. J. Mol. Sci. 2020, 21, 7400. [Google Scholar] [CrossRef]
- Huang, J.; Lu, W.; Ouyang, H.; Chen, Y.; Zhang, C.; Luo, X.; Li, M.; Shu, J.; Zheng, Q.; Chen, H.; et al. Transplantation of Mesenchymal Stem Cells Attenuates Pulmonary Hypertension by Normalizing the EndMT. Am. J. Respir. Cell Mol. Biol. 2019, 62, 49–60. [Google Scholar] [CrossRef]
- de Mendonça, L.; Felix, N.S.; Blanco, N.G.; Da Silva, J.S.; Ferreira, T.P.; Abreu, S.C.; Cruz, F.F.; Rocha, N.; Silva, P.M.; Martins, V.; et al. Mesenchymal stromal cell therapy reduces lung inflammation and vascular remodeling and improves hemodynamics in experimental pulmonary arterial hypertension. Stem Cell Res. Ther. 2017, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Martire, A.; Bedada, F.B.; Uchida, S.; Pöling, J.; Krüger, M.; Warnecke, H.; Richter, M.; Kubin, T.; Herold, S.; Braun, T. Mesenchymal stem cells attenuate inflammatory processes in the heart and lung via inhibition of TNF signaling. Basic Res. Cardiol. 2016, 111, 54. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Moore, S.; Crosby, A.; Southwood, M.; Du, X.; Tan, H.; Xie, S.; Vassallo, A.; Wood, A.J.; Wallace, E.M.; et al. Targeting HIF2α-ARNT hetero-dimerisation as a novel therapeutic strategy for Pulmonary Arterial Hypertension. Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 541900378. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Mizuno, S.; Al Hussaini, A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, L.; Zhang, Z.; Prado, E.; Fu, L.; Xu, X.; Du, L. Epigenetic Regulation and Its Therapeutic Potential in Pulmonary Hypertension. Front. Pharmacol. 2018, 9, 241. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurakula, K.; Smolders, V.F.E.D.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.-J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. https://doi.org/10.3390/biomedicines9010057
Kurakula K, Smolders VFED, Tura-Ceide O, Jukema JW, Quax PHA, Goumans M-J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines. 2021; 9(1):57. https://doi.org/10.3390/biomedicines9010057
Chicago/Turabian StyleKurakula, Kondababu, Valérie F. E. D. Smolders, Olga Tura-Ceide, J. Wouter Jukema, Paul H. A. Quax, and Marie-José Goumans. 2021. "Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence?" Biomedicines 9, no. 1: 57. https://doi.org/10.3390/biomedicines9010057
APA StyleKurakula, K., Smolders, V. F. E. D., Tura-Ceide, O., Jukema, J. W., Quax, P. H. A., & Goumans, M.-J. (2021). Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines, 9(1), 57. https://doi.org/10.3390/biomedicines9010057