Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural and Biochemical Alterations of Proteins and Lp Induced by High Glucose
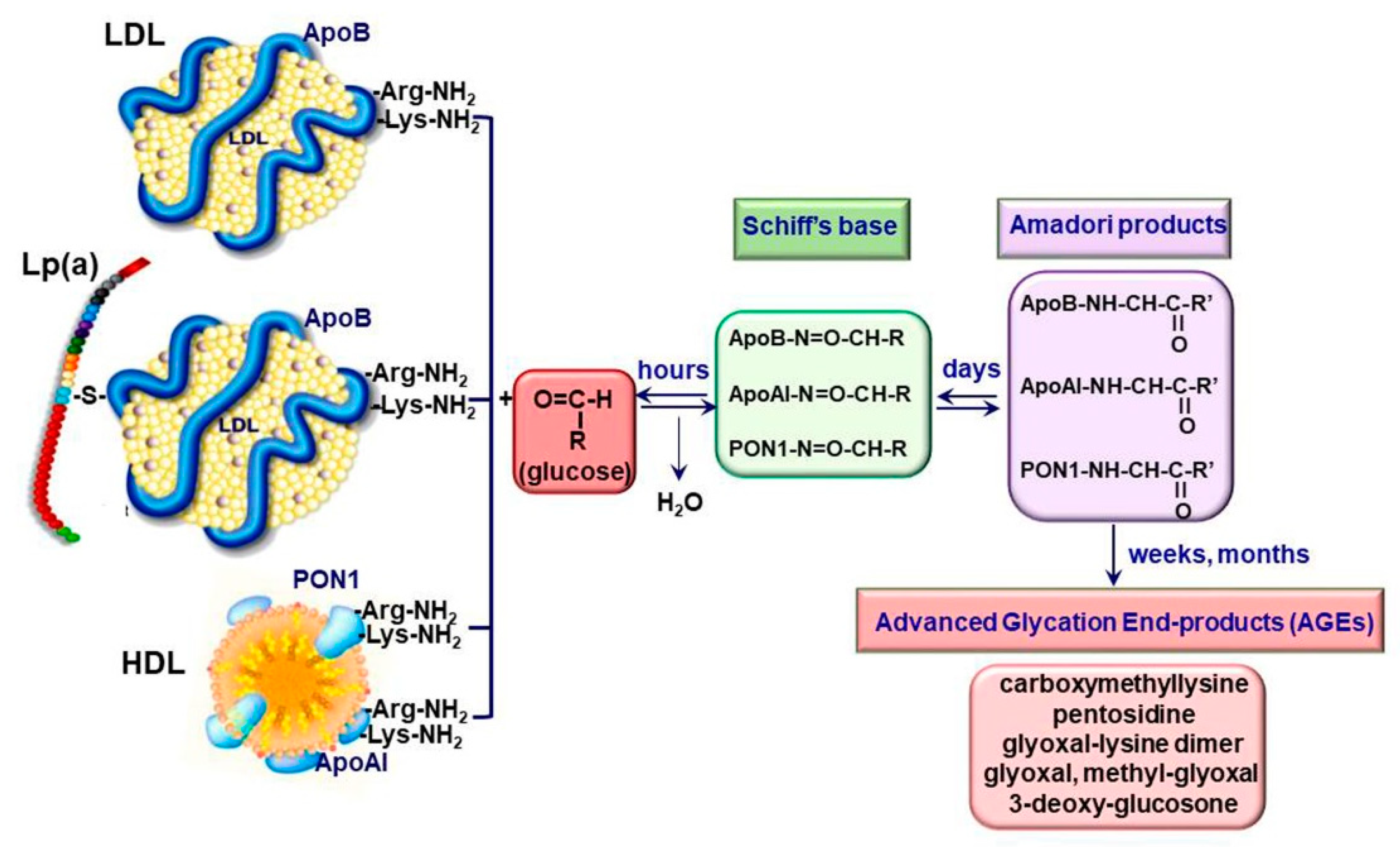
2.1. Generation of Advanced Glycation End Products
2.2. Irreversible Glycation of Lipoproteins
2.3. Receptors for AGE-Proteins
3. Endothelial Cell Dysfunction in Diabetes
4. Glycated Lipoproteins Detrimental Actions in Endothelial Cells
4.1. Reduction of Nitric Oxide Bioavailability
4.2. Induction of Oxidative Stress
4.2.1. Upregulation of the Main EC Pro-Oxidant Proteins by gLp
4.2.2. Modulation of the Activity of the Cellular Antioxidant Defense System by gLp
4.3. Activation of Endoplasmic Reticulum Stress
4.4. Stimulation of Monocytes Adhesion to Endothelial Cells
4.5. Generation of Fibrinolytic Regulators
4.6. Induction of Endothelial Cell Apoptosis
5. Glycated Lipoproteins in Diabetes
5.1. Glycated LDL Participate in Atheroma Formation
5.2. Dysfunctional HDL Are Pro-Atherogenic Particles in Diabetes
5.3. Hyperglycemia Alters miRNAs Profiles in Plasma and Lipoproteins
6. Promising Therapies to Reduce ECD in Diabetes
6.1. In Vitro Approaches to Decrease the Effects of gLp in EC
6.2. Therapies Used to Alleviate the Vascular Disorders in Diabetes
6.2.1. The Use of Hypoglycemiant Compounds
6.2.2. Therapeutic Compounds to Reduce Formation of AGE
6.2.3. Antioxidants to Decrease CVD in Diabetes
6.3. New Promising Therapies to Alleviate ECD in Diabetes
6.3.1. RAGE Inhibitors
6.3.2. MiRNA Based Therapies
6.3.3. Inhibition of Glycated-Lp(a)
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Gregg, E.W.; Williams, D.E.; Geiss, L. Changes in diabetes-related complications in the United States. N. Engl. J. Med. 2014, 371, 286–287. [Google Scholar] [CrossRef]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B.; Diabetes, C.; Complications Trial/Epidemiology of Diabetes, I.; et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef]
- Ceriello, A. The emerging challenge in diabetes: The “metabolic memory”. Vasc. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef]
- An, H.; Wei, R.; Ke, J.; Yang, J.; Liu, Y.; Wang, X.; Wang, G.; Hong, T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J. Diabetes Its Complicat. 2016, 30, 1017–1024. [Google Scholar] [CrossRef]
- Liu, T.; Gong, J.; Chen, Y.; Jiang, S. Periodic vs constant high glucose in inducing pro-inflammatory cytokine expression in human coronary artery endothelial cells. Inflamm. Res. 2013, 62, 697–701. [Google Scholar] [CrossRef]
- Liu, T.S.; Pei, Y.H.; Peng, Y.P.; Chen, J.; Jiang, S.S.; Gong, J.B. Oscillating high glucose enhances oxidative stress and apoptosis in human coronary artery endothelial cells. J. Endocrinol. Investig. 2014, 37, 645–651. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Hill, R.B. Mitochondrial regulation of diabetic vascular disease: An emerging opportunity. Transl. Res. J. Lab. Clin. Med. 2018, 202, 83–98. [Google Scholar] [CrossRef]
- Coco, C.; Sgarra, L.; Potenza, M.A.; Nacci, C.; Pasculli, B.; Barbano, R.; Parrella, P.; Montagnani, M. Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus? Int. J. Mol. Sci. 2019, 20, 2949. [Google Scholar] [CrossRef]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef]
- Kosmopoulos, M.; Drekolias, D.; Zavras, P.D.; Piperi, C.; Papavassiliou, A.G. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Mol, M.; Degani, G.; Coppa, C.; Baron, G.; Popolo, L.; Carini, M.; Aldini, G.; Vistoli, G.; Altomare, A. Advanced lipoxidation end products (ALEs) as RAGE binders: Mass spectrometric and computational studies to explain the reasons why. Redox Biol. 2019, 23, 101083. [Google Scholar] [CrossRef]
- Alique, M.; Luna, C.; Carracedo, J.; Ramirez, R. LDL biochemical modifications: A link between atherosclerosis and aging. Food Nutr. Res. 2015, 59, 29240. [Google Scholar] [CrossRef]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LDL. Curr. Opin. Lipidol. 2008, 19, 378–384. [Google Scholar] [CrossRef]
- Simionescu, M.; Popov, D.; Sima, A. Endothelial dysfunction in diabetes. In Vascular Involvement in Diabetes—Clinical, Experimental and Beyond; Cheta, D.M., Ed.; Romanian Academy Publishing House and Karger: Bucharest, Basel, 2005; pp. 15–34. [Google Scholar]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Cybulska, B.; Klosiewicz-Latoszek, L.; Penson, P.E.; Banach, M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog. Cardiovasc. Dis. 2020, 63, 219–227. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Witztum, J.L.; Fisher, M.; Pietro, T.; Steinbrecher, U.P.; Elam, R.L. Nonenzymatic glucosylation of high-density lipoprotein accelerates its catabolism in guinea pigs. Diabetes 1982, 31, 1029–1032. [Google Scholar] [CrossRef]
- Sima, A.V.; Botez, G.M.; Stancu, C.S.; Manea, A.; Raicu, M.; Simionescu, M. Effect of irreversibly glycated LDL in human vascular smooth muscle cells: Lipid loading, oxidative and inflammatory stress. J. Cell. Mol. Med. 2010, 14, 2790–2802. [Google Scholar] [CrossRef]
- Deleanu, M.; Sanda, G.M.; Stancu, C.S.; Popa, M.E.; Sima, A.V. Profiles of Fatty Acids and the Main Lipid Peroxidation Products of Human Atherogenic Low Density Lipoproteins. Rev. Chim. 2016, 67, 8–12. [Google Scholar]
- Zhang, J.; Ren, S.; Shen, G.X. Glycation amplifies lipoprotein(a)-induced alterations in the generation of fibrinolytic regulators from human vascular endothelial cells. Atherosclerosis 2000, 150, 299–308. [Google Scholar] [CrossRef]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef]
- Gordon, S.M.; Davidson, W.S.; Urbina, E.M.; Dolan, L.M.; Heink, A.; Zang, H.; Lu, L.J.; Shah, A.S. The effects of type 2 diabetes on lipoprotein composition and arterial stiffness in male youth. Diabetes 2013, 62, 2958–2967. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Wautier, J.L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef]
- Lu, Q.; Lu, L.; Chen, W.; Chen, H.; Xu, X.; Zheng, Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2015, 253, 669–680. [Google Scholar] [CrossRef]
- Egana-Gorrono, L.; Lopez-Diez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.H. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008, 283, 35507–35516. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Mompeo, B.; Popov, D.; Sima, A.; Constantinescu, E.; Simionescu, M. Diabetes-induced structural changes of venous and arterial endothelium and smooth muscle cells. J. Submicrosc. Cytol. Pathol. 1998, 30, 475–484. [Google Scholar]
- Suganya, N.; Bhakkiyalakshmi, E.; Sarada, D.V.; Ramkumar, K.M. Reversibility of endothelial dysfunction in diabetes: Role of polyphenols. Br. J. Nutr. 2016, 116, 223–246. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc. Res. 2009, 82, 371–381. [Google Scholar] [CrossRef]
- Dobi, A.; Bravo, S.B.; Veeren, B.; Paradela-Dobarro, B.; Alvarez, E.; Meilhac, O.; Viranaicken, W.; Baret, P.; Devin, A.; Rondeau, P. Advanced glycation end-products disrupt human endothelial cells redox homeostasis: New insights into reactive oxygen species production. Free Radic. Res. 2019, 53, 150–169. [Google Scholar] [CrossRef]
- Wang, C.C.; Lee, A.S.; Liu, S.H.; Chang, K.C.; Shen, M.Y.; Chang, C.T. Spironolactone ameliorates endothelial dysfunction through inhibition of the AGE/RAGE axis in a chronic renal failure rat model. BMC Nephrol. 2019, 20, 351. [Google Scholar] [CrossRef]
- Zhao, R.; Ren, S.; Moghadasain, M.H.; Rempel, J.D.; Shen, G.X. Involvement of fibrinolytic regulators in adhesion of monocytes to vascular endothelial cells induced by glycated LDL and to aorta from diabetic mice. J. Leukoc. Biol. 2014, 95, 941–949. [Google Scholar] [CrossRef]
- Toma, L.; Sanda, G.M.; Deleanu, M.; Stancu, C.S.; Sima, A.V. Glycated LDL increase VCAM-1 expression and secretion in endothelial cells and promote monocyte adhesion through mechanisms involving endoplasmic reticulum stress. Mol. Cell. Biochem. 2016, 417, 169–179. [Google Scholar] [CrossRef]
- Zhou, X.; Weng, J.; Xu, J.; Xu, Q.; Wang, W.; Zhang, W.; Huang, Q.; Guo, X. Mdia1 is Crucial for Advanced Glycation End Product-Induced Endothelial Hyperpermeability. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 45, 1717–1730. [Google Scholar] [CrossRef]
- Li, J.; Schmidt, A.M. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef]
- Caballero, A.E.; Arora, S.; Saouaf, R.; Lim, S.C.; Smakowski, P.; Park, J.Y.; King, G.L.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes 1999, 48, 1856–1862. [Google Scholar] [CrossRef]
- Carrizzo, A.; Izzo, C.; Oliveti, M.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Di Pietro, P.; Calabrese, M.; De Simone, E.; Sciarretta, S.; et al. The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. Int. J. Mol. Sci. 2018, 19, 2968. [Google Scholar] [CrossRef]
- Thorand, B.; Baumert, J.; Chambless, L.; Meisinger, C.; Kolb, H.; Doring, A.; Lowel, H.; Koenig, W.; Group, M.K.S. Elevated markers of endothelial dysfunction predict type 2 diabetes mellitus in middle-aged men and women from the general population. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 398–405. [Google Scholar] [CrossRef]
- Lim, S.C.; Caballero, A.E.; Smakowski, P.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Soluble intercellular adhesion molecule, vascular cell adhesion molecule, and impaired microvascular reactivity are early markers of vasculopathy in type 2 diabetic individuals without microalbuminuria. Diabetes Care 1999, 22, 1865–1870. [Google Scholar] [CrossRef]
- Festa, A.; D′Agostino, R., Jr.; Tracy, R.P.; Haffner, S.M.; Insulin Resistance Atherosclerosis, S. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetes 2002, 51, 1131–1137. [Google Scholar] [CrossRef]
- Vischer, U.M.; Emeis, J.J.; Bilo, H.J.; Stehouwer, C.D.; Thomsen, C.; Rasmussen, O.; Hermansen, K.; Wollheim, C.B.; Ingerslev, J. von Willebrand factor (vWf) as a plasma marker of endothelial activation in diabetes: Improved reliability with parallel determination of the vWf propeptide (vWf:AgII). Thromb. Haemost. 1998, 80, 1002–1007. [Google Scholar]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflug. Archiv Eur. J. Physiol. 2016, 468, 1125–1137. [Google Scholar] [CrossRef]
- Rabini, R.A.; Vignini, A.; Salvolini, E.; Staffolani, R.; Martarelli, D.; Moretti, N.; Mazzanti, L. Activation of human aortic endothelial cells by LDL from Type 1 diabetic patients: An in vitro study. Atherosclerosis 2002, 165, 69–77. [Google Scholar] [CrossRef]
- Artwohl, M.; Graier, W.F.; Roden, M.; Bischof, M.; Freudenthaler, A.; Waldhausl, W.; Baumgartner-Parzer, S.M. Diabetic LDL triggers apoptosis in vascular endothelial cells. Diabetes 2003, 52, 1240–1247. [Google Scholar] [CrossRef][Green Version]
- Toma, L.; Stancu, C.S.; Sanda, G.M.; Sima, A.V. Anti-oxidant and anti-inflammatory mechanisms of amlodipine action to improve endothelial cell dysfunction induced by irreversibly glycated LDL. Biochem. Biophys. Res. Commun. 2011, 411, 202–207. [Google Scholar] [CrossRef]
- Mohanan Nair, M.; Zhao, R.; Xie, X.; Shen, G.X. Impact of glycated LDL on endothelial nitric oxide synthase in vascular endothelial cells: Involvement of transmembrane signaling and endoplasmic reticulum stress. J. Diabetes Its Complicat. 2016, 30, 391–397. [Google Scholar] [CrossRef]
- Stancu, C.S.; Georgescu, A.; Toma, L.; Sanda, G.M.; Sima, A.V. Glycated low density lipoproteins alter vascular reactivity in hyperlipidemic hyperglycemic hamsters. Ann. Rom. Soc. Cell Biol. 2012, 17, 9–15. [Google Scholar]
- Dong, Y.; Wu, Y.; Wu, M.; Wang, S.; Zhang, J.; Xie, Z.; Xu, J.; Song, P.; Wilson, K.; Zhao, Z.; et al. Activation of protease calpain by oxidized and glycated LDL increases the degradation of endothelial nitric oxide synthase. J. Cell. Mol. Med. 2009, 13, 2899–2910. [Google Scholar] [CrossRef]
- Nofer, J.R.; van der Giet, M.; Tolle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Godecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Curtiss, L.K.; Witztum, J.L. Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 1985, 34, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Nakajima, T.; Miyazaki, T.; Koyama, I.; Hokari, S.; Inoue, I.; Kawai, S.; Shimomura, H.; Katayama, S.; Hara, A.; et al. Glycated high-density lipoprotein regulates reactive oxygen species and reactive nitrogen species in endothelial cells. Metab. Clin. Exp. 2003, 52, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Persegol, L.; Verges, B.; Foissac, M.; Gambert, P.; Duvillard, L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 2006, 49, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horvath, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef]
- Vaisar, T.; Couzens, E.; Hwang, A.; Russell, M.; Barlow, C.E.; DeFina, L.F.; Hoofnagle, A.N.; Kim, F. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS ONE 2018, 13, e0192616. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D′Amico, F.; Rodriguez Guzman, R.; Centofanti, F.; Doldo, E.; Cespedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Toma, L.; Stancu, C.S.; Botez, G.M.; Sima, A.V.; Simionescu, M. Irreversibly glycated LDL induce oxidative and inflammatory state in human endothelial cells; added effect of high glucose. Biochem. Biophys. Res. Commun. 2009, 390, 877–882. [Google Scholar] [CrossRef]
- Xie, X.; Zhao, R.; Shen, G.X. Impact of cyanidin-3-glucoside on glycated LDL-induced NADPH oxidase activation, mitochondrial dysfunction and cell viability in cultured vascular endothelial cells. Int. J. Mol. Sci. 2012, 13, 15867–15880. [Google Scholar] [CrossRef]
- Toma, L.; Sanda, G.M.; Niculescu, L.S.; Deleanu, M.; Stancu, C.S.; Sima, A.V. Caffeic acid attenuates the inflammatory stress induced by glycated LDL in human endothelial cells by mechanisms involving inhibition of AGE-receptor, oxidative, and endoplasmic reticulum stress. BioFactors 2017, 43, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.L. Mitochondrial Dysfunction Signature in Diabetic Vascular Endothelium. J. Clin. Exp. Pathol. 2018, 8. [Google Scholar] [CrossRef]
- Sangle, G.V.; Chowdhury, S.K.; Xie, X.; Stelmack, G.L.; Halayko, A.J.; Shen, G.X. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated low-density lipoprotein. Free Radic. Biol. Med. 2010, 48, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chowdhury, S.R.; Sangle, G.; Shen, G.X. Impact of diabetes-associated lipoproteins on oxygen consumption and mitochondrial enzymes in porcine aortic endothelial cells. Acta Biochim. Pol. 2010, 57, 393–398. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef]
- Zhao, R.; Shen, G.X. Functional modulation of antioxidant enzymes in vascular endothelial cells by glycated LDL. Atherosclerosis 2005, 179, 277–284. [Google Scholar] [CrossRef]
- Pathomthongtaweechai, N.; Chutipongtanate, S. AGE/RAGE signaling-mediated endoplasmic reticulum stress and future prospects in non-coding RNA therapeutics for diabetic nephropathy. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 131, 110655. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress? Exp. Diabetes Res. 2012, 2012, 481840. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.S.; Carnuta, M.G.; Sanda, G.M.; Toma, L.; Deleanu, M.; Niculescu, L.S.; Sasson, S.; Simionescu, M.; Sima, A.V. Hyperlipidemia-induced hepatic and small intestine ER stress and decreased paraoxonase 1 expression and activity is associated with HDL dysfunction in Syrian hamsters. Mol. Nutr. Food Res. 2015, 59, 2293–2302. [Google Scholar] [CrossRef]
- Zhao, R.; Xie, X.; Le, K.; Li, W.; Moghadasian, M.H.; Beta, T.; Shen, G.X. Endoplasmic reticulum stress in diabetic mouse or glycated LDL-treated endothelial cells: Protective effect of Saskatoon berry powder and cyanidin glycans. J. Nutr. Biochem. 2015, 26, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, X.; Feng, L.; Yang, H.; Yu, W.; Feng, T.; Wang, S.; Wang, J.; Liu, N. Glycation of paraoxonase 1 by high glucose instigates endoplasmic reticulum stress to induce endothelial dysfunction in vivo. Sci. Rep. 2017, 7, 45827. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C.; Thorpe, S.R.; Fu, M.X.; Harper, C.M.; Yoo, J.; Kim, S.M.; Wong, H.; Peters, A.L. Glycation impairs high-density lipoprotein function. Diabetologia 2000, 43, 312–320. [Google Scholar] [CrossRef]
- Carnuta, M.G.; Stancu, C.S.; Toma, L.; Sanda, G.M.; Niculescu, L.S.; Deleanu, M.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; et al. Dysfunctional high-density lipoproteins have distinct composition, diminished anti-inflammatory potential and discriminate acute coronary syndrome from stable coronary artery disease patients. Sci. Rep. 2017, 7, 7295. [Google Scholar] [CrossRef]
- Morgantini, C.; Natali, A.; Boldrini, B.; Imaizumi, S.; Navab, M.; Fogelman, A.M.; Ferrannini, E.; Reddy, S.T. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011, 60, 2617–2623. [Google Scholar] [CrossRef]
- Kearney, K.; Tomlinson, D.; Smith, K.; Ajjan, R. Hypofibrinolysis in diabetes: A therapeutic target for the reduction of cardiovascular risk. Cardiovasc. Diabetol. 2017, 16, 34. [Google Scholar] [CrossRef]
- Ren, S.; Lee, H.; Hu, L.; Lu, L.; Shen, G.X. Impact of diabetes-associated lipoproteins on generation of fibrinolytic regulators from vascular endothelial cells. J. Clin. Endocrinol. Metab. 2002, 87, 286–291. [Google Scholar] [CrossRef]
- Shen, G.X. Impact and mechanism for oxidized and glycated lipoproteins on generation of fibrinolytic regulators from vascular endothelial cells. Mol. Cell. Biochem. 2003, 246, 69–74. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, S.; Sun, D.; Shen, G.X. Influence of glycation on LDL-induced generation of fibrinolytic regulators in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.M.; Halayko, A.J.; Stelmack, G.L.; Zhu, F.; Zhao, R.; Hillier, C.T.; Shen, G.X. Effects of oxidized and glycated low-density lipoproteins on transcription and secretion of plasminogen activator inhibitor-1 in vascular endothelial cells. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2006, 15, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Shen, G.X. Involvement of heat shock factor-1 in glycated LDL-induced upregulation of plasminogen activator inhibitor-1 in vascular endothelial cells. Diabetes 2007, 56, 1436–1444. [Google Scholar] [CrossRef]
- Sangle, G.V.; Zhao, R.; Mizuno, T.M.; Shen, G.X. Involvement of RAGE, NADPH oxidase, and Ras/Raf-1 pathway in glycated LDL-induced expression of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Endocrinology 2010, 151, 4455–4466. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Shen, G.X. Impact of antioxidants and HDL on glycated LDL-induced generation of fibrinolytic regulators from vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell. Mol. Life Sci. CMLS 2017, 74, 4387–4403. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chen, H.H.; Huang, M.T.; Raya, J.L.; Yang, J.H.; Chen, C.H.; Gaubatz, J.W.; Pownall, H.J.; Taylor, A.A.; Ballantyne, C.M.; et al. Pro-apoptotic low-density lipoprotein subfractions in type II diabetes. Atherosclerosis 2007, 193, 283–291. [Google Scholar] [CrossRef]
- Li, X.L.; Li, B.Y.; Cheng, M.; Yu, F.; Yin, W.B.; Cai, Q.; Zhang, Z.; Zhang, J.H.; Wang, J.F.; Zhou, R.H.; et al. PIMT prevents the apoptosis of endothelial cells in response to glycated low density lipoproteins and protective effects of grape seed procyanidin B2. PLoS ONE 2013, 8, e69979. [Google Scholar] [CrossRef]
- Yin, W.; Li, B.; Li, X.; Yu, F.; Cai, Q.; Zhang, Z.; Wang, J.; Zhang, J.; Zhou, R.; Cheng, M.; et al. Critical role of prohibitin in endothelial cell apoptosis caused by glycated low-density lipoproteins and protective effects of grape seed procyanidin B2. J. Cardiovasc. Pharmacol. 2015, 65, 13–21. [Google Scholar] [CrossRef]
- Matsunaga, T.; Iguchi, K.; Nakajima, T.; Koyama, I.; Miyazaki, T.; Inoue, I.; Kawai, S.; Katayama, S.; Hirano, K.; Hokari, S.; et al. Glycated high-density lipoprotein induces apoptosis of endothelial cells via a mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2001, 287, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Kim, J.Y.; Choi, I.; Kim, J.R.; Won, K.C.; Cho, K.H. Fructated apolipoprotein A-I exacerbates cellular senescence in human umbilical vein endothelial cells accompanied by impaired insulin secretion activity and embryo toxicity. Biochem. Cell Biol. Biochim. Biol. Cell. 2016, 94, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Makita, Z.; Koschinsky, T.; Cerami, A.; Vlassara, H. Lipid advanced glycosylation: Pathway for lipid oxidation in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 6434–6438. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.; Stancu, C. Modified lipoproteins accumulate in human coronary atheroma. J. Cell. Mol. Med. 2002, 6, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Tames, F.J.; Mackness, M.I.; Arrol, S.; Laing, I.; Durrington, P.N. Non-enzymatic glycation of apolipoprotein B in the sera of diabetic and non-diabetic subjects. Atherosclerosis 1992, 93, 237–244. [Google Scholar] [CrossRef]
- Simionescu, M.; Popov, D.; Sima, A.; Hasu, M.; Costache, G.; Faitar, S.; Vulpanovici, A.; Stancu, C.; Stern, D.; Simionescu, N. Pathobiochemistry of combined diabetes and atherosclerosis studied on a novel animal model. The hyperlipemic-hyperglycemic hamster. Am. J. Pathol. 1996, 148, 997–1014. [Google Scholar] [PubMed]
- Sima, A.; Popov, D.; Starodub, O.; Stancu, C.; Cristea, C.; Stern, D.; Simionescu, M. Pathobiology of the heart in experimental diabetes: Immunolocalization of lipoproteins, immunoglobulin G, and advanced glycation endproducts proteins in diabetic and/or hyperlipidemic hamster. Lab. Investig. J. Tech. Methods Pathol. 1997, 77, 3–18. [Google Scholar]
- Hwang, S.W.; Lee, Y.M.; Aldini, G.; Yeum, K.J. Targeting Reactive Carbonyl Species with Natural Sequestering Agents. Molecules 2016, 21, 280. [Google Scholar] [CrossRef]
- von Eckardstein, A.; Widmann, C. High-density lipoprotein, beta cells, and diabetes. Cardiovasc. Res. 2014, 103, 384–394. [Google Scholar] [CrossRef]
- Younis, N.H.; Soran, H.; Charlton-Menys, V.; Sharma, R.; Hama, S.; Pemberton, P.; Elseweidy, M.M.; Durrington, P.N. High-density lipoprotein impedes glycation of low-density lipoprotein. Diabetes Vasc. Dis. Res. 2012, 10, 152–160. [Google Scholar] [CrossRef]
- Meneses, M.J.; Silvestre, R.; Sousa-Lima, I.; Macedo, M.P. Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 4049. [Google Scholar] [CrossRef]
- Femlak, M.; Gluba-Brzozka, A.; Cialkowska-Rysz, A.; Rysz, J. The role and function of HDL in patients with diabetes mellitus and the related cardiovascular risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [PubMed]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Xie, S.; Li, J.; Tian, R.; Peng, Y.Y. Myeloperoxidase-mediated oxidation targets serum apolipoprotein A-I in diabetic patients and represents a potential mechanism leading to impaired anti-apoptotic activity of high density lipoprotein. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 441, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Craciun, E.C.; Leucuta, D.C.; Rusu, R.L.; David, B.A.; Cret, V.; Dronca, E. Paraoxonase-1 activities in children and adolescents with type 1 diabetes mellitus. Acta Biochim. Pol. 2016, 63, 511–515. [Google Scholar] [CrossRef]
- Mastorikou, M.; Mackness, B.; Liu, Y.; Mackness, M. Glycation of paraoxonase-1 inhibits its activity and impairs the ability of high-density lipoprotein to metabolize membrane lipid hydroperoxides. Diabet. Med. J. Br. Diabet. Assoc. 2008, 25, 1049–1055. [Google Scholar] [CrossRef]
- Boemi, M.; Leviev, I.; Sirolla, C.; Pieri, C.; Marra, M.; James, R.W. Serum paraoxonase is reduced in type 1 diabetic patients compared to non-diabetic, first degree relatives; influence on the ability of HDL to protect LDL from oxidation. Atherosclerosis 2001, 155, 229–235. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Dullaart, R.P.; Otvos, J.D.; James, R.W. Serum paraoxonase-1 activity is more closely related to HDL particle concentration and large HDL particles than to HDL cholesterol in Type 2 diabetic and non-diabetic subjects. Clin. Biochem. 2014, 47, 1022–1027. [Google Scholar] [CrossRef]
- Tabara, Y.; Arai, H.; Hirao, Y.; Takahashi, Y.; Setoh, K.; Kawaguchi, T.; Kosugi, S.; Ito, Y.; Nakayama, T.; Matsuda, F.; et al. Different inverse association of large high-density lipoprotein subclasses with exacerbation of insulin resistance and incidence of type 2 diabetes: The Nagahama study. Diabetes Res. Clin. Pract. 2017, 127, 123–131. [Google Scholar] [CrossRef]
- Manjunatha, S.; Distelmaier, K.; Dasari, S.; Carter, R.E.; Kudva, Y.C.; Nair, K.S. Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metab. Clin. Exp. 2016, 65, 1421–1431. [Google Scholar] [CrossRef]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Sartori, C.H.; Oliveira, E.R.; Nakandakare, E.R.; Stefano, J.T.; Giannella-Neto, D.; Correa-Giannella, M.L.; Passarelli, M. Advanced glycated albumin isolated from poorly controlled type 1 diabetes mellitus patients alters macrophage gene expression impairing ABCA-1-mediated reverse cholesterol transport. Diabetes/Metab. Res. Rev. 2013, 29, 66–76. [Google Scholar] [CrossRef] [PubMed]
- McEneny, J.; Daniels, J.A.; McGowan, A.; Gunness, A.; Moore, K.; Stevenson, M.; Young, I.S.; Gibney, J. A Cross-Sectional Study Demonstrating Increased Serum Amyloid A Related Inflammation in High-Density Lipoproteins from Subjects with Type 1 Diabetes Mellitus and How this Association Was Augmented by Poor Glycaemic Control. J. Diabetes Res. 2015, 2015, 351601. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Roxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef]
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and miR-92a Identified in Circulating HDL Discriminate between Stable and Vulnerable Coronary Artery Disease Patients. PLoS ONE 2015, 10, e0140958. [Google Scholar] [CrossRef]
- Simionescu, N.; Niculescu, L.S.; Sanda, G.M.; Margina, D.; Sima, A.V. Analysis of circulating microRNAs that are specifically increased in hyperlipidemic and/or hyperglycemic sera. Mol. Biol. Rep. 2014, 41, 5765–5773. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Li, L.; Su, B.; Yang, L.; Fan, W.; Yin, Q.; Chen, L.; Cui, T.; Zhang, J.; et al. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: Implications for glomerular endothelial injury. BMC Nephrol. 2014, 15, 142. [Google Scholar] [CrossRef]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Simionescu, N.; Niculescu, L.S.; Carnuta, M.G.; Sanda, G.M.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. Hyperglycemia Determines Increased Specific MicroRNAs Levels in Sera and HDL of Acute Coronary Syndrome Patients and Stimulates MicroRNAs Production in Human Macrophages. PLoS ONE 2016, 11, e0161201. [Google Scholar] [CrossRef]
- Florijn, B.W.; Duijs, J.; Levels, J.H.; Dallinga-Thie, G.M.; Wang, Y.; Boing, A.N.; Yuana, Y.; Stam, W.; Limpens, R.; Au, Y.W.; et al. Diabetic Nephropathy Alters the Distribution of Circulating Angiogenic MicroRNAs Among Extracellular Vesicles, HDL, and Ago-2. Diabetes 2019, 68, 2287–2300. [Google Scholar] [CrossRef]
- Chiang, C.K.; Wang, C.C.; Lu, T.F.; Huang, K.H.; Sheu, M.L.; Liu, S.H.; Hung, K.Y. Involvement of Endoplasmic Reticulum Stress, Autophagy, and Apoptosis in Advanced Glycation End Products-Induced Glomerular Mesangial Cell Injury. Sci. Rep. 2016, 6, 34167. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R.; et al. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012, 35, 1364–1379. [Google Scholar] [CrossRef]
- Wang, C.P.; Lorenzo, C.; Habib, S.L.; Jo, B.; Espinoza, S.E. Differential effects of metformin on age related comorbidities in older men with type 2 diabetes. J. Diabetes Its Complicat. 2017, 31, 679–686. [Google Scholar] [CrossRef]
- Lee, K.T.; Yeh, Y.H.; Chang, S.H.; See, L.C.; Lee, C.H.; Wu, L.S.; Liu, J.R.; Kuo, C.T.; Wen, M.S. Metformin is associated with fewer major adverse cardiac events among patients with a new diagnosis of type 2 diabetes mellitus: A propensity score-matched nationwide study. Medicine 2017, 96, e7507. [Google Scholar] [CrossRef]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef]
- Fei, Y.; Tsoi, M.F.; Cheung, B.M.Y. Cardiovascular outcomes in trials of new antidiabetic drug classes: A network meta-analysis. Cardiovasc. Diabetol. 2019, 18, 112. [Google Scholar] [CrossRef]
- Rahbar, S.; Natarajan, R.; Yerneni, K.; Scott, S.; Gonzales, N.; Nadler, J.L. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin. Chim. Acta Int. J. Clin. Chem. 2000, 301, 65–77. [Google Scholar] [CrossRef]
- Haddad, M.; Knani, I.; Bouzidi, H.; Berriche, O.; Hammami, M.; Kerkeni, M. Plasma Levels of Pentosidine, Carboxymethyl-Lysine, Soluble Receptor for Advanced Glycation End Products, and Metabolic Syndrome: The Metformin Effect. Dis. Markers 2016, 2016, 6248264. [Google Scholar] [CrossRef]
- Pereira, A.; Fernandes, R.; Crisostomo, J.; Seica, R.M.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14357. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.; Santos-Silva, D.; Rodrigues, T.; Matafome, P.; Crisostomo, J.; Sena, C.; Goncalves, L.; Seica, R. Pyridoxamine reverts methylglyoxal-induced impairment of survival pathways during heart ischemia. Cardiovasc. Ther. 2013, 31, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.M.; Soro-Paavonen, A.; Sheehy, K.; Li, J.; Calkin, A.C.; Koitka, A.; Rajan, S.N.; Brasacchio, D.; Allen, T.J.; Cooper, M.E.; et al. Delayed intervention with AGE inhibitors attenuates the progression of diabetes-accelerated atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 2011, 54, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Liang, J.T.; Tsai, P.S.; Wu, M.S.; Hsu, K.L. Prevention of arterial stiffening by pyridoxamine in diabetes is associated with inhibition of the pathogenic glycation on aortic collagen. Br. J. Pharmacol. 2009, 157, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef]
- Tam, H.L.; Shiu, S.W.; Wong, Y.; Chow, W.S.; Betteridge, D.J.; Tan, K.C. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis 2010, 209, 173–177. [Google Scholar] [CrossRef]
- Fukushima, Y.; Daida, H.; Morimoto, T.; Kasai, T.; Miyauchi, K.; Yamagishi, S.; Takeuchi, M.; Hiro, T.; Kimura, T.; Nakagawa, Y.; et al. Relationship between advanced glycation end products and plaque progression in patients with acute coronary syndrome: The JAPAN-ACS sub-study. Cardiovasc. Diabetol. 2013, 12, 5. [Google Scholar] [CrossRef]
- Cuccurullo, C.; Iezzi, A.; Fazia, M.L.; De Cesare, D.; Di Francesco, A.; Muraro, R.; Bei, R.; Ucchino, S.; Spigonardo, F.; Chiarelli, F.; et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2716–2723. [Google Scholar] [CrossRef]
- Quade-Lyssy, P.; Kanarek, A.M.; Baiersdorfer, M.; Postina, R.; Kojro, E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J. Lipid Res. 2013, 54, 3052–3061. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Rahimi, M.; Yousefi, B.; Shafiei-Irannejad, V. New potentials for 3-hydroxy-3-methyl-glutaryl-coenzymeA reductase inhibitors: Possible applications in retarding diabetic complications. J. Cell. Physiol. 2019, 234, 19393–19405. [Google Scholar] [CrossRef]
- Collins, R.; Armitage, J.; Parish, S.; Sleigh, P.; Peto, R.; Heart Protection Study Collaborative, G. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 2003, 361, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H.; et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Ramos, R.; Comas-Cufi, M.; Marti-Lluch, R.; Ballo, E.; Ponjoan, A.; Alves-Cabratosa, L.; Blanch, J.; Marrugat, J.; Elosua, R.; Grau, M.; et al. Statins for primary prevention of cardiovascular events and mortality in old and very old adults with and without type 2 diabetes: Retrospective cohort study. Bmj 2018, 362, k3359. [Google Scholar] [CrossRef] [PubMed]
- Balbi, M.E.; Tonin, F.S.; Mendes, A.M.; Borba, H.H.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Antioxidant effects of vitamins in type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, J.; Kong, D.; Yang, C.; Yu, H.; Pan, Q.; Liu, W.; Ding, Y.; Liu, H. The Effect of Antioxidant Vitamins on Patients With Diabetes and Albuminuria: A Meta-Analysis of Randomized Controlled Trials. J. Ren. Nutr. Off. J. Counc. Ren. Nutr. Natl. Kidney Found. 2020, 30, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Baziar, N.; Nasli-Esfahani, E.; Djafarian, K.; Qorbani, M.; Hedayati, M.; Mishani, M.A.; Faghfoori, Z.; Ahmaripour, N.; Hosseini, S. The Beneficial Effects of Alpha Lipoic Acid Supplementation on Lp-PLA2 Mass and Its Distribution between HDL and apoB-Containing Lipoproteins in Type 2 Diabetic Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Oxidative Med. Cell. Longev. 2020, 2020, 5850865. [Google Scholar] [CrossRef] [PubMed]
- Alem, M.M. Allopurinol and endothelial function: A systematic review with meta-analysis of randomized controlled trials. Cardiovasc. Ther. 2018, 36, e12432. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C.; et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef]
- Montero, D.; Walther, G.; Stehouwer, C.D.; Houben, A.J.; Beckman, J.A.; Vinet, A. Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. An Off. J. Int. Assoc. Study Obes. 2014, 15, 107–116. [Google Scholar] [CrossRef]
- Altunina, N.V.; Lizogub, V.G.; Bondarchuk, O.M. Alpha-Lipoic Acid as a Means of Influence on Systemic Inflammation in Type 2 Diabetes Mellitus Patients with Prior Myocardial Infarction. J. Med. Life 2020, 13, 32–36. [Google Scholar] [CrossRef]
- Stitt, A.W.; He, C.; Friedman, S.; Scher, L.; Rossi, P.; Ong, L.; Founds, H.; Li, Y.M.; Bucala, R.; Vlassara, H. Elevated AGE-modified ApoB in sera of euglycemic, normolipidemic patients with atherosclerosis: Relationship to tissue AGEs. Mol. Med. 1997, 3, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, M.; Zhang, Z.; Yu, Y.; Chen, Q.; Zhang, W.; Zhao, X. Blockade of receptor for advanced glycation end products protects against systolic overload-induced heart failure after transverse aortic constriction in mice. Eur. J. Pharmacol. 2016, 791, 535–543. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef]
- Watanabe, M.; Toyomura, T.; Wake, H.; Liu, K.; Teshigawara, K.; Takahashi, H.; Nishibori, M.; Mori, S. Differential contribution of possible pattern-recognition receptors to advanced glycation end product-induced cellular responses in macrophage-like RAW264.7 cells. Biotechnol. Appl. Biochem. 2020, 67, 265–272. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, M.; Xu, Q.; Liang, J.; Chen, R.; Xiao, Y.; Fang, M.; Chen, L. Effect of the Diabetic Environment On the Expression of MiRNAs in Endothelial Cells: Mir-149-5p Restoration Ameliorates the High Glucose-Induced Expression of TNF-alpha and ER Stress Markers. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 120–135. [Google Scholar] [CrossRef]
- Chen, Q.; Qiu, F.; Zhou, K.; Matlock, H.G.; Takahashi, Y.; Rajala, R.V.S.; Yang, Y.; Moran, E.; Ma, J.X. Pathogenic Role of microRNA-21 in Diabetic Retinopathy Through Downregulation of PPARalpha. Diabetes 2017, 66, 1671–1682. [Google Scholar] [CrossRef]
- Icli, B.; Wu, W.; Ozdemir, D.; Li, H.; Haemmig, S.; Liu, X.; Giatsidis, G.; Cheng, H.S.; Avci, S.N.; Kurt, M.; et al. MicroRNA-135a-3p regulates angiogenesis and tissue repair by targeting p38 signaling in endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5599–5614. [Google Scholar] [CrossRef]
- Venkat, P.; Cui, C.; Chopp, M.; Zacharek, A.; Wang, F.; Landschoot-Ward, J.; Shen, Y.; Chen, J. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke 2019, 50, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Gencer, B.; Kronenberg, F.; Stroes, E.S.; Mach, F. Lipoprotein(a): The revenant. Eur. Heart J. 2017, 38, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toma, L.; Stancu, C.S.; Sima, A.V. Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines 2021, 9, 18. https://doi.org/10.3390/biomedicines9010018
Toma L, Stancu CS, Sima AV. Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines. 2021; 9(1):18. https://doi.org/10.3390/biomedicines9010018
Chicago/Turabian StyleToma, Laura, Camelia Sorina Stancu, and Anca Volumnia Sima. 2021. "Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies" Biomedicines 9, no. 1: 18. https://doi.org/10.3390/biomedicines9010018
APA StyleToma, L., Stancu, C. S., & Sima, A. V. (2021). Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines, 9(1), 18. https://doi.org/10.3390/biomedicines9010018