Positron Emission Tomography for Response Evaluation in Microenvironment-Targeted Anti-Cancer Therapy

Abstract
1. Introduction
2. Glucose Metabolism of Cancer and FDG-PET
3. Quantitative Parameters for Response Evaluation with PET
4. Therapeutic Monitoring with FDG-PET
5. Machine Learning for Imaging Cancer Heterogeneity and Interpretation
6. Radiomics for Diagnosis and Assessment of Current Therapy
7. Cancer Metabolomics as a Target of Therapy and Response Evaluation with PET
8. Response Evaluation of Immune Checkpoint Inhibitor Therapy with PET
9. Tumor Microenvironment and Cancer Stem Cells as a Target of Therapy
10. Biochemical Properties of C-X-C Chemokine Receptor Type 4 and Cancer Immunotherapy
11. CXCR4-Directed Imaging and Anti-Cancer Therapy with α Particle Radiation
12. Response Evaluation of Novel Therapeutics with Molecular Imaging
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oriuchi, N.; Higuchi, T.; Ishikita, T.; Miyakubo, M.; Hanaoka, H.; Iida, Y.; Endo, K. Present role and future prospect of positron emission tomography in clinical oncology. Cancer Sci. 2006, 97, 1291–1297. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Sano, K.; Nishizawa, S.; Nakamura, M.; Ogasawara, T.; Sadato, N.; Yonekura, Y. FDG-PET for prediction of tumour aggressiveness and response to intra-arterial chemotherapy and radiotherapy in head and neck cancer. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 63–71. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Kernstine, K.H.; Faubert, B.; Do, Q.N.; Rogers, T.D.; Hensley, C.T.; Cai, J.; Torrealba, J.; Oliver, D.; Wachsmann, J.W.; Lenkinski, R.E.; et al. Does tumor FDG-PET avidity represent enhanced glycolytic metabolism in non-small cell lung cancer? Ann. Thorac. Surg. 2020, 109, 1019–1025. [Google Scholar] [CrossRef]
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Navdeep, S.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Hundshammer, C.; Braeuer, M.; Müller, C.A.; Hansen, A.E.; Schillmaier, M.; Düwel, S.; Feuerecker, B.; Glaser, S.J.; Haase, A.; Weichert, W.; et al. Simultaneous characterization of tumor cellularity and the Warburg effect with PET, MRI and hyperpolarized 13C-MRSI. Theranostics 2018, 8, 4765–4780. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Long, H.; Hu, Y.; Qiu, X.; Chen, X.; Li, Z.; Fan, D.; Ma, B.; Fan, Q. Expression of MDR1, HIF-1α and MRP1 in sacral chordoma and chordoma cell line CM-319. J. Exp. Clin. Cancer Res. 2010, 29, 158. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Rev. Cancer 2016, 16, 613–614. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef]
- Kaira, K.; Endo, M.; Abe, M.; Nakagawa, K.; Ohde, Y.; Okumura, T.; Takahashi, T.; Murakami, H.; Tsuya, A.; Nakamura, Y.; et al. Biologic correlation of 2-[18F]-fluoro-2-deoxy-d-glucose uptake on positron emission tomography in thymic epithelial tumors. J. Clin. Oncol. 2010, 28, 3746–3753. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Lodge, M.A.; Chaudhry, M.A.; Wahl, R.L. Noise considerations for PET quantification using maximum and peak standardized uptake value. J. Nucl. Med. 2012, 53, 1041–1047. [Google Scholar] [CrossRef]
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.G.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: Version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef] [PubMed]
- Guler, O.C.; Torun, N.; Yildirim, B.A.; Onal, C. Pretreatment metabolic tumour volume and total lesion glycolysis are not independent prognosticators for locally advanced cervical cancer patients treated with chemoradiotherapy. Br. J. Radiol. 2018, 91, 20170552. [Google Scholar] [CrossRef] [PubMed]
- Lodge, M.A. Repeatability of SUV in Oncologic 18F-FDG PET. J. Nucl. Med. 2017, 58, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, F.; Apostolova, I.; Oehme, L.; Kotzerke, J.; Jvan den Hoff, J. Test-Retest variability in lesion SUV and lesion SUR in 18F-FDG PET: An analysis of data from two prospective multicenter trials. J. Nucl. Med. 2017, 58, 1770–1775. [Google Scholar] [CrossRef]
- Blautzik, J.; Grelich, L.; Schramm, N.; Henkel, R.; Bartenstein, P.; Pfluger, T. What and how should we measure in paediatric oncology FDG-PET/CT? Comparison of commonly used SUV metrics for differentiation between paediatric tumours. EJNMMI Res. 2019, 9, 115. [Google Scholar] [CrossRef]
- Tang, L.; Wang, X.-J.; Baba, H.; Giganti, F. Gastric cancer and image-derived quantitative parameters: Part 2—A critical review of DCE-MRI and 18F-FDG PET/CT findings. Eur. Radiol. 2020, 30, 247–260. [Google Scholar] [CrossRef]
- Endoh, H.; Yamamoto, R.; Ichikawa, A.; Shiozawa, S.; Nishizawa, N.; Satoh, Y.; Oriuchi, N. Prognostic impact of preoperative FDG-PET positive lymph nodes in lung cancer. Int. J. Clin. Oncol. 2020, in press. [Google Scholar] [CrossRef]
- Chen, K.Y.; Cypess, A.M.; Laughlin, M.R.; Haft, C.R.; Hu, H.H.; Bredella, M.A.; Enerbäck, S.; Kinahan, P.E.; van Marken Lichtenbelt, W.; Lin, F.I.; et al. Brown adipose reporting criteria in imaging studies (BARCIST 1.0): Recommendations for standardized FDG-PET/CT experiments in Humans. Cell Metab. 2016, 24, 210–222. [Google Scholar] [CrossRef]
- Burger, I.A.; Casanova, R.; Steiger, S.; Husmann, L.; Stolzmann, P.; Huellner, M.W.; Curioni, A.; Hillinger, S.C.; Schmidtlein, C.R.; Soltermann, A. 18F-FDG PET/CT of non-small cell lung carcinoma under neoadjuvant chemotherapy: Background-Based adaptive-volume metrics outperform TLG and MTV in predicting histopathologic response. J. Nucl. Med. 2016, 57, 849–854. [Google Scholar] [CrossRef]
- Minamimoto, R.; Takeda, Y.; Hotta, M.; Toyohara, J.; Nakajima, K.; Naka, G.; Sugiyama, H. 18F-FDG and 11C-4DST PET/CT for evaluating response to platinum-based doublet chemotherapy in advanced non-small cell lung cancer: A prospective study. EJNMMI Res. 2019, 9, 4. [Google Scholar] [CrossRef]
- Higuchi, T.; Fujimoto, Y.; Ozawa, H.; Bun, A.; Fukui, R.; Miyagawa, Y.; Imamura, M.; Kitajima, K.; Yamakado, K.; Miyoshi, Y. Significance of metabolic tumor volume at baseline and reduction of mean standardized uptake value in 18F-FDG-PET/CT imaging for predicting pathological complete response in breast cancers treated with preoperative chemotherapy. Ann. Surg. Oncol. 2019, 26, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cheon, G.J.; Kang, S.Y.; Paeng, J.C.; Kang, K.W.; Lee, D.S.; Chung, J.K. Prognostic value of simultaneous 18F-FDG PET/MRI using a combination of metabolo-volumetric parameters and apparent diffusion coefficient in treated head and neck cancer. EJNMMI Res. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, J.; Li, X.; Xie, L.; Qin, L.; Peng, F.; Cheng, M. Prognostic value of metabolic tumor volume and total lesion glycolysis from 18F-FDG PET/CT in lymph node metastases and risk stratification of endometrial carcinoma. J. Gynecol. Oncol. 2019, 30, e89. [Google Scholar] [CrossRef] [PubMed]
- Im, H.J.; Pak, K.; Cheon, G.J.; Kang, K.W.; Kim, S.J.; Kim, I.J.; Chung, J.K.; Kim, E.E.; Lee, D.S. Prognostic value of volumetric parameters of (18)F-FDG PET in non-small-cell lung cancer: A meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Young, H.; Baum, R.; Cremerius, U.; Herholz, K.; Hoekstra, O.; Lammertsma, A.A.; Pruim, J.; Price, P. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: Review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur. J. Cancer 1999, 35, 1773–1782. [Google Scholar] [CrossRef]
- Wahl, R.L.; Jacene, H.; Kasamon, Y.; Lodge, M.A. From RECIST to PERCIST: Evolving considerations for PET response criteria in solid tumors. J. Nucl. Med. 2009, 50, 122S–150S. [Google Scholar] [CrossRef]
- Sunaga, N.; Oriuchi, N.; Kaira, K.; Yanagitani, N.; Tomizawa, Y.; Hisada, T.; Ishizuka, T.; Endo, K.; Mori, M. Usefulness of FDG-PET for early prediction of the response to gefitinib in non-small cell lung cancer. Lung Cancer 2008, 59, 203–210. [Google Scholar] [CrossRef]
- Desar, I.M.; van Herpen, C.M.; van Laarhoven, H.W.; Barentsz, J.O.; van der Graaf, W.T. Beyond RECIST: Molecular and functional imaging techniques for evaluation of response to targeted therapy. Cancer Treat Rev. 2009, 35, 309–321. [Google Scholar] [CrossRef]
- Milano, A.; Perri, F.; Ciarmiello, A.; Caponigro, F. Targeted-Therapy and imaging response: A new paradigm for clinical evaluation? Rev. Recent Clin. Trials 2011, 6, 259–265. [Google Scholar] [CrossRef]
- Abdel-Wahab, N.; Shah, M.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E. Use of immune checkpoint inhibitors in the treatment of patients with cancer and preexisting autoimmune disease: A systematic review. Ann. Intern. Med. 2018, 168, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Higuchi, T.; Naruse, I.; Arisaka, Y.; Tokue, A.; Altan, B.; Suda, S.; Mogi, A.; Shimizu, K.; Sunaga, N.; et al. Metabolic activity by 18F-FDG-PET/CT is predictive of early response after nivolumab in previously treated NSCLC. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sachpekidis, C.S.; Hassel, J.C.; Dimitrakopoulou-Strauss, A. 18F-FDG PET/CT reveals disease remission in a patient with Ipilimumab refractory advanced melanoma treated with Pembrolizumab. Clin. Nucl. Med. 2016, 41, 156–158. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Ehlerding, E.B.; Hernandez, R.; Rekoske, B.T.; Graves, S.A.; Sun, H.; Liu, G.; McNeel, D.G.; Barnhart, T.E.; Cai, W. Preclinical pharmacokinetics and biodistribution studies of 89Zr-labeled Pembrolizumab. J. Nucl. Med. 2017, 58, 162–168. [Google Scholar] [CrossRef]
- Tanaka, F.; Takenaka, K.; Oyanagi, H.; Fujinaga, T.; Otake, Y.; Yanagihara, K.; Ito, H.; Wada, H. Skip mediastinal nodal metastases in non-small cell lung cancer. Eur. J. Cardio-Thorac. Surg. 2004, 25, 1114–1120. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Z.; Li, Y.; Chen, Z.; Lu, P.; Wang, W.; Liu, W.; Yu, L. Comparison of machine learning methods for classifying mediastinal lymph node metastasis of non-small cell lung cancer from 18F-FDG PET/CT images. EJNMMI Res. 2017, 7, 11. [Google Scholar] [CrossRef]
- Erickson, B.J.; Korfiatis, P.; Akkus, Z.; Kline, T.L. Machine learning for medical imaging. Radiographics 2017, 37, 505–515. [Google Scholar] [CrossRef]
- Endoh, H.; Yamamoto, R.; Ichikawa, A.; Shiozawa, S.; Nishizawa, N.; Satoh, Y.; Oriuchi, N. Clinicopathological significance of false-positive lymph node status on 18F-FDG PET in lung cancer. Clin. Lung Cancer 2020. [Google Scholar] [CrossRef]
- Choy, G.; Khalilzadeh, O.; Michalski, M.; Do, S.; Samir, A.E.; Pianykh, O.S.; Geis, J.R.; Pandharipande, P.V.; Brink, J.A.; Dreyer, K.J. Current applications and future impact of machine learning in radiology. Radiology 2018, 288, 318–328. [Google Scholar] [CrossRef]
- Betancur, J.; Rubeaux, M.; Fuchs, T.A.; Otaki, Y.; Arnson, Y.; Slipczuk, L.; Benz, D.C.; Germano, G.; Dey, D.; Lin, C.J.; et al. Automatic valve plane localization in myocardial perfusion SPECT/CT by machine learning: Anatomic and clinical validation. J. Nucl. Med. 2017, 58, 961–967. [Google Scholar] [CrossRef]
- Kolossváry, M.; Park, J.; Bang, J.I.; Zhang, J.; Lee, J.M.; Paeng, J.C.; Merkely, B.; Narula, J.; Kubo, T.; Akasaka, T.; et al. Identification of invasive and radionuclide imaging markers of coronary plaque vulnerability using radiomic analysis of coronary computed tomography angiography. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.M.; Levin, D.C.; Parker, L.; Cavanaugh, B.; Frangos, A.J.; Sunshine, J.H. How widely is computer-aided detection used in screening and diagnostic mammography? J. Am. Coll. Radiol. 2010, 7, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.J.; Abraham, L.; Taplin, S.H.; Geller, B.M.; Carney, P.A.; D’Orsi, C.; Elmore, J.G.; Barlow, W.E. Breast cancer surveillance consortium. effectiveness of computer-aided detection in community mammography practice. J. Natl. Cancer Inst. 2011, 103, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.L.; Siegel, E.L. Legal ramifications of computer-aided detection in mammography. J. Am. Coll. Radiol. 2015, 12, 572–574. [Google Scholar] [CrossRef]
- Halabi, S.S.; Prevedello, L.M.; Kalpathy-Cramer, J.; Mamonov, A.B.; Bilbily, A.; Cicero, M.; Pan, I.; Araújo Pereira, L.; Sousa, R.T.; Abdala, N.; et al. The RSNA pediatric bone age machine learning challenge. Radiology 2019, 290, 498–503. [Google Scholar] [CrossRef]
- Chan, S.; Siegel, E.L. Will machine learning end the viability of radiology as a thriving medical specialty? Br. J. Radiol. 2019, 92, 20180416. [Google Scholar] [CrossRef]
- Public Workshop—Evolving Role of Artificial Intelligence in Radiological Imaging; Comments of the American Colloge of Radiology. Available online: https://www.acr.org/-/media/ACR/NOINDEX/Advocacy/acr rsna comments fda-ai-evolvingrole-ws 6-30-2020.pdf (accessed on 20 August 2020).
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef]
- Nioche, C.; Orlhac, F.; Boughdad, S.; Reuzé, S.; Goya-Outi, J.; Robert, C.; Pellot-Barakat, C.; Soussan, M.; Frouin, F.; Buvat, I. LIFEx: A freeware for radiomic feature calculation in multimodality imaging to accelerate advances in the characterization of tumor heterogeneity. Cancer Res. 2018, 78, 4786–4789. [Google Scholar] [CrossRef]
- Orlhac, F.; Nioche, C.; Soussan, M.; Buvat, I. Understanding changes in tumor texture indices in PET: A comparison between visual assessment and index values in simulated and patient data. J. Nucl. Med. 2017, 58, 387–392. [Google Scholar] [CrossRef]
- Pyka, T.; Gempt, J.; Hiob, D.; Ringel, F.; Schlegel, J.; Bette, S.; Wester, H.J.; Meyer, B.; Förster, S. Textural analysis of pre-therapeutic [18F]-FET-PET and its correlation with tumor grade and patient survival in high-grade gliomas. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 133–141. [Google Scholar] [CrossRef]
- Hatt, M.; Tixier, F.; Visvikis, D. Cheze le rest C. Radiomics in PET/CT: More than meets the eye? J. Nucl. Med. 2017, 58, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Fornacon-Wood, I.; Faivre-Finn, C.; O’Connor, J.P.B.; Price, G.J. Radiomics as a personalized medicine tool in lung cancer: Separating the hope from the hype. Lung Cancer 2020, 146, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Avanzo, M.; Stancanello, J.; El Naqa, I. Beyond imaging: The promise of radiomics. Phys. Med. 2017, 38, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Parmar, C.; Grossmann, P.; Quackenbush, J.; Lambin, P.; Bussink, J.; Mak, R.; Aerts, H.J.W.L. Exploratory study to identify radiomics classifiers for lung cancer histology. Front. Oncol. 2016, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.A.; Thornton, A.; Lu, H.; Tam, H.; Wallitt, K.; Rodgers, N.; Scarsbrook, A.; McDermott, G.; Cook, G.J.; Landau, D.; et al. Discovery of pre-therapy 2-deoxy-2-18F-fluoro-d-glucose positron emission tomography-based radiomics classifiers of survival outcome in non-small-cell lung cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Crispin-Ortuzar, M.; Apte, A.; Grkovski, M.; Oh, J.H.; Lee, N.Y.; Schöder, H.; Humm, J.L.; Deasy, J.O. Predicting hypoxia status using a combination of contrast-enhanced computed tomography and [18F]-Fluorodeoxyglucose positron emission tomography radiomics features. Radiother. Oncol. 2018, 127, 36–42. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.; Guo, W.; Drukker, K.; Lan, L.; Giger, M.L.; Ji, Y. Deciphering genomic underpinnings of quantitative MRI-based radiomic phenotypes of invasive breast carcinoma. Sci. Rep. 2015, 5, 17787. [Google Scholar] [CrossRef]
- Zinn, P.O.; Singh, S.K.; Kotrotsou, A.; Hassan, I.; Thomas, G.; Luedi, M.M.; Elakkad, A.; Elshafeey, N.; Idris, T.; Mosley, J.; et al. A co-clinical radiogenomic validation study—Conserved magnetic resonance radiomic appearance of Periostin expressing Glioblastoma in patients and xenograft models. Clin. Cancer Res. 2018, 24, 6288–6299. [Google Scholar] [CrossRef]
- Lu, H.; Arshad, M.; Thornton, A.; Avesani, G.; Cunnea, P.; Curry, E.; Kanavati, F.; Liang, J.; Nixon, K.T.; Williams, S.T.; et al. A mathematical-descriptor of tumor-mesoscopic-structure from computed-tomography images annotates prognostic- and molecular-phenotypes of epithelial ovarian cancer. Nat. Commun. 2019, 10, 764. [Google Scholar] [CrossRef]
- Diehn, M.; Nardini, C.; Wang, D.S.; McGovern, S.; Jayaraman, M.; Liang, Y.; Aldape, K.; Cha, S.; Kuo, M.D. Identification of noninvasive imaging surrogates for brain tumor gene-expression modules. Proc. Natl. Acad. Sci. USA 2008, 105, 5213–5218. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, J.; Balagurunathan, Y.; Li, Q.; Garcia, A.L.; Stringfield, O.; Ye, Z.; Gillies, R.J. Radiomic features are associated with EGFR mutation status in lung adenocarcinomas. Clin. Lung Cancer 2016, 17, 441–448. [Google Scholar] [CrossRef]
- Yoon, H.J.; Sohn, I.; Cho, J.H.; Lee, H.Y.; Kim, J.H.; Choi, Y.L.; Kim, H.; Lee, G.; Lee, K.S.; Kim, J. Decoding tumor phenotypes for ALK, ROS1, and RET fusions in lung adenocarcinoma using a radiomics approach. Medicine (Baltimore) 2015, 94, e1753. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Zalcman, G.; Crino, L.; Biondani, P.; Barlesi, F.; Filleron, T.; Dingemans, A.M.C.; Léna, H.; Monnet, I.; Rothschild, S.I.; et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: Results from the EUROS1 cohort. J. Clin. Oncol. 2015, 33, 992–999. [Google Scholar] [CrossRef]
- Li, H.; Zhang, R.; Wang, S.; Fang, M.; Zhu, Y.; Hu, Z.; Dong, D.; Shi, J.; Tian, J. CT-Based Radiomic Signature as a Prognostic Factor in Stage IV ALK-Positive Non-small-cell Lung Cancer Treated With TKI Crizotinib: A Proof-of-Concept Study. Front. Oncol. 2020, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.; Rose, C.J.; Waterton, J.C.; Carano, R.A.; Parker, G.J.; Jackson, A. Imaging intratumor heterogeneity: Role in therapy response, resistance, and clinical outcome. Clin. Cancer Res. 2015, 21, 249–257. [Google Scholar] [CrossRef]
- Park, S.; Ha, S.; Lee, S.H.; Paeng, J.C.; Keam, B.; Kim, T.M.; Kim, D.W.; Heo, D.S. Intratumoral heterogeneity characterized by pretreatment PET in non-small cell lung cancer patients predicts progression-free survival on EGFR tyrosine kinase inhibitor. PLoS ONE 2018, 13, e0189766. [Google Scholar] [CrossRef]
- Nie, K.; Shi, L.; Chen, Q.; Hu, X.; Jabbour, S.K.; Yue, N.; Niu, T.; Sun, X. Rectal cancer: Assessment of neoadjuvant chemo-radiation outcome based on radiomics of multi-parametric MRI. Clin. Cancer Res. 2016, 22, 5256–5263. [Google Scholar] [CrossRef]
- Oikonomou, A.; Khalvati, F.; Tyrrell, P.N.; Haider, M.A.; Tarique, U.; Jimenez-Juan, L.; Tjong, M.C.; Poon, I.; Eilaghi, A.; Lisa Ehrlich, L. Radiomics analysis at PET/CT contributes to prognosis of recurrence and survival in lung cancer treated with stereotactic body radiotherapy. Sci. Rep. 2018, 8, 4003. [Google Scholar] [CrossRef]
- Gnep, K.; Fargeas, A.; Gutierrez-Carvajal, R.E.; Commandeur, F.; Mathieu, R.; Ospina, J.D.; Rolland, Y.; Rohou, T.; Vincendeau, S.; Hatt, M. Haralick textural features on T2 -weighted MRI are associated with biochemical recurrence following radiotherapy for peripheral zone prostate cancer. J. Magn. Reson. Imaging 2017, 45, 103–117. [Google Scholar] [CrossRef]
- Liu, J.; Mao, Y.; Li, Z.; Zhang, D.; Zhang, Z.; Hao, S.; Li, B. Use of texture analysis based on contrast-enhanced MRI to predict treatment response to chemoradiotherapy in nasopharyngeal carcinoma. J. Magn. Reson. Imaging 2016, 44, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Aerts, H.J. The potential of radiomic-based phenotyping in precision medicine: A review. JAMA Oncol. 2016, 2, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Gotz, M.; Muschelli, J.; Wick, A.; Neuberger, U.; Shinohara, R.T.; Sill, M.; Nowosielski, M.; Schlemmer, H.P.; Radbruch, A. Large-scale radiomic profiling of recurrent glioblastoma identifies an imaging predictor for stratifying anti-angiogenic treatment response. Clin. Cancer Res. 2016, 22, 5765–5771. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Limkin, E.J.; Vakalopoulou, M.; Dercle, L.; Champiat, S.; Han, S.R.; Verlingue, L.; Brandao, D.; Lancia, A.; Ammari, S. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti- PD-L1 immunotherapy: An imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018, 19, 1180–1191. [Google Scholar] [CrossRef]
- Moran, A.; Daly, M.E.; Yip, S.S.F.; Yamamoto, T. Radiomics-based assessment of radiation-induced lung injury after stereotactic body radiotherapy. Clin. Lung Cancer 2017, 18, e425–e431. [Google Scholar] [CrossRef] [PubMed]
- Mattonen, S.A.; Palma, D.A.; Johnson, C.; Louie, A.V.; Landis, M.; Rodrigues, G.; Chan, I.; Etemad-Rezai, R.; Yeung, T.P.C.; Senan, S. Detection of local cancer recurrence after stereotactic ablative radiation therapy for lung cancer: Physician performance versus radiomic assessment. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, L.V.; Noordzij, W.; Brouwer, C.L.; Boellaard, R.; Burgerhof, J.G.M.; Langendijk, J.A.; Sijtsema, N.M.; Steenbakkers, R.J.H.M. 18F-FDG PET image biomarkers improve prediction of late radiation-induced xerostomia. Radiother. Oncol. 2018, 126, 89–95. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, D.; Oh, A.; Mahmud, D.L.; et al. A novel l-asparaginase with low L-glutaminase coactivity is highly efficacious against both T and B cell acute lymphoblastic leukemias in vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef]
- Fung, M.K.L.; Chan, G.C.F. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017, 10, 144. [Google Scholar] [CrossRef]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.-C.; Vandekeere, S.; Bouche, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Pauleit, D.; Stoffels, G.; Schaden, W.; Hamacher, K.; Bauer, F.; Tellmann, L.; Herzog, H.; Bröer, S.; Coenen, H.H.; Langen, K.J. PET with O-(2-18F fluoroethyl)-L-tyrosine in peripheral tumors: First clinical results. J. Nucl. Med. 2005, 46, 411–416. [Google Scholar] [PubMed]
- Jager, P.L.; Groen, H.J.M.; van der Leest, A.; van Putten, J.W.; Pieterman, R.M.; de Vries, E.G.; Piers, D.A. L-3-123I-iodo-amethyl-L-tyrosine SPECT in non-small cell lung cancer: Preliminary observations. J. Nucl. Med. 2001, 42, 579–585. [Google Scholar] [PubMed]
- Kaira, K.; Oriuchi, N.; Otani, Y.; Shimizu, K.; Tanaka, S.; Imai, H.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Ishizuka, T. Fluorine-18-α-methyltyrosine positron emission tomography for diagnosis and staging of lung cancer: A clinicopathologic study. Clin. Cancer Res. 2007, 13, 6369–6378. [Google Scholar] [CrossRef]
- Tomiyoshi, K.; Inoue, T.; Higuchi, T.; Ahmed, K.; Sarwar, M.; Alyafei, S.; Zhang, H.; Matsubara, K.; Endo, K.; Yang, D. Metabolic studies of [18F-alpha-methyl]tyrosine in mice bearing colorectal carcinoma LS-180. Anti-Cancer Drugs 1999, 10, 329–336. [Google Scholar] [CrossRef]
- Wei, L.; Tominaga, H.; Ohgaki, R.; Wiriyasermkul, P.; Hagiwara, K.; Okuda, S.; Kaira, K.; Oriuchi, N.; Nagamori, S.; Kanai, Y. Specific transport of 3-fluoro-l-α-methyl-tyrosine by LAT1 explains its specificity to malignant tumors in imaging. Cancer Sci. 2016, 107, 347–352. [Google Scholar] [CrossRef]
- Suzuki, S.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Sohda, M.; Yokobori, T.; Miyazaki, T.; Oriuchi, N.; Tominaga, H.; Kanai, Y.; et al. Biological significance of fluorine-18-α-methyltyrosine (FAMT) uptake on PET in patients with oesophageal cancer. Br. J. Cancer 2014, 110, 1985–1991. [Google Scholar] [CrossRef]
- Kaira, K.; Higuchi, T.; Sunaga, N.; Arisaka, Y.; Hisada, T.; Tominaga, H.; Oriuchi, N.; Asao, T.; Tsushima, Y.; Yamada, M. Usefulness of 18F-α-Methyltyrosine PET for therapeutic monitoring of patients with advanced Lung Cancer. Anti-Cancer Res. 2016, 36, 6481–6490. [Google Scholar] [CrossRef]
- Shimizu, K.; Kaira, K.; Higuchi, T.; Hisada, T.; Yokobori, T.; Oyama, T.; Asao, T.; Tsushima, Y.; Shirabe, K. Relationship between tumor immune markers and Fluorine-18-α-Methyltyrosine ([18F]FAMT) uptake in PAtients with Lung Cancer. Mol. Imaging Biol. 2020, 22, 1078–1086. [Google Scholar] [CrossRef]
- Pascal Häfliger, P.; Charles, R.P. The L-Type amino acid transporter LAT1—An emerging target in cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef]
- Okano, N.; Kawai, K.; Yamauchi, Y.; Kobayashi, T.; Naruge, D.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, L-type amino acids transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed]
- Egler, R.A.; Ahuja, S.P.; Matloub, Y. l-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J. Pharmacol. Pharmacother. 2016, 7, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.; Luengo, A.; Mayers, J.M.; Wyckoff, J.; Del Rosario, A.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mouron, S.; Munoz, J.; Gomez-Lopez, G.; Jimenez-Renard, V.; Mulero, F.; et al. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Chiou, V.L.; Burotto, M. Pseudoprogression and immune-related response in solid tumors. J. Clin. Oncol. 2015, 33, 3541–3543. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Jagannathan, J.P.; Krajewski, K.M.; O’Regan, K.; Hatabu, H.; Shapiro, G.; Ramaiya, N.H. Personalized tumor response assessment in the era of molecular medicine: Cancer-Specific and therapy-specific response criteria to complement pitfalls of RECIST. AJR Am. J. Roentgenol. 2012, 198, 737–745. [Google Scholar] [CrossRef]
- Nishino, M.; Giobbie-Hurder, A.; Gargano, M.; Suda, M.; Ramaiya, N.H.; Hodi, F.S. Developing a common language for tumor response to immunotherapy: Immune-Related response criteria using unidimensional measurements. Clin. Cancer Res. 2013, 19, 3936–3943. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, R.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef]
- Natarajan, A.; Mayer, A.T.; Reeves, R.E.; Nagamine, C.M.; Gambhir, S.S. Development of novel ImmunoPET tracers to image human PD-1 checkpoint expression on tumor-infiltrating lymphocytes in a humanized mouse model. Mol. Imaging Biol. 2017, 19, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, S.; Hobo, W.; Molkenboer-Kuenen, J.D.; Olive, D.; Oyen, W.J.D.; Dolstra, H.; Boerman, O.C. Noninvasive imaging of tumor PD-L1 expression using radiolabeled anti-PD-L1 antibodies. Cancer Res. 2015, 75, 2928–2936. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Caraballo, A.; Mahmood, U. Quantitative CD3 PET imaging predicts tumor growth response to anti-CTLA-4 therapy. J. Nucl. Med. 2016, 57, 1607–1611. [Google Scholar] [CrossRef]
- Mayer, A.T.; Natarajan, A.; Gordon, S.R.; Maute, R.L.; McCracken, M.N.; Ring, A.M.; Weissman, I.L.; Gambhir, S.S. Practical Immuno-PET radiotracer design considerations for human immune checkpoint imaging. J. Nucl. Med. 2017, 58, 538–546. [Google Scholar] [CrossRef]
- Maute, R.L.; Gordon, S.R.; Mayer, A.T.; McCracken, M.N.; Natarajan, A.; Ring, N.G.; Kimura, R.; Tsai, J.M.; Manglik, A.; Kruse, A.C.; et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E6506–E6514. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Inoue, M.; Itasaka, S.; Hirota, K.; Morinibu, A.; Shinomiya, K.; Zeng, L.; Ou, G.; Zhu, Y.; Yoshimura, M.; et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat. Commun. 2012, 3, 783. [Google Scholar] [CrossRef]
- Pantel, K.; Speicher, M.R. The biology of circulating tumor cells. Oncogene 2016, 35, 1216–1224. [Google Scholar] [CrossRef]
- Francart, M.E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial–mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev. Dyn. 2018, 247, 432–450. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. (Berlin) 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Li, J.; Ai, Y.; Wang, L.; Bu, P.; Sharkey, C.C.; Wu, Q.; Wun, B.; Roy, S.; Shen, X.; King, M.R. Targeted drug delivery to circulating tumor cells via platelet membrane-functionalized particles. Biomaterials 2016, 76, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Y.; Zhong, J. Side population cells and drug resistance in breast cancer. Mol. Med. Rep. 2015, 11, 4297–4302. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, B.; Zhang, Z.; Liu, X.; Qi, X.; Zhao, J.; Jiang, Y.; Zhai, H.; Ji, Y.; Luo, D. Stem cell-like circulating tumor cells indicate poor prognosis in gastric cancer. Biomed. Res. Int. 2014, 2014, 981261. [Google Scholar] [CrossRef]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.O.; Johansen, T.; Juhász, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 67. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Oh, M.; Tanaka, T.; Kobayashi, M.; Furukawa, T.; Mori, T.; Kudo, T.; Fujieda, S.; Fujibayashi, Y. Radio-copper-labeled Cu-ATSM: An indicator of quiescent but clonogenic cells under mild hypoxia in a Lewis lung carcinoma model. Nucl. Med. Biol. 2009, 36, 419–426. [Google Scholar] [CrossRef]
- Yoshii, Y.; Furukawa, T.; Kiyono, Y.; Watanabe, R.; Waki, A.; Mori, T.; Yoshii, H.; Oh, M.; Asai, T.; Okazawa, H.; et al. Copper-64-diacetyl-bis (N4-methylthiosemicarbazone) accumulates in rich regions of CD133+ highly tumorigenic cells in mouse colon carcinoma. Nucl. Med. Biol. 2010, 37, 395–404. [Google Scholar] [CrossRef]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L.A. CD133 as a biomarker for putative cancer stem cells in solid tumours: Limitations, problems and challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Gaedicke, S.; Braun, F.; Prasad, S.; Machein, M.; Firat, E.; Hettich, M.; Gudihal, R.; Zhu, X.; Klingner, K.; Schüler, J. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E692–E701. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/XCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.F.; Clarke, C.; Stordal, B.; Carey, M.S.; Broaddus, R.; Gallagher, W.M.; Crown, J.; Mills, G.; Bryan, T.; Hennessy, B.T.; et al. OvMark: A user-friendly system for the identification of prognostic biomarkers in publically available ovarian cancer gene expression datasets. Mol. Cancer 2014, 13, 241. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019, 35, 588–602. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.R.; et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef]
- Eckert, F.; Schilbach, K.; Klumpp, L.; Bardoscia, L.; Sezgin, E.C.; Schwab, M.; Zips, D.; Huber, S.M. Potential Role of CXCR4 Targeting in the Context of Radiotherapy and Immunotherapy of Cancer. Front. Immunol. 2018, 9, 3018. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, B.; Liang, Y.; Reeves, P.M.; Qu, X.; Ran, C.; Liu, Q.; Callahan, M.V.; Sluder, A.E.; Gelfand, J.A.; et al. Dual blockade of CXCL12-CXCR4 and PD-1–PD-L1 pathways prolongs survival of ovarian tumor–bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J. 2019, 33, 6596–6608. [Google Scholar] [CrossRef]
- D’Alterio, C.; Buoncervello, M.; Ieranò, C.; Napolitano, M.; Portella, L.; Rea, G.; Barbieri, A.; Luciano, A.; Scognamiglio, G.; Tatangelo, F.; et al. Targeting CXCR4 potentiates anti-PD-1 efficacy modifying the tumor microenvironment and inhibiting neoplastic PD-1. J. Exp. Clin. Cancer Res. 2019, 38, 432. [Google Scholar] [CrossRef]
- Singh, P.; Mohammad, K.S.; Pelus, L.M. CXCR4 expression in the bone marrow microenvironment is required for hematopoietic stem and progenitor cell maintenance and early hematopoietic regeneration after myeloablation. Stem Cells 2020, 38, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.J.; Keller, U.; Schottelius, M.; Beer, A.; Philipp-Abbrederis, K.; Hoffmann, F.; Šimeček, J.; Gerngross, C.; Lassmann, M.; Herrmann, K.; et al. Disclosing the CXCR4 expression in lymphoproliferative diseases by targeted molecular imaging. Theranostics 2015, 5, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Vag, T.; Gerngross, C.; Herhaus, P.; Eiber, M.; Philipp-Abbrederis, K.; Graner, F.P.; Ettl, J.; Keller, U.; Wester, H.J.; Schwaiger, M. First experience with chemokine receptor CXCR4-targeted PET imaging of patients with solid cancers. J. Nucl. Med. 2016, 57, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Abraham, M.; Avivi, I.J.; Rowe, J.M.; Beider, K.; Wald, H.; Tiomkin, L.; Ribakovsky, L.; Riback, Y.; Ramati, Y.; et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin. Cancer Res. 2014, 20, 469–479. [Google Scholar] [CrossRef]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1 alpha)-CXCR4/CXCR7 pathway inhibition: An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef]
- Broussas, M.; Boute, N.; Akla, B.; Berger, S.; Beau-Larvor, C.; Champion, T.; Robert, A.; Beck, A.J.; Haeuw, J.F.; Goetsch, L.; et al. A new anti-CXCR4 antibody that blocks the CXCR4/SDF-1 axis and mobilizes effector cells. Mol. Cancer Ther. 2016, 15, 1890–1899. [Google Scholar] [CrossRef]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hanscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [177Lu]pentixather: Comprehensive preclinical characterization of a first CXCR4-directed endoradiotherapeutic agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D.; Szajek, L.; Farber, J.M.; Kiesewetter, D.O. 64Cu-AMD3100: A novel imaging agent for targeting chemokine receptor CXCR4. Bioorg. Med. Chem. 2009, 17, 1486–1493. [Google Scholar] [CrossRef]
- Burke, B.P.; Miranda, C.S.; Lee, R.E.; Renard, I.; Nigam, S.; Clemente, G.S.; D’Huys, T.; Ruest, T.; Domarkas, J.; Thompson, J.A.; et al. 64Cu PET imaging of the CXCR4 chemokine receptor using a cross-bridged Cyclam bis-tetraazamacrocyclic antagonist. J. Nucl. Med. 2020, 61, 123–128. [Google Scholar] [CrossRef]
- Poschenrieder, A.; Osl, T.; Schottelius, M.; Hoffmann, F.; Wirtz, M.; Schwaiger, M.; Wester, H.J. First 18F-labeled pentixafor-based imaging agent for PET imaging of CXCR4 expression in vivo. Tomography 2016, 2, 85–93. [Google Scholar] [CrossRef]
- Sehedic, D.; Chourpa, I.; Tetaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindre, F.; et al. Locoregional confinement and major clinical benefit of 188Re-Loaded CXCR4-targeted nanocarriers in an orthotopic human to mouse model of glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Lewington, V.; Shore, N.; Kratochwil, C.; Levy, M.; Lindén, O.; Noordzij, W.; Park, J.; Saad, F. Targeted alpha therapy, an emerging class of cancer agents: A review. JAMA Oncol. 2018, 4, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.-K.V.; Press, O. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical Experience with a Particle–Emitting 211At: Treatment of Recurrent Brain Tumor Patients with 211At-Labeled Chimeric Antitenascin Monoclonal Antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar] [CrossRef]
- Andersson, H.; Cederkrantz, E.; Bäck, T.; Divgi, C.; Elgqvist, J.; Himmelman, J.; Horvath, G.; Jacobsson, L.; Jensen, H.; Lindegren, S.; et al. Intraperitoneal a-Particle Radioimmunotherapy of Ovarian Cancer Patients: Pharmacokinetics and Dosimetry of 211At-MX35 F(ab)2—A Phase I Study. J. Nucl. Med. 2009, 50, 1153–1160. [Google Scholar] [CrossRef]
- Oriuchi, N.; Aoki, M.; Ukon, N.; Washiyama, K.; Shimoyama, S.; Nishijima, K.; Takahashi, K.; Ito, H.; Ikezoe, T.; Songji, Z. Possibility of cancer stem cells-targeted radioimmunotherapy for acute myelogenous leukemia with 211At-CXCR4 monoclonal antibody. Sci. Rep. 2020, 10, 6810. [Google Scholar] [CrossRef]
- Ehlerding, E.B.; England, C.G.; McNeel, D.G.; Cai, W. Molecular imaging of immunotherapy targets in cancer. J. Nucl. Med. 2016, 57, 1487–1492. [Google Scholar] [CrossRef]
- Ahrens, E.T.; Bulte, J.W. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 2013, 13, 755–763. [Google Scholar] [CrossRef]
- Yongtao, Z.; Jiongwei, H.; Tongming, Z.; Ronggang, L.; Zhifu, W.; Fukai, M.; Jianhong, Z. Stem Cell Tracking Technologies for Neurological Regenerative Medicine Purposes. Stem Cells Int. 2017, 2017, 2934149. [Google Scholar] [CrossRef]
- Zeelen, C.; Paus, C.; Draper, D.; Heskamp, S.; Signore, A.; Galli, F.; Griessinger, C.M.; Aarntzen, E.H. In vivo imaging of tumor-infiltrating immune cells: Implications for cancer immunotherapy. Q. J. Nucl. Med. Mol. Imaging 2018, 62, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.; Mileshkin, L.; Wall, D.; Bartholeyns, J.; Thompson, M.; Coverdale, J.; Lau, E.; Wong, J.; Eu, P.; Hicks, R.J.; et al. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol. Immunother. 2007, 56, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pandey, M.K.; Demirhan, Y.E.; Nesbitt, J.J.; Crespo-Diaz, R.J.; Terzic, A.; Behfar, A.; DeGrado, T.R. Novel 89Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Meller, B.; Frohn, C.; Brand, J.M.; Lauer, I.; Schelper, L.F.; von Hof, K.; Kirchner, H.; Richter, E.; Baehre, M. Monitoring of a new approach of immunotherapy with allogenic 111In-labelled NK cells in patients with renal cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 403–407. [Google Scholar] [CrossRef]
- Melder, R.J.; Brownell, A.L.; Shoup, T.M.; Brownell, G.L.; Jain, R.K. Imaging of activated natural killer cells in mice by positron emission tomography: Preferential uptake in tumors. Cancer Res. 1993, 53, 5867–5871. [Google Scholar]
- Quillien, V.; Moisan, A.; Lesimple, T.; Leberre, C.; Toujas, L. Biodistribution of 111indium-labeled macrophages infused intravenously in patients with renal carcinoma. Cancer Immunol. Immunother. 2001, 50, 477–482. [Google Scholar] [CrossRef]
- Weissleder, R.; Nahrendorf, M.; Pittet, M.J. Imaging macrophages with nanoparticles. Nat. Mater. 2014, 13, 125–138. [Google Scholar] [CrossRef]
- Srinivas, M.; Boehm-Sturm, P.; Figdor, C.G.; de Vries, I.J.; Hoehn, M. Labeling cells for in vivo tracking using 19F MRI. Biomaterials 2012, 33, 8830–8840. [Google Scholar] [CrossRef]
- Pérez-Medina, C.; Tang, J.; Abdel-Atti, D.; Hogstad, B.; Merad, M.; Fisher, E.A.; Fayad, Z.A.; Lewis, J.S.; Mulder, W.J.; Reiner, T. PET Imaging of Tumor-Associated Macrophages with 89Zr-Labeled High-Density Lipoprotein Nanoparticles. J. Nucl. Med. 2015, 56, 1272–1277. [Google Scholar] [CrossRef]
- Schniering, J.; Benešová, M.; Brunner, M.; Haller, S.; Cohrs, S.; Frauenfelder, T.; Vrugt, B.; Feghali-Bostwick, C.; Schibli, R.; Distler, O.; et al. 18F-AzaFol for Detection of Folate Receptor-β Positive Macrophages in Experimental Interstitial Lung Disease-A Proof-of-Concept Study. Front. Immunol. 2019, 10, 2724. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Galli, F.; Suman, V.J.; Nevala, W.K.; Paulsen, A.M.; Hung, J.C.; Gansen, D.N.; Erickson, L.A.; Marchetti, P.; Wiseman, G.A.; et al. Non-invasive visualization of tumor infiltrating lymphocytes in patients with metastatic melanoma undergoing immune checkpoint inhibitor therapy: A pilot study. Oncotarget 2018, 9, 30268–30278. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, R.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Immuno-PET of Murine T Cell Reconstitution Postadoptive Stem Cell Transplantation Using Anti-CD4 and Anti-CD8 Cys-Diabodies. J. Nucl. Med. 2015, 56, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, R.; Escuin-Ordinas, H.; Mok, S.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Witte, O.N.; Ribas, A.; Wu, A.M. An Effective Immuno-PET imaging method to monitor CD8-Dependent responses to immunotherapy. Cancer Res. 2016, 76, 73–82. [Google Scholar] [CrossRef]
- Seo, J.W.; Tavaré, R.; Mahakian, L.M.; Silvestrini, M.T.; Tam, S.; Ingham, E.S.; Salazar, F.B.; Borowsky, A.D.; Wu, A.M.; Ferrara, K.W. CD8+ T-Cell Density Imaging with 64Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols. Clin. Cancer Res. 2018, 24, 4976–4987. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Boisgard, R.; Siquier-Pernet, K.; Decaudin, D.; Dollé, F.; Tavitian, B. Differential expression of the 18 kDa translocator protein (TSPO) by neoplastic and inflammatory cells in mouse tumors of breast cancer. Mol. Pharm 2011, 8, 823–832. [Google Scholar] [CrossRef]
- Griessinger, C.; Maurer, A.; Kesenheimer, C.; Kehlbach, R.; Reischl, G.; Ehrlichmann, W.; Bukala, D.; Harant, M.; Cay, F.; Brück, J.; et al. 64Cu antibody-targeting of the T cell receptor and subsequent internalization enables in vivo tracking of lymphocytes by PET. Proc. Natl. Acad. Sci. USA 2015, 112, 1161–1166. [Google Scholar] [CrossRef]
- Galli, F.; Brück, J.; Rapisarda, A.S.; Stabile, H.; Malviya, G.; Manni, I.; Bonanno, E.; Piaggio, G.; Gismondi, A.; Santoni, A.; et al. In vivo imaging of natural killer cell trafficking in tumors. J. Nucl. Med. 2015, 56, 1575–1580. [Google Scholar] [CrossRef]


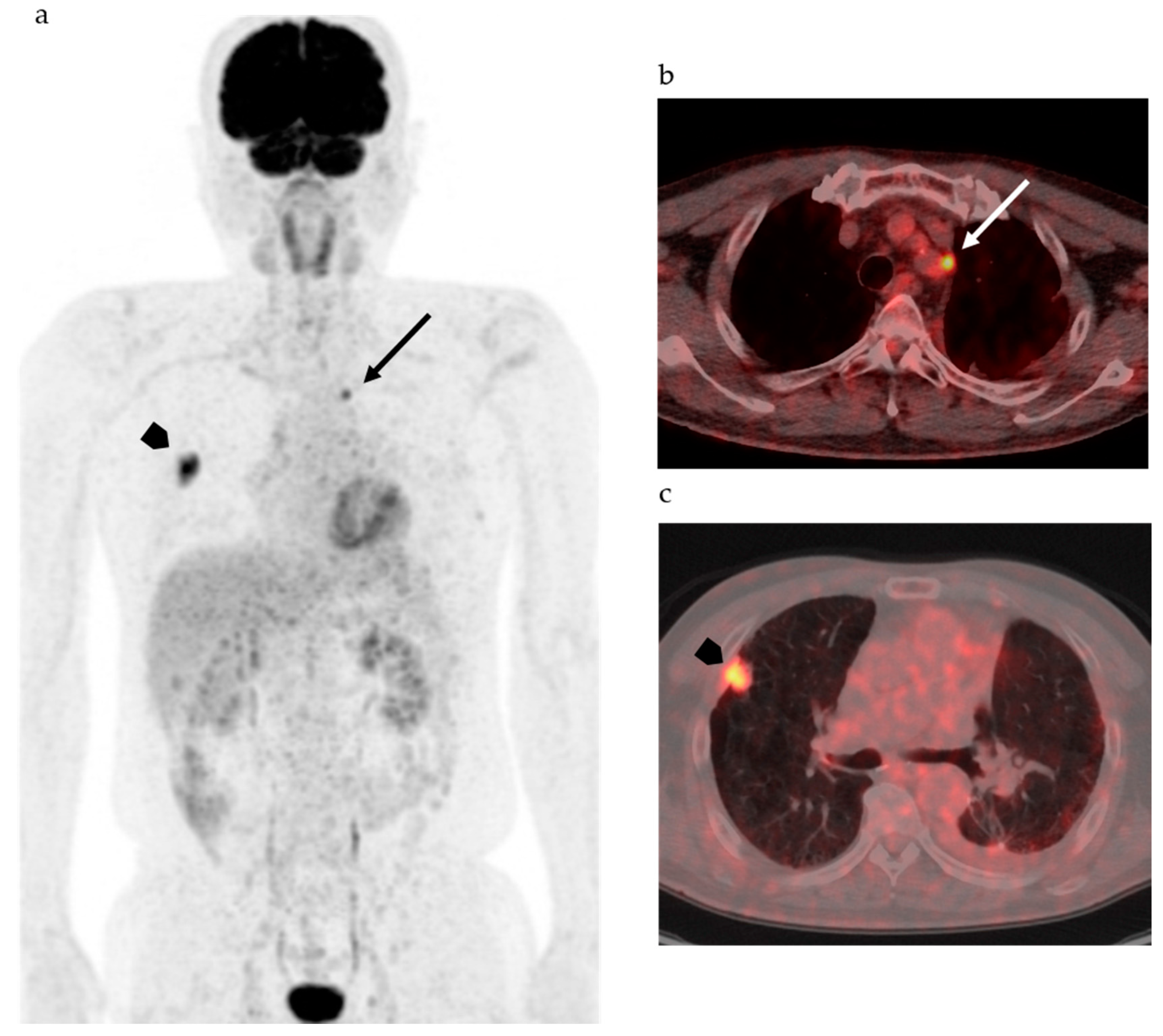{kind=link}
| Conventional and Immune-Related Tumor Response Criteria | ||||||
|---|---|---|---|---|---|---|
| Criteria | Measurement | CR | PR | PD | Confirmation of PD | New Lesion |
| RECIST 1.1 (2009) | Unidimensional (LD for non-nodal lesions; LPD for LN) | Disappearance of all target lesions <10 mm for any pathological LN | ≥30% reduction | ≥20% and ≥5 mm increase, new lesion, or nontarget PD | Not required | Defines PD |
| irRECIST (2013) | Unidimensional (LD for non-nodal lesions; LPD for LN) | Disappearance of all target lesions | ≥30% reduction | ≥20% and ≥5 mm increase, new lesion, or nontarget PD | Required on consecutive studies at least 4 weeks apart | Does not define PD; measurements of new lesions included in the total tumor burden |
| iRECIST (2017) | Unidimensional (LD for non-nodal lesions; LPD for LN) | Disappearance of all target lesions | ≥30% reduction | ≥20% and ≥5 mm increase, new lesion, or nontarget PD | Required at the next assessment 4–8 weeks later | Defines unconfirmed PD; confirms PD if additional new lesions or size increase (≧5 mm for the sum of new target or any increase in new nontarget lesions) are noted on the next assessment |
| FDG-PET Response Evaluation Criteria | ||||||
| Criteria | Measurement | CMR | PMR | PMD | Confirmation of PMD | New Lesion |
| EORTC (2000) | SUVmax | Complete resolution of FDG uptake in all lesions | > 25% reduction in the sum of SUVmax after more than one cycle of treatment | > 25% increase in the sum of SUVmax or appearance of new lesions | Not required | Defines PD |
| PERCIST (2009) | SUVpeak | Complete resolution of FDG uptake in all lesions | ≥ 30% reduction of the peak lean body mass SUV (SULpeak) and an absolute drop of 0.8 SULpeak units | > 30% increase in the SULpeak and an absolute increase of 0.8 SULpeak, or appearance of new lesions | Not required | Defines PD |
| Radiopharmaceutical for Therapy | Radiation | Half-Life | Radiopharmaceutical for Diagnosis |
|---|---|---|---|
| 177Lu-DOTA-TATE | Beta ray (β− particle) | 78 h | 68Ga-DOTA-TATE |
| 213Bi-DOTA-TOC | Alpha ray (He2+ particle) | 0.76 h | 68Ga-DOTA-TOC |
| 177Lu-PSMA | Beta ray (β− particle) | 78 h | 68Ga-PSMA |
| 225Ac-PSMA | Alpha ray (He2+ particle) | 10 d | 68Ga-PSMA |
| Target | Radiolabeling Agent | Application/Mechanism | References |
|---|---|---|---|
| T lymphocytes | 111In-oxine, 89Zr-oxine | Tumor infiltration | [171,174,175] |
| 18F-FDG | Cytokine production | ||
| SPIO | |||
| NK cells | 111In-oxine, 89Zr-oxine | Tumor infiltration | [171,176,177] |
| 18F-FDG, 11C-methyl iodide | NK cell homing | ||
| SPIO | |||
| Macrophages | 111In-oxine, 89Zr-nanoparticles | Tumor infiltration | [171,178,179,180,181,182] |
| 18F-FDG | Tumor-associated macrophages | ||
| SPIO, 19F-perfluorocarbon | |||
| Interleukin-2 | Iodine-123, Technetium-99 m, Fluorine-18 | Interleukin-2 receptors on T cells | [183] |
| Anti-CD8 cys-diabody | Zirconium-89, Copper-64 | CD8+ T cells | [184,185,186] |
| Anti-CD8 mAb | |||
| PK11195 | Carbon-11 | Tumor-associated macrophages, Translocator protein | [187] |
| Anti-TCR mAb | Copper-64 | Tumor infiltration of T cells | [188] |
| Anti-CD56 mAb | Technetium-99 m | NK cells | [189] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oriuchi, N.; Sugawara, S.; Shiga, T. Positron Emission Tomography for Response Evaluation in Microenvironment-Targeted Anti-Cancer Therapy. Biomedicines 2020, 8, 371. https://doi.org/10.3390/biomedicines8090371
Oriuchi N, Sugawara S, Shiga T. Positron Emission Tomography for Response Evaluation in Microenvironment-Targeted Anti-Cancer Therapy. Biomedicines. 2020; 8(9):371. https://doi.org/10.3390/biomedicines8090371
Chicago/Turabian StyleOriuchi, Noboru, Shigeyasu Sugawara, and Tohru Shiga. 2020. "Positron Emission Tomography for Response Evaluation in Microenvironment-Targeted Anti-Cancer Therapy" Biomedicines 8, no. 9: 371. https://doi.org/10.3390/biomedicines8090371
APA StyleOriuchi, N., Sugawara, S., & Shiga, T. (2020). Positron Emission Tomography for Response Evaluation in Microenvironment-Targeted Anti-Cancer Therapy. Biomedicines, 8(9), 371. https://doi.org/10.3390/biomedicines8090371