1. Introduction
Neutrophils are one of the most abundant leukocytes in blood circulation. In humans around 50–70% [
1] and in pigs around 15–72% [
2,
3,
4] circulating leukocytes are neutrophils. Furthermore, they are one of the first cells infiltrating into infected tissue [
1]. Neutrophils have different intra- and extra-cellular mechanisms to eliminate microbes [
5]. One intracellular mechanism is phagocytosis where neutrophils engulf the microbe in the phagosomal machinery and kill the microbe with different antimicrobial components, for example, myeloperoxidase, neutrophil elastase or antimicrobial peptides [
6,
7]. These different components can kill a wide variety of microorganisms. Furthermore, in the phagosome, the production of reactive oxygen species (ROS) is initiated and a decrease in pH occurs, leading to a powerful antimicrobial activity [
6]. Furthermore, neutrophils can release these antimicrobial components from vesicles/granules to the extracellular space. This process is called degranulation of the neutrophil [
5]. Another extracellular mechanism for controlling pathogens is the release of neutrophil extracellular traps (NETs) [
8]. In this mechanism, neutrophils release DNA fibers decorated with antimicrobial molecules. Pathogens can be killed or at least immobilized inside these NETs and therefore the pathogen spreading is prevented [
5,
6]. Despite being such important cells and having different functions in the control of invading microorganisms, neutrophils have been reported to be short-living cells. There are reports that neutrophils have a circulating half-life of less than 8 h in humans [
9,
10,
11]. When used ex vivo in culture, the half-life of the neutrophil was shown to be less than 24 h [
12]. A single study reported that neutrophils could have a half-life of around 5 days [
12]. However, this brought some controversy [
13] and finally a contradictory study was conducted that disproved the theory of a half-life of approximately 5 days. It was again described that neutrophils have a half-life of less than 24 h [
14].
Given neutrophils’ important role in immune response and their use in research, it is necessary to understand their viability and function ex vivo. When working with fresh blood to isolate neutrophils, how long the blood can be stored for before the isolation procedure begins is a topic of continuous debate. In the case of animal experiments or clinical studies, it is not always possible to directly harvest neutrophils from blood of animals or humans due to technical reasons. For this reason, the aim of this study was to investigate neutrophil viability and antimicrobial activity after being stored in whole blood for 24 h at room temperature when compared to fresh blood. This time-point was furthermore chosen as this would allow an over-night express shipping of blood to the respective lab.
3. Discussion
The differentiation and maturation of neutrophils are complex processes which take around 14 days in the bone marrow [
33]. Despite this, neutrophils have a very short life-span in blood circulation, estimated to be less than 24 h [
14]. However, this topic is controversially discussed, and some authors describe long-living neutrophils [
11,
12]. Thus, it is conceivable that blood cells die during storage of whole blood when they are planned to be used for ex vivo experiments. In clinical studies, it is of interest to work with material from patients, but an immediate processing of blood samples is not always possible, and time-consuming shipping/transport may be needed. Therefore, it is of interest to investigate if storing whole blood for 24 h has an impact on neutrophil harvest, viability, and activity.
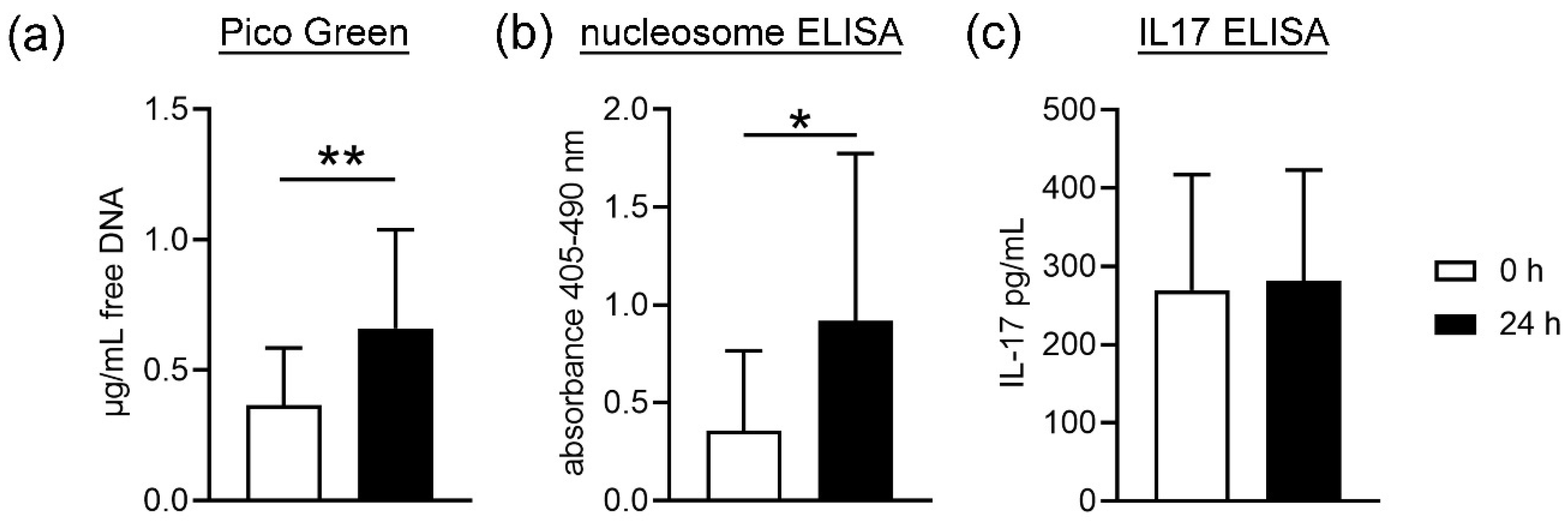
First, cell death of blood cells was analyzed by studying free DNA and nucleosome in plasma. NET formation is one of the proinflammatory death pathways of neutrophils that involves cell membrane lysis and releases free DNA [
34]. In this mechanism, the neutrophil releases a DNA backbone decorated with antimicrobial components [
5,
6]. The quantification of free DNA is a marker used to detect and quantify NETs [
35]. Nevertheless, free DNA and nucleosome are not exclusively NET markers, as necrosis also result in free DNA. As shown in
Figure 1, we found a significant increase in free DNA and nucleosome in the plasma samples from stored blood compared to fresh blood. This could be due to a higher cell death or membrane lysis from all blood cells, including neutrophils. As IL17 is next to other markers such as MPO or neutrophil elastase, only one NET marker [
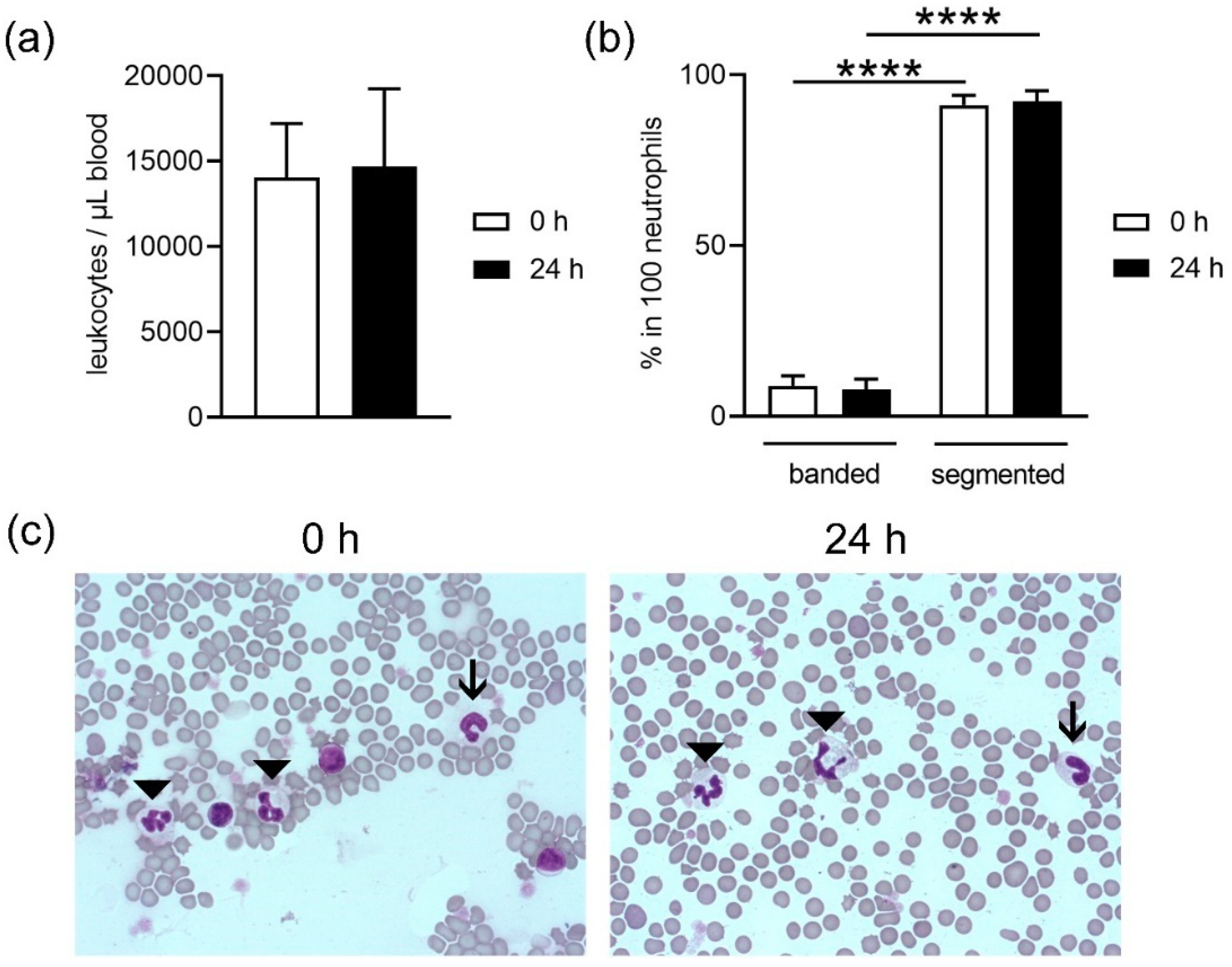
18] and did not differ in plasma from fresh and stored blood; thus, it may be hypothesized that the higher amount of free DNA does not originate from NET-releasing cells. Further analysis is needed to confirm this hypothesis. These data are confirmed by counting neutrophils after density gradient-based separation and harvest of neutrophils from fresh compared to stored blood. Slightly less neutrophils are harvested from stored blood compared to fresh blood, indicating cell death during storage time or isolation. In contrast, no significant difference between fresh and stored blood was detected for white blood cell counts (WBC) (
Figure 2a), but the analysis did not differentiate between living and dead.
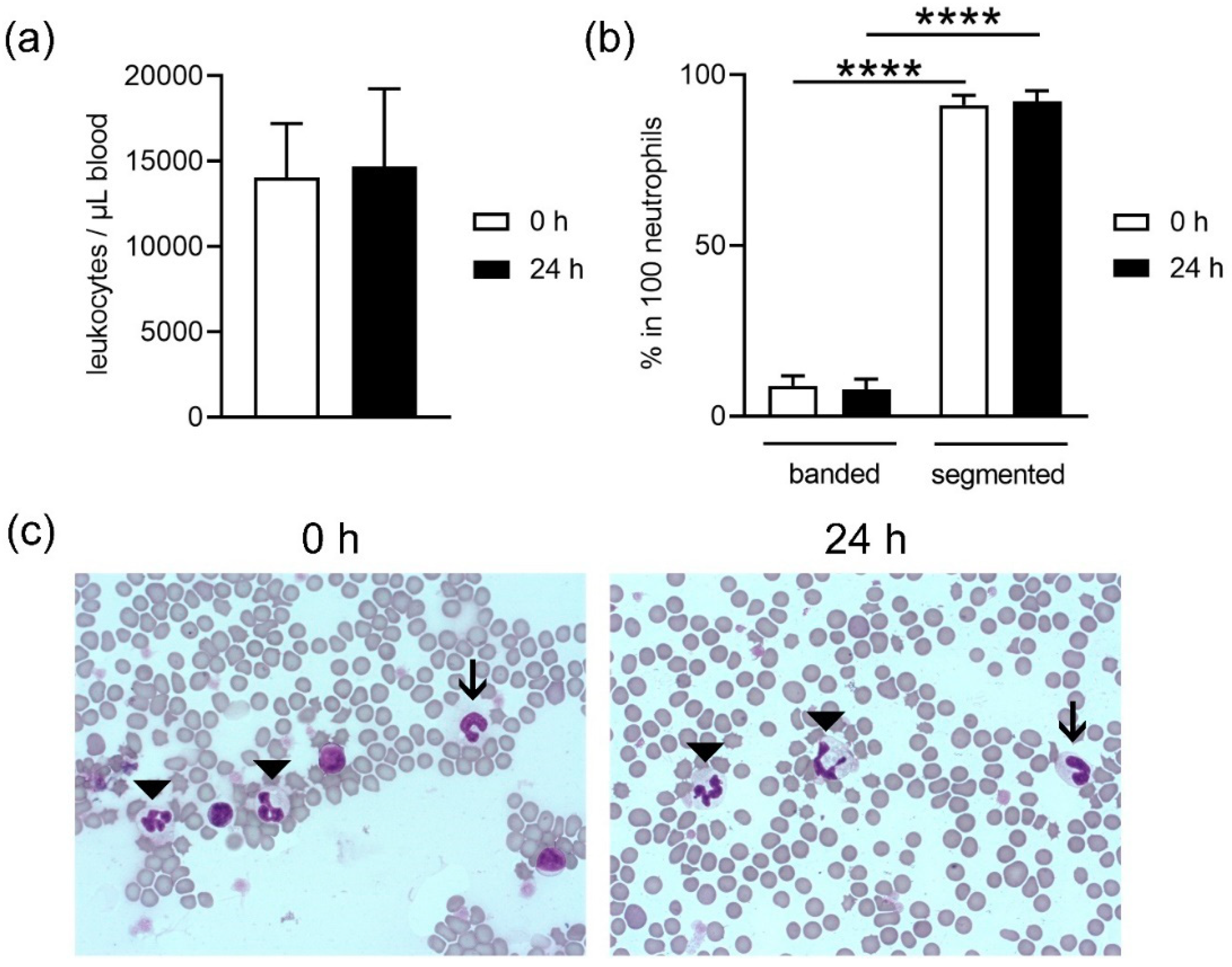
Neutrophils originate in the bone marrow from hematopoietic stem cells. Here, differentiation of cells occurs until the first progenitor of neutrophils is achieved, the promyelocyte. In the next stages, the cells develop granules, differentiation continues and the stage of band neutrophils is reached. These mature further and develop a segmented nucleus. This is the reason why they are also called polymorphonuclear granulocytes. Segmented neutrophils are the final stage of maturity [
6,
33,
36]. Most of the neutrophils circulating in blood are mature neutrophils (segmented), but also band neutrophils can be found in a lower amount [
1,
2,
3,
4]. It is possible to find higher amounts of immature neutrophils circulating in blood due to acute infection or inflammation, where cells are needed quickly and, therefore, there is no time to finish maturation in the bone marrow [
36]. In this study we used healthy blood donors and, as such, we expected to find a higher number of segmented neutrophils (
Figure 2b,c). No differences were found in maturation of the band neutrophils between fresh or stored blood. Since the ratio did not shift, it can be speculated that segmented neutrophils do not exclusively die during storage.
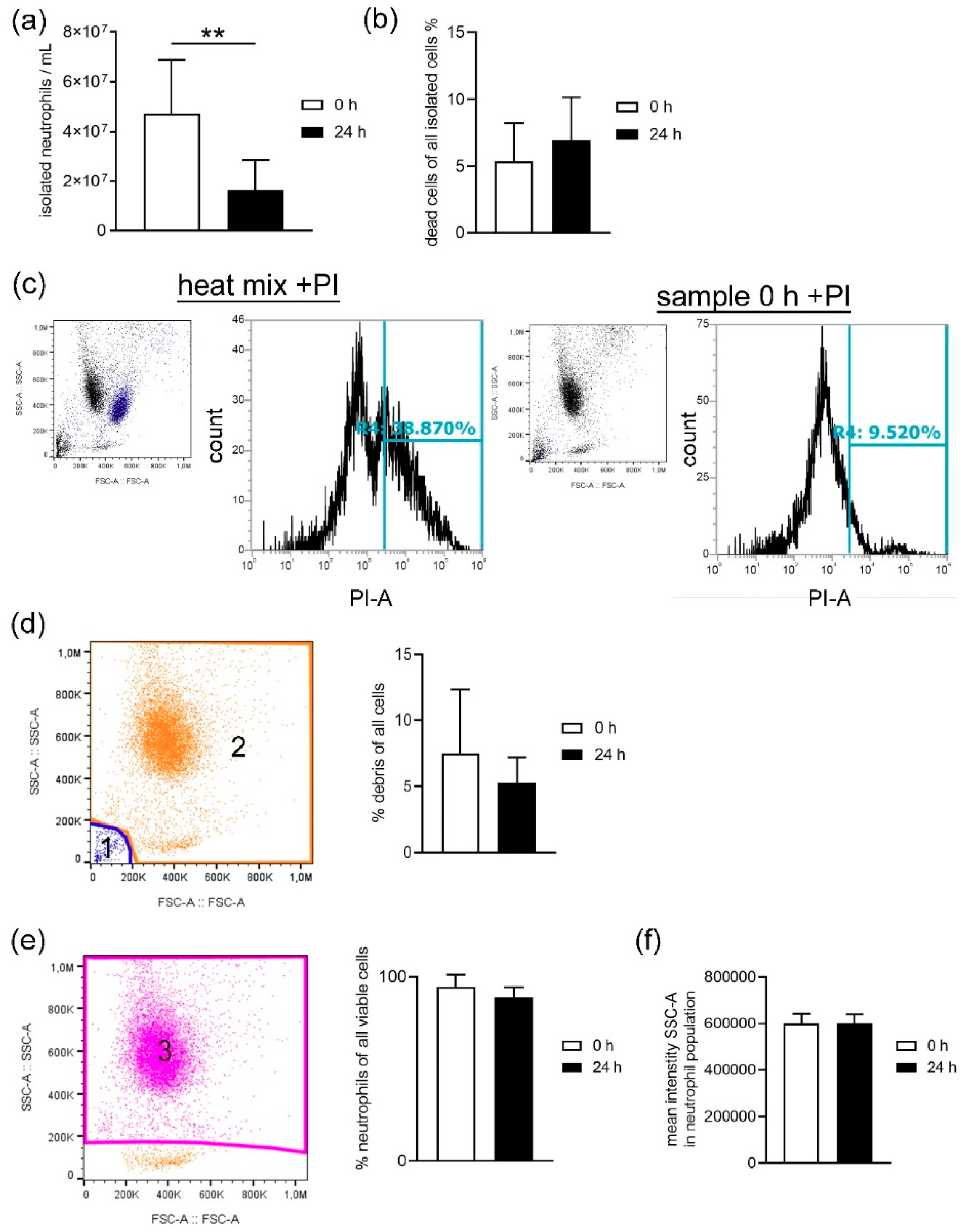
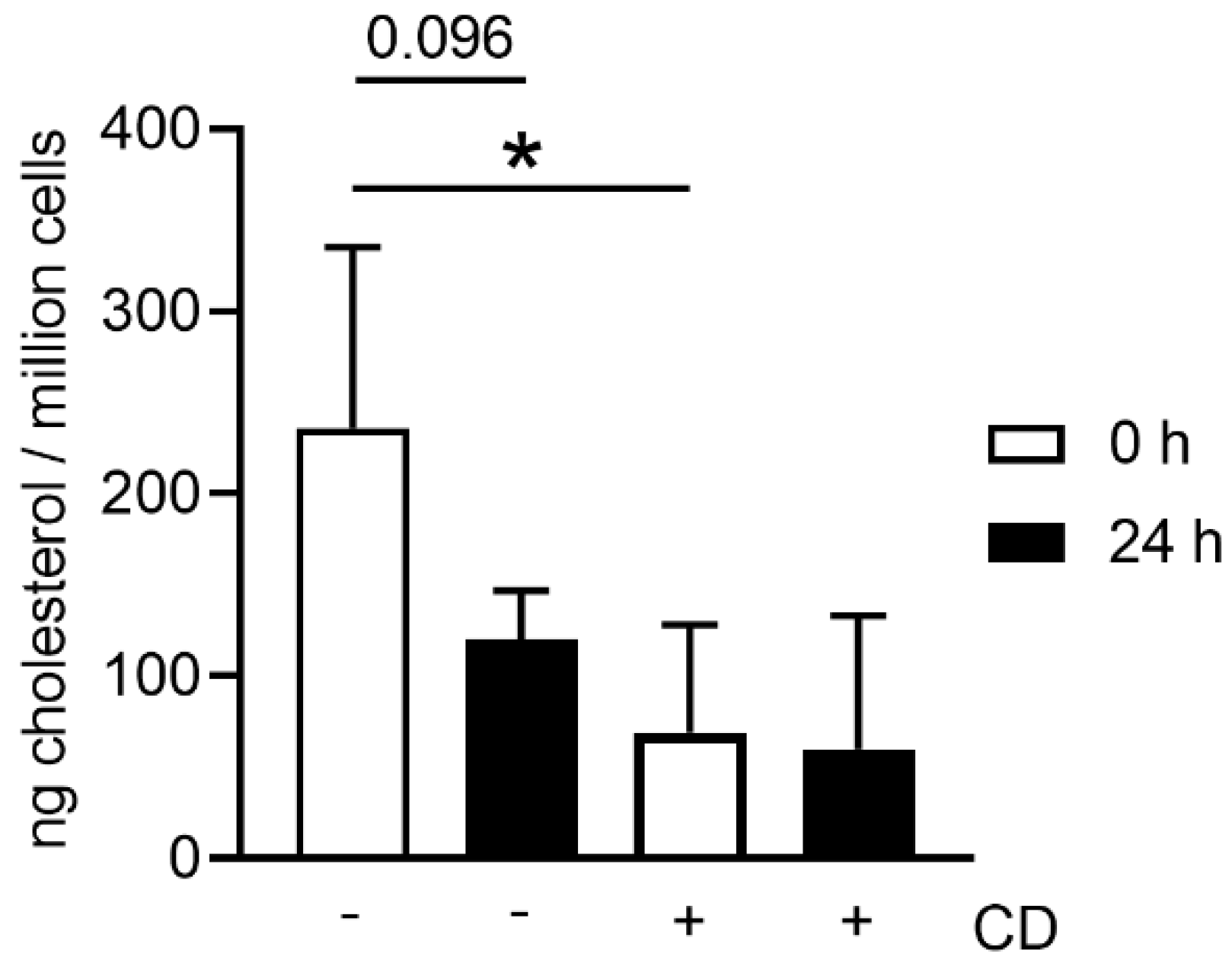
When harvesting neutrophils from fresh or stored blood by density gradient centrifugation, the number of dead cells was slightly higher in neutrophils isolated from stored blood (
Figure 3b), confirming the increased free DNA level shown for whole blood in
Figure 1. Even though the number of leukocytes counted in whole blood by Leuko-TIC was roughly equal (
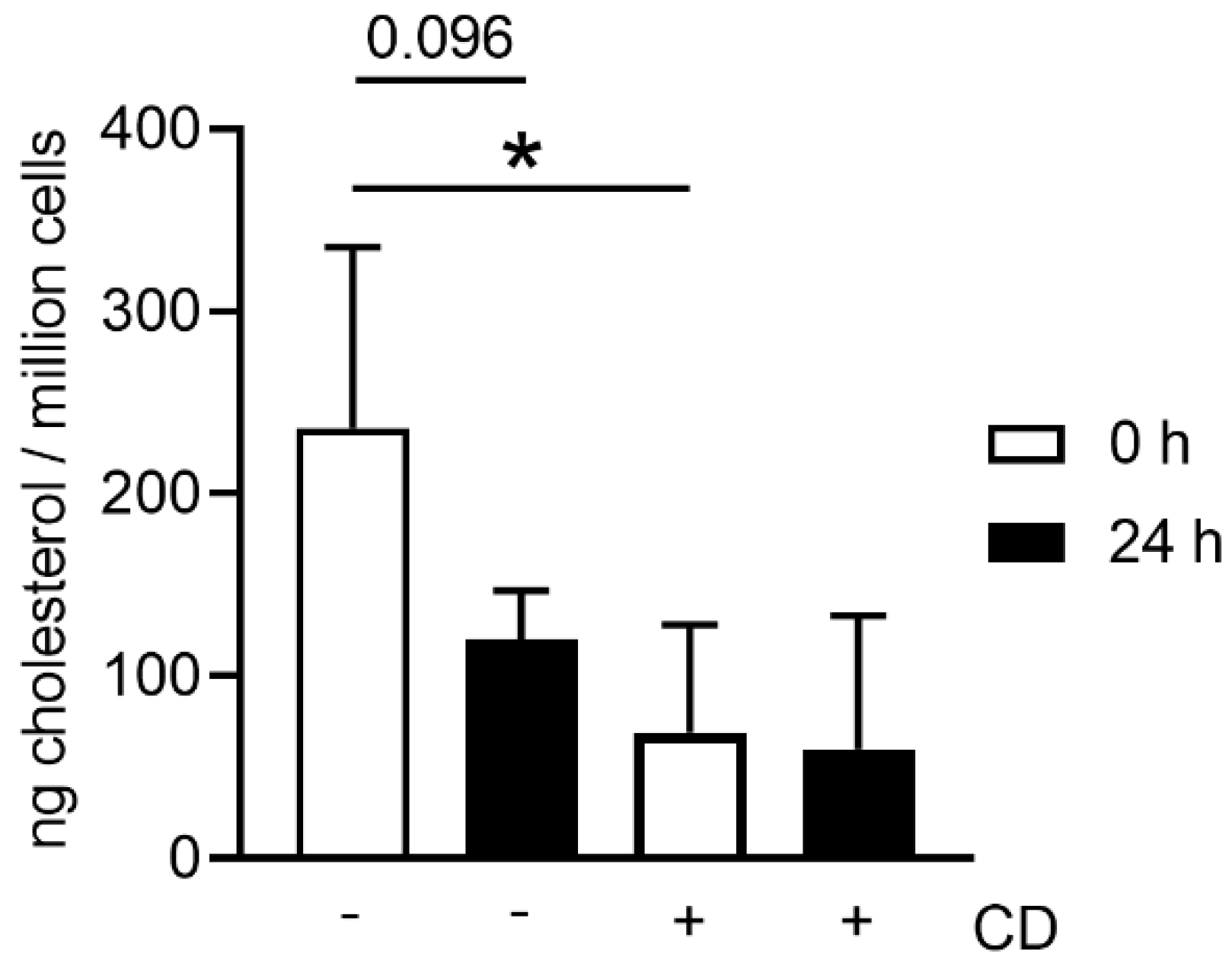
Figure 2a), it seems that neutrophils from stored blood are more fragile. Therefore, it could be hypothesized that the steps during neutrophil isolation (e.g., the erythrocyte lysis step) do destroy more neutrophils from stored blood. One explanation for cells being more fragile is the lower content of cholesterol. Cholesterol is part of the neutrophil cell membrane and is involved in the process of adhesion and transmigration [
37]. Our findings of lower cholesterol content in neutrophils isolated from stored blood (
Figure 7) matches the hypothesis that neutrophils become more fragile and sensitive to rupture after storage. This would explain the significant higher number of isolated neutrophils from fresh blood compared to stored blood (
Figure 3a). Furthermore, this aligns with the finding of more free DNA in the plasma of stored blood. An explanation could be that either the storage does not allow the cells to be preserved or their life time has ended, as previously described [
14]. For this reason, it would be interesting to evaluate under other storage conditions, such as another temperature or storage time, if the number of neutrophils that can be isolated could be higher.
Despite this discussion, it is important to note that the amount that can be achieved from stored blood is enough for experiments to study neutrophil functions ex vivo. Therefore, we were interested to find out if these remaining neutrophils have normal activity and still could be used for ex vivo investigations.
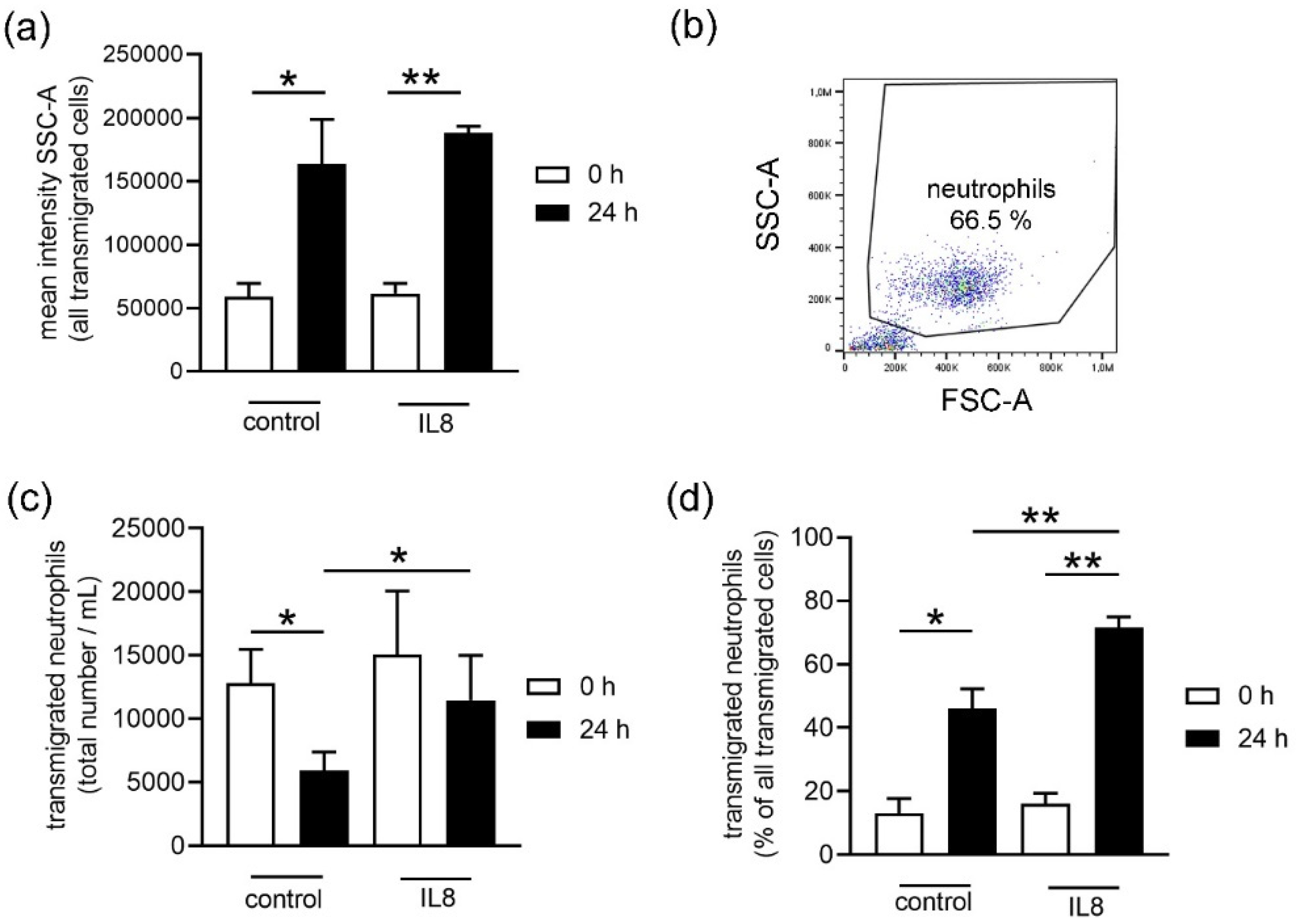
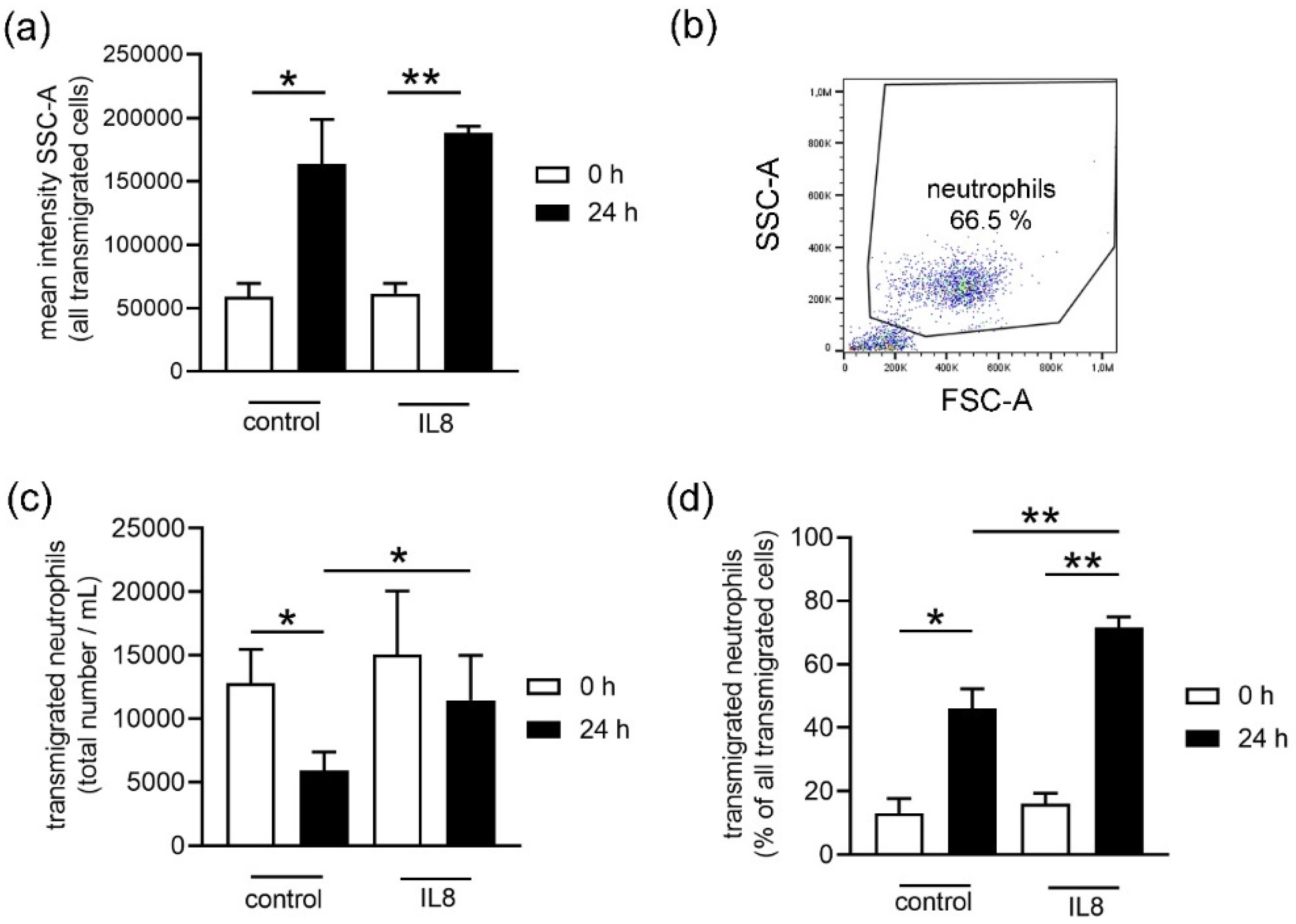
Initially, when an infection or inflammation occurs in the body, neutrophils are recruited by chemoattractant molecules. One well described example is IL8: this chemokine can activate neutrophils, attracts them to the site of infection, and promotes their adhesion to the endothelium to finally transmigrate into the infected tissue [
5,
6]. Interestingly, the surviving neutrophils showed a higher transmigration rate in response to IL8 in stored blood compared to fresh blood (
Figure 4b). Only neutrophils from stored blood transmigrated significantly more to the IL8 stimulus when compared to the unstimulated control group (
Figure 4b). This suggests that storage could either increase a more specific reaction to stimulation or decrease the transmigration of other cells from stored blood or a combination of both.
Some metabolic reactions in cells produce reactive oxygen species (ROS), which are reactive molecules and free radicals derived from oxygen [
38]. It is known that once neutrophils are activated or stimulated, they are able to produce ROS as molecules with antimicrobial potential [
6]. When ROS is in contact with the pathogen it can cause damage in the cell membrane, nucleic acid and proteins [
6]. These molecules can also be involved in cell activation and can enhance killing processes in neutrophils. For example, after neutrophils are stimulated with PMA, IL8 or a pathogen, the NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase) complex in the neutrophil is activated and produces ROS. This enhances the reaction in the granules, combining the neutrophil elastase and myeloperoxidase with the chromatin, and finally they are released as NETs [
15,
24,
39]. As ROS can be found in higher amounts when neutrophils are activated, we used ROS as a parameter for neutrophil activity. Indeed, ROS production was found in neutrophils from fresh and stored blood. This demonstrates that neutrophils are still active despite prior storage of blood (
Figure 5). However, when neutrophils from stored blood are stimulated, they have slightly higher ROS activity compared to neutrophils derived from fresh blood. This again could be related to the cholesterol level of the cell since ROS-producing NADPH oxidase is found anchored in cholesterol-rich microdomains. In good correlation with our data, it has been reported that a reduced cholesterol content of the cell may lead to a higher enzymatic activity of the NADPH oxidase [
40]. Similarly, we here show that neutrophils derived from stored blood show a decreased cholesterol level and at the same time increased ROS production.
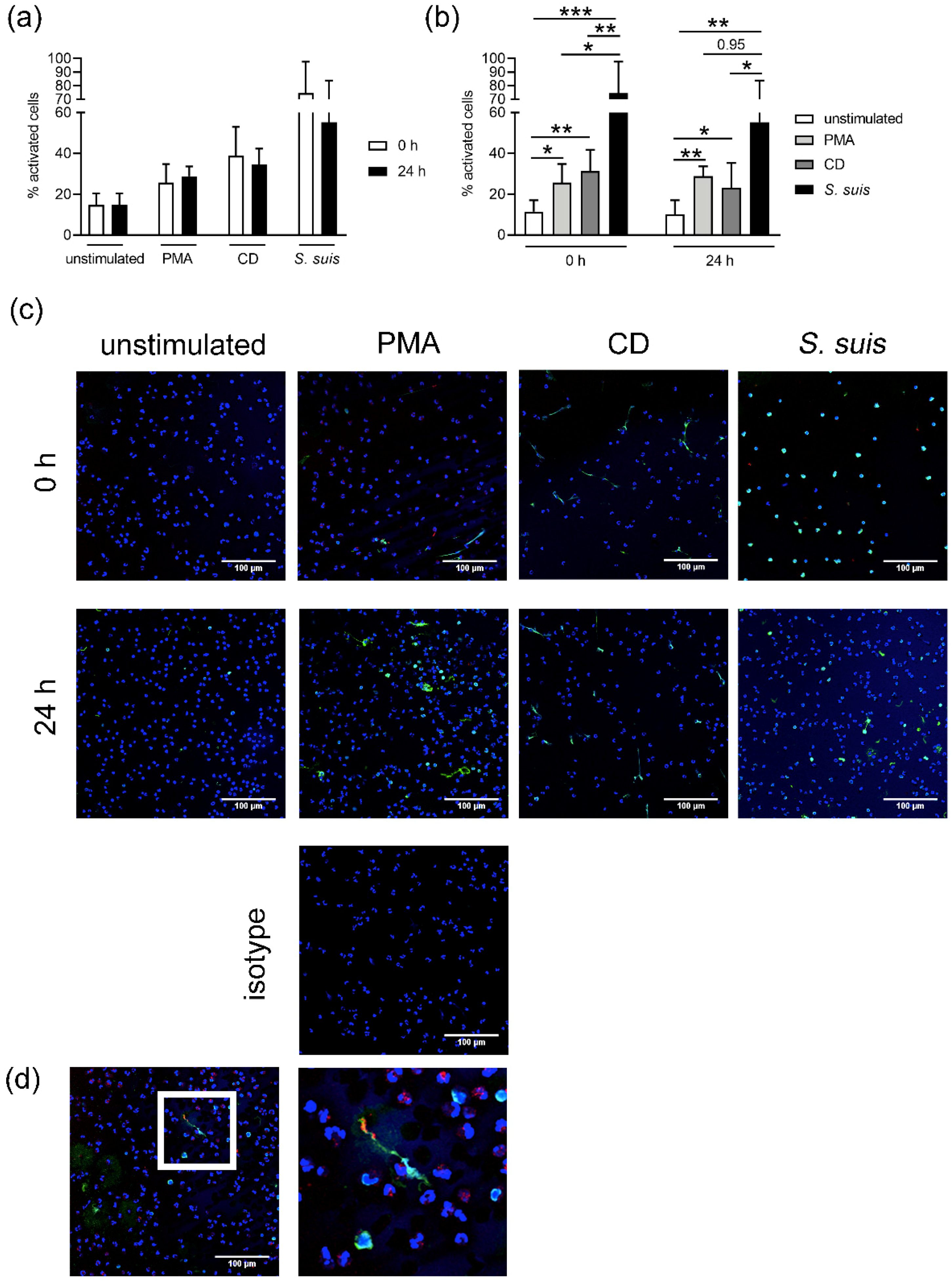
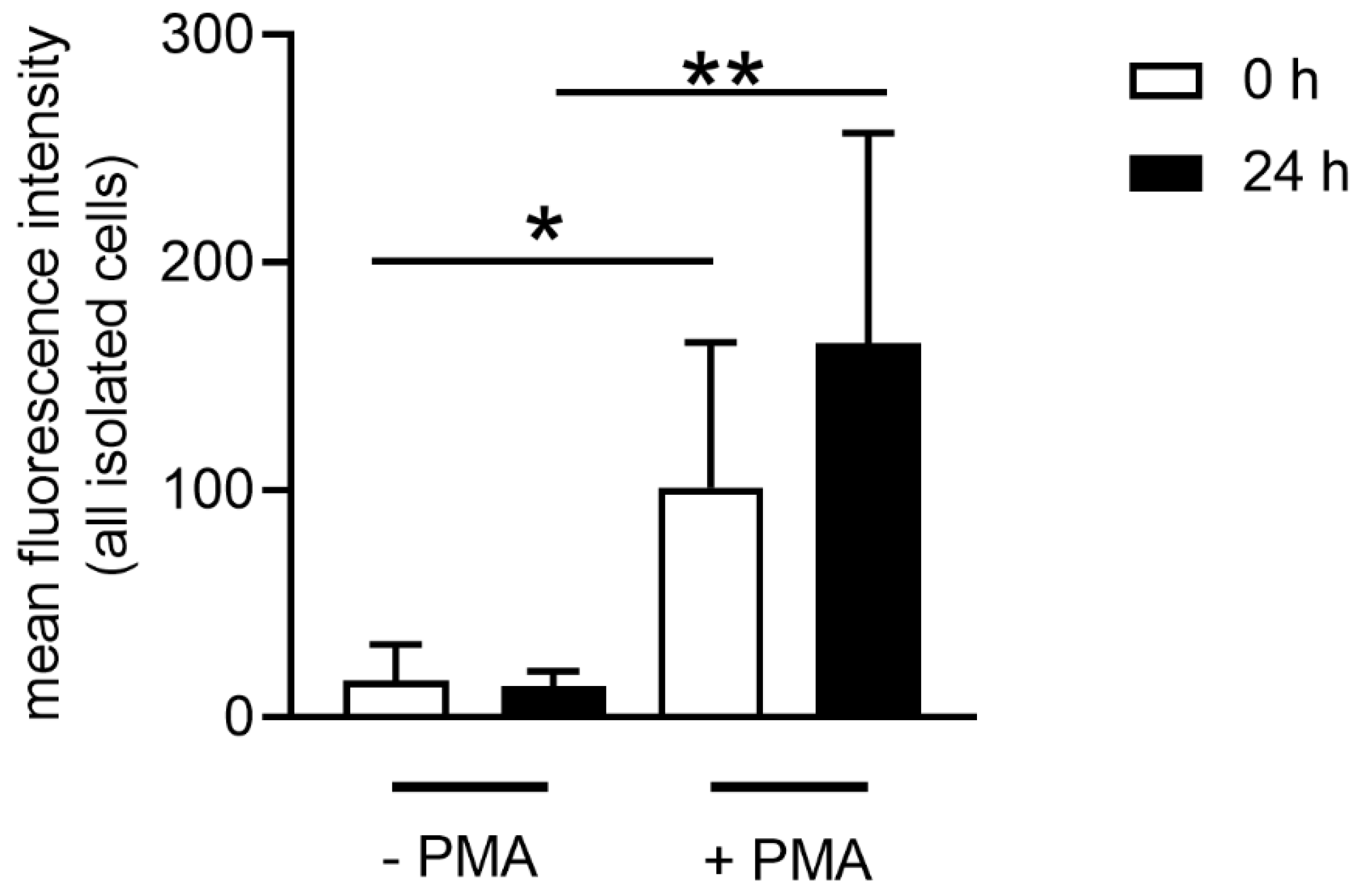
As previously described, neutrophils can be stimulated to release NETs in the presence of pathogens, PMA, IL8, CD and others by different pathways [
15,
24,
27,
39,
41]. We confirmed that both neutrophils from fresh and stored blood release NETs after incubation with
S. suis, PMA or CD. However, the level of NET release varied depending on the stimulus (
Figure 6). When neutrophils are stimulated with CD, NET release can occur in a short period of time (around 30 min) [
27,
41]. We identified the tendency of slightly higher NET release in neutrophils from fresh blood after stimulation with CD, which depletes cholesterol. In contrast with cholesterol, which mediates ROS-independent NET formation, PMA can induce NETs by activating the NADPH oxidase pathway and ROS production [
15,
39]. Neutrophils from fresh and stored blood release NETs and produce ROS after PMA stimulation. However, a slightly higher ROS production and NET release was observed in the neutrophils from stored blood, which aligns with higher ROS activity of those cells (
Figure 5 and
Figure 6).
Since neutrophils were isolated from porcine blood,
S.
suis was used as a natural stimulus for NET release. This pathogen is described as a zoonotic agent causing meningitis in pigs and it has been seen that neutrophils release NETs upon infection of this pathogen [
42]. A higher release of NETs was found in neutrophils from fresh blood after stimulation with
S. suis (
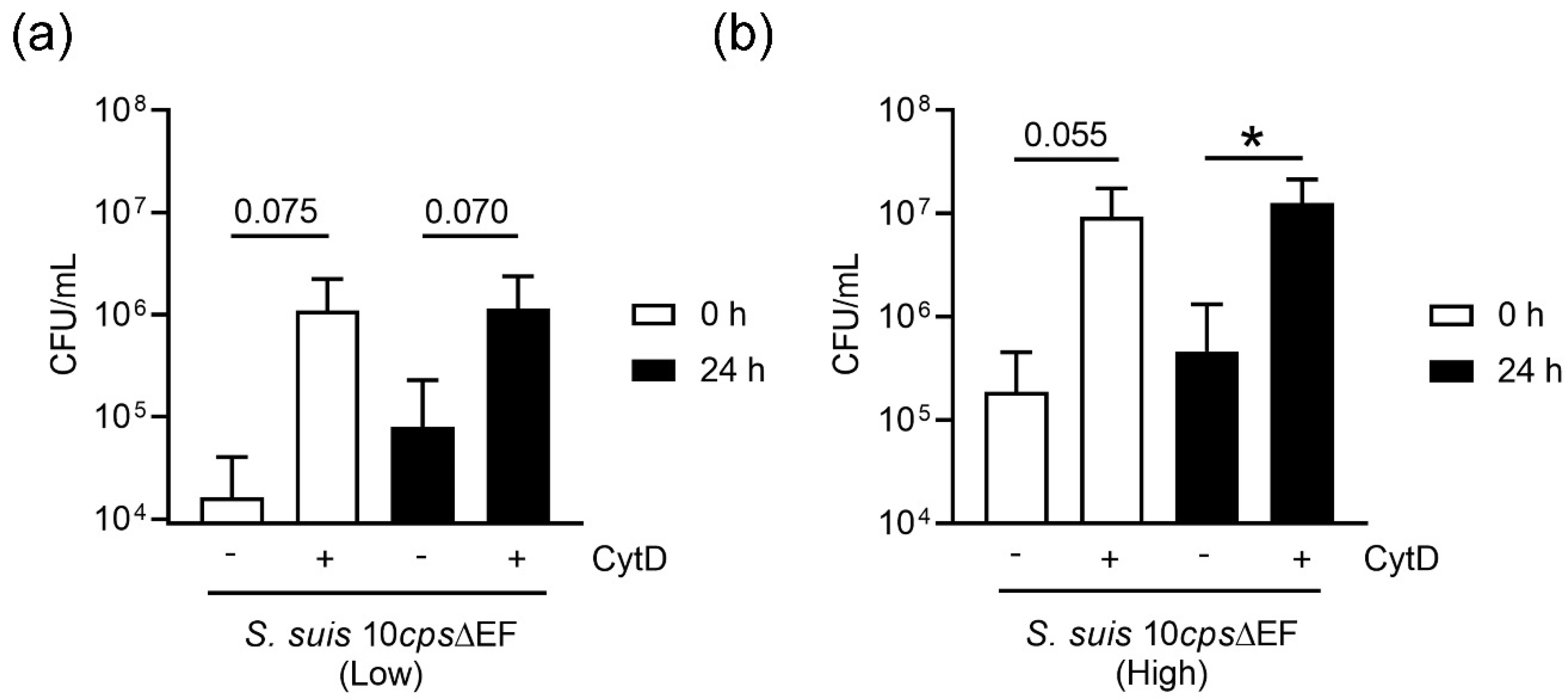
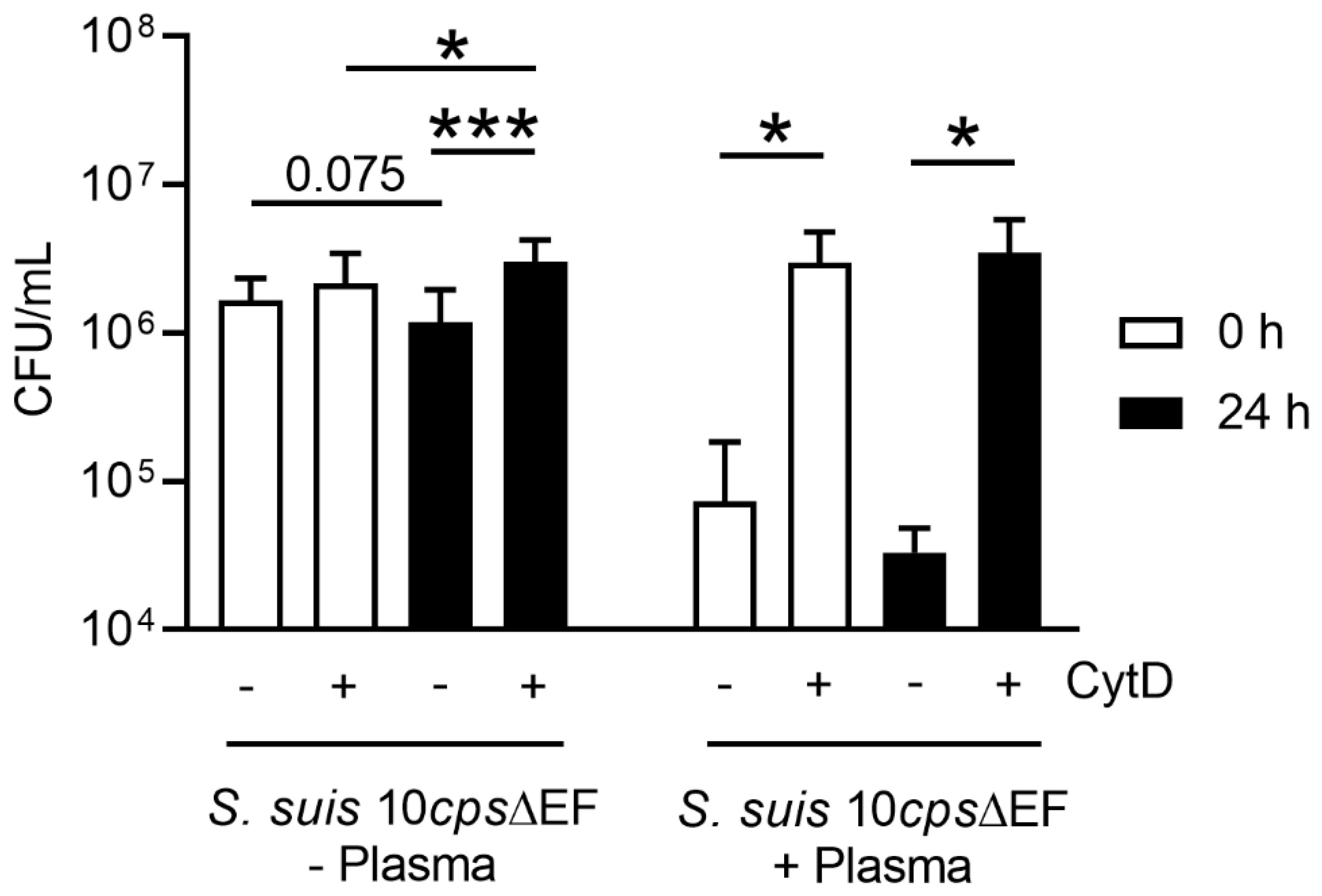
Figure 6). This correlates with the data obtained in the neutrophil killing assay, especially when plasma is absent (
Figure 9). Neutrophils from fresh and stored blood still have efficient phagocytic activity. It is well known [
43] that neutrophils considerably improve pathogen elimination due to opsonizing antibodies or complements present in plasma and/or serum. Nevertheless, the overall phenotype might completely depend on the pathogen that is used for experiments. It could be possible that this tendency may change depending on the pathogen. As an example, NETs are known to act antimicrobial against
S. suis, but in contrast pathogens such as
Actinobacillus (A.) pleuropneumoniae have been reported to induce NETs and benefit from them as a nutrient source (NAD or adenosine) for growth [
19]. Thus, future experiments need to verify if neutrophils from stored blood still behave similarly when other pathogens are used for the assays.
In summary, our data show that surviving neutrophils harvested from 24 h stored blood still have comparable or even enhanced antimicrobial activity, as seen in the case of ROS production, phagocytosis or NET formation in response to a selected bacterial pathogen. Nevertheless, it is recommended to analyze exact cell counts and viability of cells, since storage seems to influence the survival of some blood cell populations, including neutrophils. As components of dying cells can be released into the whole blood, it is recommended to critically consider using stored whole blood in ex vivo killing assays. These released components could interfere with bacterial growth and/or killing by neutrophils. However, for all experiments, similar storage times are always recommended during sets of experiments, since storage might slightly impact the antimicrobial efficiency of isolated cells. If shorter storage or other storage conditions (e.g., temperature) do influence the analyzed parameters in a comparable manner, they should be analyzed in future studies.
4. Materials and Methods
4.1. Collection and Storage of the Blood Samples
The collection of blood from healthy pigs was registered at the Lower Saxonian State Office for Consumer Protection and Food Safety (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, No. 33.9-42502-05-18A302). It was conducted in line with the recommendations of the German Society for Laboratory Animal Science (Gesellschaft für Versuchstierkunde) and the German Veterinary Association for the Protection of Animals (Tierärztliche Vereinigung für Tierschutz e. V.) (
http://www.gv-solas.de).
The donor pigs were kept in the Clinic for Swine or the Research Center for Emerging Infections and Zoonoses from the University of Veterinary Medicine Hannover, Germany. A total of 30 mL of fresh blood was collected from healthy pigs in S-Monovette®® Lithium-Heparin 9 mL tubes (Sarstedt, Nümbrecht Germany). Half of the blood was analyzed immediately (fresh blood, 0 h) and the rest of the blood was stored in the dark at room temperature for 24 h (stored blood, 24 h). Room temperature was chosen to avoid cold shock of the blood cells.
The same assays were performed with fresh and stored blood for a paired analysis.
4.2. Measurement of Free DNA, Histone Fragments and IL17 in Plasma
Plasma samples were collected from the fresh and stored blood. Therefore, 1 mL of the heparinized blood was centrifuged (2100× g, 15 min at 20 °C), and the plasma was collected and stored at −20 °C until the respective assays were performed.
A Quant-iT ™ PicoGreen ™ assay (Thermo Fisher, P11496 Invitrogen, Carlsbad, CA, USA) was used, as described previously [
35], to determine the amount of free DNA in the plasma samples. The amount of DNA in each sample was calculated based on the standard curve.
The number of histone-associated-DNA-fragments (mono- and oligo-nucleosomes) were quantified with a cell death detection ELISA PLUS kit (Roche Diagnostics GmbH, Mannheim, Germany). The assay was conducted following the manufacturer’s instructions.
The determination of IL17 was analyzed with porcine IL17 ELISA (IL-17 pig ELISA Kit, ab193732 Abcam® Berlin, Germany) following the manufacturer’s instructions.
4.3. Blood Smear Analysis
Blood smears were made, air dried and stained with HAEMA quick stain (DIFF Quick, Labor + Technik, Eberhard Lehmann GmbH, Germany, No. LT001), following the manufacturer’s instructions. In the stained smears, the amount of segmented and band neutrophils (in 100 neutrophils) were counted using a light microscope (LEICA DM IL LED, HI Plan I 40×/0.50 PH2 objective) and the percentage in each sample was calculated. Example pictures were made with a Zeiss Axio Imager M2 microscope with a Plan-Apochromat 63×/1.4 Oil DIC ∞/0.17 objective.
4.4. Counting of White Blood Cells
White blood cells (WBCs) present in the whole blood tubes (S-Monovette®® EDTA K3/Forensic 9 mL, Sarstedt, Nümbrecht Germany) were determined with the Leuko-TIC®® kit (Bioanalytic GmbH, Umkirch, Germany). The samples were analyzed following the manufacturer’s instructions. The cells were counted in a Neubauer chamber with a light microscope (Leica DM IL LED, HI Plan I 10×/0.25 PH1 objective).
4.5. Bacterial Strain and Growth Conditions
Streptococcus (
S.)
suis cps type 2 strain 10 [
32] (NET induction) and
Streptococcus (
S.)
suis 10
cpsΔ
EF (used in the killing assay as a mutant strain without a capsule that is sensitive to phagocytosis), kindly provided by Hilde Smith (Wageningen, GE, The Netherlands) [
32], were used in this study. Therefore, working cryostocks were produced.
S.
suis strains were grown on a Columbia blood agar plate (Columbia Agar with 7% Sheep Blood; Thermo Scientific
TM PB5008A, Waltham, MA, USA) at 37 °C without CO
2 (20–24 h). Two colonies were inoculated in 10 mL Todd Hewitt Broth (THB) ((BD Bacto™ Dehydrated Culture Media: Todd Hewitt Broth; Becton Dickinson, 249240, Franklin Lakes, NJ, USA) and incubated at 37 °C for around 16 h in a melting ice-bath to delay start of growth. A 1:50 dilution from the overnight culture was conducted in pre-warmed THB (total 50 mL) and incubated at 37 °C until it reached an optical density of OD
600nm ~ 0.85 ± 0.05 (
S.
suis 10
cpsΔ
EF) and OD
600nm = 1.15 ± 0.05 (
S. suis cps type 2 strain 10), the late exponential growth phase. Immediately, the culture was mixed with glycerol (final concentration of 15%) and aliquots in 1.5 mL tubes were shock frosted in liquid nitrogen. The cryostocks were stored at −80 °C until they were used and only thawed once.
4.6. Whole Blood Killing Assay
A total of 500 μL blood was mixed in a 1.5 mL tube with
S. suis 10
cpsΔ
EF (high infection dose = 1.5 × 10
6 CFU/mL or low infection dose = 1.5 × 10
5 CFU/mL). Samples were incubated with and without 10 μg/mL (final concentration) of cytochalasin D (Sigma-Aldrich, Munich, Germany C2618), a fungal toxin that inhibits phagocytosis. Samples were incubated for 2 h on a rotator (7 rpm) at 37 °C. To determine the CFU/mL, serious dilutions at the time points 0 and 2 h were plated on Columbia blood agar plates (Columbia Agar with 7% Sheep Blood; Thermo Scientific
TM PB5008A, Waltham, MA, USA) and incubated for 20–24 h at 37 °C. The survival factor (SF) was calculated with the formula SF
2h = CFU
2h/CFU
0h [
44].
4.7. Neutrophil Isolation
Porcine neutrophils were purified from the whole blood using a density gradient with Biocoll (1.077 g/mL, Biochrom, L 6115, Berlin, Germany) as previously described [
19,
35]. The final cell pellet was resuspended in 1 mL cold Roswell Park Memorial Institute (RPMI) 1640 Medium, without phenol red (Thermo Fisher, 11835063 Gibco™ RPMI 1640 Medium, no phenol red, Carlsbad, CA, USA). The cells were stained with trypan blue (1680.1 Carl Roth
®®, Karlsruhe, Germany) and counted in a Neubauer chamber. The cell number was adjusted with RPMI to the needed cell concentration depending on the experiment.
4.8. Analysis of Isolated Neutrophils (Viability and ROS Production)
Purified neutrophils were analyzed for purity, granularity, viability and ROS production by flow cytometry analysis using Attune®® NxT Acoustic Focusing Flow Cytometer, Invitrogen (Laser 488 nm (50 mW), filter BL1 = 530/30 (ROS), BL2 = 590/40 (PI)). The acquisition volume was set to 100 μL, the acquisition speed was set to 100 μL/min and a total of 10,000 events were recorded. Cells were adjusted to 5 × 105 cells/mL in 200 μL.
The purity of neutrophils was analyzed after exclusion of debris in all viable cells based on FSC-A and SSC-A settings. Example picture’s for the gating strategy are presented in
Figure 3d,e.
The number of dead cells in all isolated cells was detected with propidium iodide (PI) staining (1.0 mg/mL in water, P4864 Sigma-Aldrich, Munich, Germany) following the manufacturer’s instructions. A dead control (10 min, 70 °C heat block) and a 1:2 diluted dead:living cell control was used to adjust the flow cytometer settings. The gating strategy is presented in
Figure 3c.
To determine ROS production in isolated neutrophils, cells were incubated with and without phorbol 12-myristate 13-acetate (PMA, 25 nM final concentration, 524,400 Sigma Aldrich, Munich, Germany). Intracellular ROS production was measured by immediately adding 2′,7′- dichlorofluorosceindiacetate (DCFH-DA, 10 μM) to each sample. All samples were incubated at 37 °C and 5% CO2 for 30 min. Respective background controls (without DCFH-DA) were included in all assays. The mean green fluorescence intensity of all cells (X-Mean of BL-1) was recorded by flow cytometer as a relative measure of ROS production. The gating strategy excluded debris and gates were set to the neutrophil population. Data were analyzed with FlowJoTM10.6.1 software (Ashland, OR, USA).
4.9. Neutrophil Killing Assay
In each well of 48 well plates (Greiner Bio-One, 677102, Kremsmünster, Austria), 2 × 105/100 μL porcine neutrophils were seeded and infected with S. suis 10cpsΔEF (MOI = 1). Samples were incubated with and without 10% of autologous porcine plasma and 10 μg/mL (final concentration) of cytochalasin D (Sigma-Aldrich, Munich, Germany C2618). RPMI medium was added to complete the volume of each well to 190 μL. The plates were centrifuged (370× g, 5 min) and were incubated for 2 h at 37 °C, 5% CO2. The survival factor (SF) was determined as described in 4.6.
4.10. Transmigration of Neutrophils from Whole Blood
In each well of 24 well plates (Greiner Bio-One, Kremsmünster, Austria), 1 mL RPMI was added with and without IL-8 (recombinant porcine IL-8/CXCL8 protein (535-IN-025 R&D Systems, Bio-Techne GmbH, MN, USA), final concentration 100 ng/mL). In each well, a sterile transwell filter with 3 μm pore size (thincert cell culture insert 662631, Greiner Bio-One, Kremsmünster, Austria) was added. Inside the transwell, 400 μL whole blood was added and the plate was incubated for 4 h at 37 °C, 5%CO
2. At the end of the incubation, the transwell filters were taken out from the wells. Each well was mixed 10 times by pipetting up and down 1 mL. From each well, 500 μL was fixed with paraformaldehyde (4% final concentration). The fixed cells were analyzed based on forward (FSC) and sideward (SSC) scatter using Attune
®® NxT Acoustic Focusing Flow Cytometer (Invitrogen, Carlsbad, CA, USA). Each sample was conducted in triplicates per individual run. Data were analyzed with FlowJo
TM10.6.1 software (Ashland, OR, USA) by drawing a gate to exclude the debris and on the individual cell type populations based on FSC-A and SSC-A, as presented in
Figure 4b.
4.11. NET Induction in Isolated Neutrophils
Cover slips (8 mm; Thermo Fisher Scientific (Bremen) GmbH) for the NET induction were used in 48 well plates and they were coated in accordance with the manufacturer’s instructions with poly-L-lysine (0.01% solution P4707, Sigma Aldrich, Munich, Germany) and handled afterwards as previously described [
19]. In each well, 2 × 10
5 neutrophils/100 μL were seeded. As a negative control, RPMI medium was added. As a positive control, 100 μL methyl-β-cyclodextrin (10 mM final concentration, C4555 Sigma Aldrich, Munich, Germany) or 100 μL phorbol 12-myristate 13-acetate (PMA, 25 nM final concentration, 524,400 Sigma Aldrich, Munich, Germany) was added. The neutrophils were infected with
S. suis cps type 2 strain 10 (MOI = 2). The plates were centrifuged (370×
g, 5 min) and incubated at 37 °C, 5% CO
2. After 3 h of incubation, the samples were fixed with paraformaldehyde (4% final concentration) and the plates were wrapped with parafilm and stored at 4 °C until the staining was conducted.
4.12. NET Staining
NETs were stained as previously described [
45]. After the permeabilization and the blocking of the samples, these were incubated with the first antibodies: a mouse monoclonal antibody (IgG2a) against DNA/histone 1 (MAB3864; Millipore 0.55 mg/mL diluted 1:1000, Billerica, MA, USA) and a rabbit anti-human myeloperoxidase (A039829-2 Agilent, Santa Clara, CA, USA, 3.2 mg, 1:337.5), for 1 h at room temperature. For the isotype controls murine IgG2a (from murine myeloma, M5409-0.2 mg/mL, 1:364 Sigma Aldrich, Munich, Germany) and rabbit IgG (from rabbit serum, Sigma Aldrich, Munich, Germany, I5006, 1.16 mg, 1:10,875) were used. As secondary antibodies, goat anti-mouse Alexa 488Plus (1:500, Invitrogen, Carlsbad, CA, USA) and goat anti-rabbit Alexa 633 (Thermo Scientific, 2 mg, 1:500, Waltham, MA, USA) were used and incubated for 1 h at room temperature in the dark. Afterwards, the samples were washed three times with 1× PBS (phosphate buffered saline) and one time with distilled water. The samples were stained 10 min (in the dark, room temperature) with aqueous Hoechst 33,342 (1:1000, stock 50 mg/mL, Sigma Aldrich, Munich, Germany). The slides were then washed three times with distilled water and embedded in 3–5 μL ProlongGold (without DAPI, Invitrogen, Carlsbad, CA, USA). The samples were dried over night at 4 °C and all cover slips were surrounded with clear nail polish. Samples were stored at 4 °C in the dark until analysis.
4.13. NET Quantification
For each sample, six pictures were made randomly. The pictures were taken by a Leica TCS SP5 AOBS confocal inverted-base fluorescence microscope with an HCX PL APO 40× 0.75–1.25 oil immersion objective. The cells present in the pictures were counted manually using ImageJ software (version 1.52q, National Institute of Health, USA). The total number of neutrophils and positive neutrophils (activated or NET-releasing) were counted. A neutrophil was counted as positive if an evident off-shoot of DNA was visible or if at least two of the following criteria were found: enlarged nucleus, decondensed nucleus or blurry rim. The percentage of NET-positive neutrophils was calculated. An average from the six pictures form each sample was made.
4.14. Cholesterol Analysis
In 1.5 mL tubes, 3 × 10
6 isolated neutrophils in 100 μL RPMI were added and stimulated with methyl-ß-cyclodextrin (10 mM final concentration, C4555 Sigma Aldrich, Munich, Germany) or not stimulated. For control, RPMI medium was used. The final volume was 200 μL. The samples were incubated for 180 min at 37 °C, 5% CO
2 without closing the lid completely. After the incubation, the samples were centrifuged (400×
g, 5 min). The supernatant was discarded carefully by pipetting. The pellet was washed twice with 500 μL of 1× PBS (Lipopolysaccharides (LPS) free) and centrifuged (400×
g, 5 min). The final pellet was resuspended in 500 μL pure SIGMA water (for RNA/DNA work) and the tubes were stored at −20 °C until the cholesterol analysis. The lipid isolation and cholesterol and oxysterol analysis was performed as previously described [
41,
46].
4.15. Statistical Analysis
Data were analyzed using Excel 2016 or 2018 (Microsoft) and GraphPad Prism 8.3 or 8.4.1(460) (GraphPad Software). Data were analyzed with one-tailed paired or unpaired Student’s t-test and presented with mean ± SD and the differences between groups were analyzed as described in the figure legends (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}