NADPH Oxidases and Their Role in Atherosclerosis

 , , and
, , and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. NOX Family Members of the Vascular System
2.1. NOX1
2.2. NOX2
2.3. NOX4
2.4. NOX5
3. Atherosclerosis: A Brief Overview of Pathogenesis
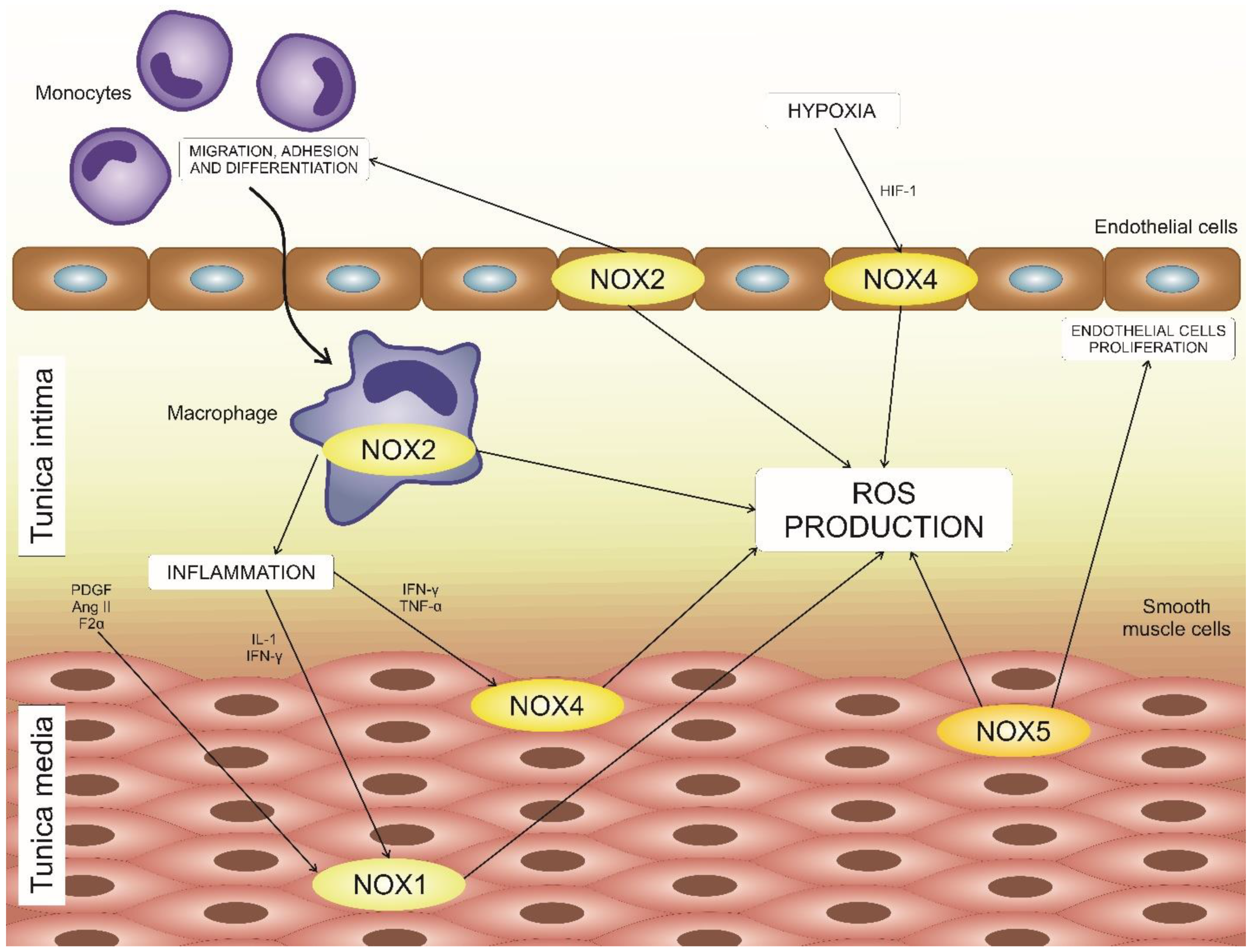
4. NADPH Oxidases in Atherosclerosis Development
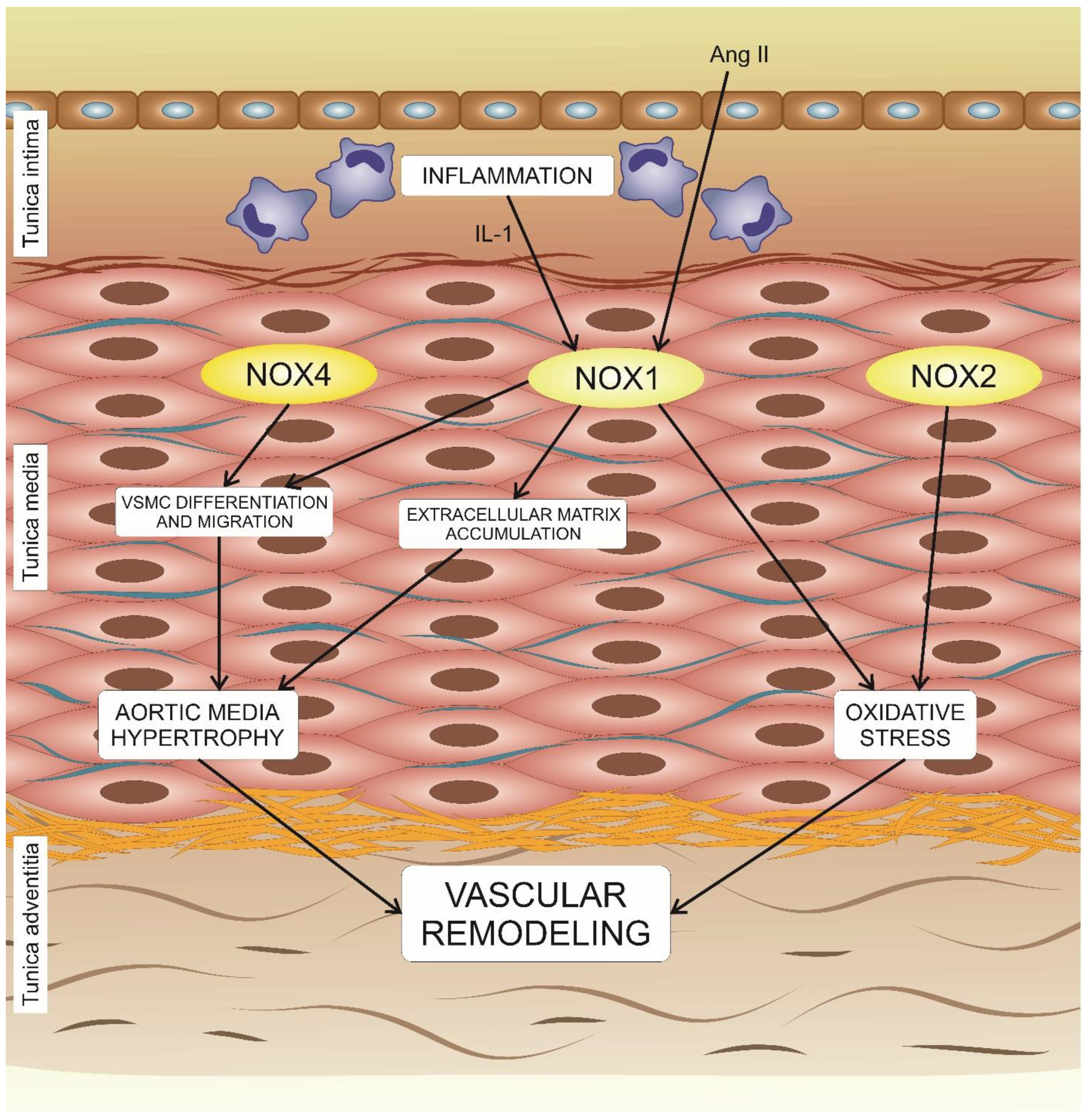
5. The Effects of NADPH Oxidases on Vascular Remodeling
6. Available Therapies and Future Perspectives
7. Conclusions
Funding
Conflicts of Interest
References
- Bedard, K.; Krause, K. The NOX Family of ROS-generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Rueckschloss, U.; Duerrschmidt, N.; Morawietz, H. NADPH oxidase in endothelial cells: Impact on atherosclerosis. Antioxid. Redox Signal. 2003, 5, 171–180. [Google Scholar] [CrossRef]
- Bryk, D.; Olejarz, W.; Zapolska-Downar, D. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postepy. Hig. Med. Dosw. (Online) 2017, 71, 57–68. [Google Scholar] [CrossRef]
- Manea, S.A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Ago, T.; Kitazono, T.; Kuroda, J.; Kumai, Y.; Kamouchi, M.; Ooboshi, H.; Wakisaka, M.; Kawahara, T.; Rokutan, K.; Ibayashi, S.; et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke 2005, 36, 1040–1046. [Google Scholar] [CrossRef]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef]
- Katsuyama, M.; Fan, C.; Yabe-Nishimura, C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: Induction of NOX1 by PGF2alpha. J. Biol. Chem. 2002, 277, 13438–13442. [Google Scholar] [CrossRef]
- Wingler, K.; Wunsch, S.; Kreutz, R.; Rothermund, L.; Paul, M.; Schmidt, H.H. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med. 2001, 31, 1456–1464. [Google Scholar] [CrossRef]
- Martín, A.; Pérez-Girón, J.V.; Hernanz, R.; Palacios, R.; Briones, A.M.; Fortuño, A.; Zalba, G.; Salaices, M.; Alonso, M.J. Peroxisome proliferator-activated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J. Hypertens. 2012, 30, 315–326. [Google Scholar] [CrossRef]
- Aguado, A.; Fischer, T.; Rodríguez, C.; Manea, A.; Martínez-González, J.; Touyz, R.M.; Hernanz, R.; Alonso, M.J.; Dixon, D.A.; Briones, A.M.; et al. HuR is required for NOX-1 but not NOX-4 regulation by inflammatory stimuli in vascular smooth muscle cells. J. Hypertens. 2016, 34, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, R.; Martínez-Revelles, S.; Palacios, R.; Martín, A.; Cachofeiro, V.; Aguado, A.; García-Redondo, L.; Barrús, M.T.; de Batista, P.R.; Briones, A.M.; et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br. J. Pharmacol. 2015, 172, 3159–3176. [Google Scholar] [CrossRef] [PubMed]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 2010, 396, 901–907. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. 2), S170–S180. [Google Scholar] [CrossRef]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH Oxidases in Vascular Pharmacology. Vascul Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef]
- Kawano, M.; Miyamoto, K.; Kaito, Y.; Sumimoto, H.; Tamura, M. Noxa1 as a moderate activator of Nox2-based NADPH oxidase. Arch. Biochem. Biophys. 2012, 519, 1–7. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Lu, X.; Murphy, T.C.; Nanes, M.S.; Hart, M. PPAR{gamma} Regulates Hypoxia-Induced Nox4 Expression in Human Pulmonary Artery Smooth Muscle Cells Through NF-{kappa}B. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L559–L566. [Google Scholar] [CrossRef]
- Fernandez, I.; Martin-Garrido, A.; Zhou, D.W.; Clempus, R.E.; Seidel-Rogol, B.; Valdivia, A.; Lassègue, B.; Garcia, A.J.; Griendling, K.K.; San Martin, A. Hic-5 Mediates TGFβ-Induced Adhesion in Vascular Smooth Muscle Cells by a Nox4-Dependent Mechanism. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1198–1206. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target 2015, 23, 888–896. [Google Scholar] [CrossRef]
- Katsuyama, M.; Matsuno, K.; Yabe-Nishimura, C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J. Clin. Biochem. Nutr. 2012, 50, 9–22. [Google Scholar] [CrossRef]
- Siuda, D.; Zechner, U.; El Hajj, N.; Prawitt, D.; Langer, D.; Xia, N.; Horke, S.; Pautz, A.; Kleinert, H.; Förstermann, U.; et al. Transcriptional regulation of Nox4 by histone deacetylases in human endothelial cells. Basic Res. Cardiol. 2012, 107, 283. [Google Scholar] [CrossRef]
- Manea, S.; Todirita, A.; Raicu, M.; Manea, A. C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J. Cell. Mol. Med. 2014, 18, 1467–1477. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell. 2010, 21, 2087–2096. [Google Scholar] [CrossRef]
- Paneni, F.; Osto, E.; Costantino, S.; Mateescu, B.; Briand, S.; Coppolino, G.; Perna, E.; Mocharla, P.; Akhmedov, A.; Kubant, R.; et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation 2013, 127, 1229–1240. [Google Scholar] [CrossRef]
- Bedard, K.; Jaquet, V.; Krause, K.H. NOX5: From basic biology to signaling and disease. Free Radic. Biol. Med. 2012, 52, 725–734. [Google Scholar] [CrossRef]
- Pandey, D.; Patel, A.; Patel, V.; Chen, F.; Qian, J.; Wang, Y.; Barman, S.A.; Venema, R.C.; Stepp, D.W.; Rudic, D.; et al. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1919–H1928. [Google Scholar] [CrossRef]
- Jay, D.B.; Papaharalambus, C.A.; Seidel-Rogol, B.; Dikalova, A.E.; Lassègue, B.; Griendling, K.K. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 329–335. [Google Scholar] [CrossRef]
- Jha, J.C.; Watson, A.M.D.; Mathew, G.; de Vos, L.C.; Jandeleit-Dahm, K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin. Sci. (London) 2017, 131, 981–990. [Google Scholar] [CrossRef]
- Schaftenaar, F.; Frodermann, V.; Kuiper, J.; Lutgens, E. Atherosclerosis: The interplay between lipids and immune cells. Curr. Opin. Lipidol. 2016, 27, 209–215. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Kwak, B.R.; Bäck, M.; Bochaton-Piallat, M.L.; Caligiuri, G.; Daemen, M.J.; Davies, P.F.; Hoefer, I.E.; Holvoet, P.; Jo, H.; Krams, R.; et al. Biomechanical factors in atherosclerosis: Mechanisms and clinical implications. Eur. Heart J. 2014, 35, 3013–3020. [Google Scholar] [CrossRef]
- Legein, B.; Temmerman, L.; Biessen, E.A.; Lutgens, E. Inflammation and immune system interactions in atherosclerosis. Cell. Mol. Life Sci. 2013, 70, 3847–3869. [Google Scholar] [CrossRef]
- Ketelhuth, D.F.; Bäck, M. The role of matrix metalloproteinases in atherothrombosis. Curr. Atheroscler. Rep. 2011, 13, 162–169. [Google Scholar] [CrossRef]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.; Sobenin, I.A.; Orekhov, A.N. The atherogenic role of circulating modified lipids in atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL particles activate inflammatory pathways in monocyte-derived macrophages: Transcriptome analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar]
- Feig, J.E.; Feig, J.L.; Kini, A.S. Statins, Atherosclerosis Regression and HDL: Insights From Within the Plaque. Int. J. Cardiol. 2015, 189, 168–171. [Google Scholar] [CrossRef]
- Antonopulos, A.S.; Margaritis, M.; Lee, R.; Channon, K.; Antoniades, C. Statins as anti-inflammatory agents in atherogenesis: Molecular mechanisms and lessons from the recent clinical trials. Curr. Pharm. Des. 2012, 18, 1519–1530. [Google Scholar] [CrossRef]
- Sukhova, G.K.; Williams, J.K.; Libby, P. Statins reduce inflammation in atheroma of nonhuman primates independent of effects on serum cholesterol. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Aquila, S. The role of oxidative stress and autophagy in atherosclerosis. Oxid. Med. Cell. Longev. 2015, 2015, 130315. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. (London) 2005, 109, 217–226. [Google Scholar] [CrossRef]
- Schröder, K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid. Redox Signal. 2014, 20, 2043–2058. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.; Touyz, R.M.; Cooper, M.E.; et al. Reactive oxygen species can provide atheroprotection via NOX4-Dependent inhibition of inflammation and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef]
- Lee, M.Y.; San Martin, A.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Hakim, Z.S.; Madamanchi, N.R.; Rojas, M.; Madamanchi, C.; Runge, M.S. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2714–2721. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, Y.; Han, X.; Zhang, Y.; Zhang, X.; Gao, Y.; Zhang, J.; Chu, L.; Zhao, S. Potential mechanisms underlying the protective effects of salvianic acid A against atherosclerosis in vivo and vitro. Biomed. Pharmacother. 2019, 109, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J.; Barman, S.A. Clarity on the Isoform-Specific Roles of NADPH Oxidases and NADPH Oxidase-4 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, E.; Gray, S.P.; Kennedy, K.; Szyndralewiez, C.; Lyle, A.N.; Lassègue, B.; Griendling, K.K.; Cooper, M.E.; Schmidt, H.H.H.W.; Jandeleit-Dahm, K.A.M. NOX4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic. Biol. Med. 2016, 97, 556–567. [Google Scholar] [CrossRef]
- Nishimura, A.; Ago, T.; Kuroda, J.; Arimura, K.; Tachibana, M.; Nakamura, K.; Wakisaka, Y.; Sadoshima, J.; Iihara, K.; Kitazono, T. Detrimental role of pericyte Nox4 in the acute phase of brain ischemia. J. Cereb. Blood Flow Metab. 2016, 36, 1143–1154. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H282–H296. [Google Scholar] [CrossRef]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Sumida, A.; Canugovi, C.; Lozhkin, A.; Hayami, T.; Madamanchi, N.R.; Runge, M.S. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol. 2019, 21, 101063. [Google Scholar] [CrossRef]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Schmidt, H.H.; Lassègue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef]
- Meijles, D.N.; Pagano, P.J. Nox and Inflammation in the Vascular Adventitia. Hypertension 2016, 67, 14–19. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Li, L.; Zhou, H.J.; Li, F.; Zhang, H.; Yu, L.; Chen, Y.; Min, W. Endothelial AIP1 Regulates Vascular Remodeling by Suppressing NADPH Oxidase-2. Front. Physiol. 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J. Opportunity Nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Griendling, K.K.; Harrison, D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Madamanchi, N.R.; Niu, X.L.; Molnar, K.C.; Runge, M.; Szyndralewiez, C.; Page, P.; Runge, M.S. NADPH oxidases regulate cd44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J. Biol. Chem. 2010, 285, 26545–26557. [Google Scholar] [CrossRef]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic Potential of NADPH Oxidase ¼ Inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, B.; Quesada, I.; Militello, R.; Amaya, C.; Miatello, R.; Cruzado, M.; Castro, C. Tempol Attenuates Atherosclerosis Associated With Metabolic Syndrome via Decreased Vascular Inflammation and NADPH-2 Oxidase Expression. Free Radic. Res. 2014, 48, 526–533. [Google Scholar] [CrossRef]
- Maqbool, A.; Watt, N.T.; Haywood, N.; Viswambharan, H.; Skromna, A.; Makava, N.; Visnagri, A.; Shawer, H.M.; Bridge, K.; Muminov, S.K.; et al. Divergent Effects of Genetic and Pharmacological Inhibition of Nox2 NADPH Oxidase on Insulin Resistance-Related Vascular Damage. Am. J. Physiol. Cell Physiol. 2020, 319, C64–C74. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. https://doi.org/10.3390/biomedicines8070206
Poznyak AV, Grechko AV, Orekhova VA, Khotina V, Ivanova EA, Orekhov AN. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines. 2020; 8(7):206. https://doi.org/10.3390/biomedicines8070206
Chicago/Turabian StylePoznyak, Anastasia V., Andrey V. Grechko, Varvara A. Orekhova, Victoria Khotina, Ekaterina A. Ivanova, and Alexander N. Orekhov. 2020. "NADPH Oxidases and Their Role in Atherosclerosis" Biomedicines 8, no. 7: 206. https://doi.org/10.3390/biomedicines8070206
APA StylePoznyak, A. V., Grechko, A. V., Orekhova, V. A., Khotina, V., Ivanova, E. A., & Orekhov, A. N. (2020). NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines, 8(7), 206. https://doi.org/10.3390/biomedicines8070206