NLPR3 Inflammasomes and Their Significance for Atherosclerosis

 , and
, and 
{kind=link}
{kind=link}
Abstract
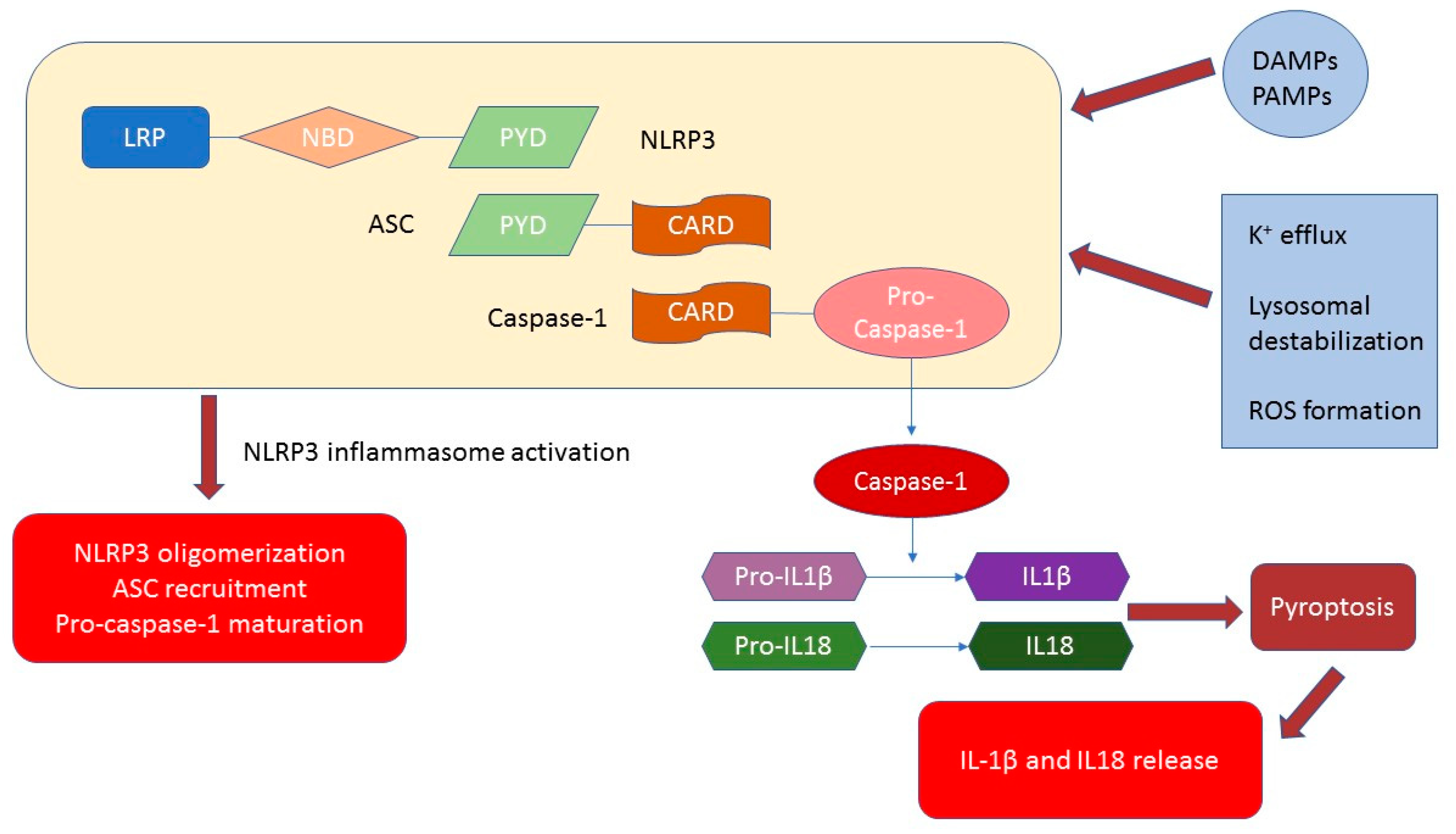
1. NLRP3 Inflammasome
2. Significance for Atherosclerosis
2.1. Relation with Mitochondrial Damage
2.2. NLRP3 Inflammasome and Lipids
2.3. Proinflammatory Cytokines
3. Targeting NLRP3 Inflammasome for the Treatment of Atherosclerosis
3.1. Statins
3.2. Metformin
3.3. Phytocompounds
3.4. Clinical Trials Targeting NLRP3 Pathway in Atherosclerotic Patients
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-W.; Lee, M.-S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharmacal Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Dorfleutner, A.; Chu, L.; Stehlik, C. Inhibiting the inflammasome: One domain at a time. Immunol. Rev. 2015, 265, 205–216. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.L.; Haley, B.; Roose-Girma, M.; Phung, Q.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Groslambert, M.; Py, B. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Py, B.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediat. Inflamm. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Diabetic Macrovascular Complications. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxidative Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Altaf, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Heart Vessel. 2015, 31, 1218–1229. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Investig. 2017, 97, 922–934. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Lou, X.; Zou, Z.; Mou, N.; Wu, W.; Huang, X.; Tan, H. Folic acid attenuates hyperhomocysteinemia-induced glomerular damage in rats. Microvasc. Res. 2013, 89, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the nlrp3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-M.; Zhang, Q. Homocysteine-impaired angiogenesis is associated with VEGF/VEGFR inhibition. Front. Biosci. 2012, 4, 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.-L. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, 154–162. [Google Scholar] [CrossRef]
- Wang, L.; Qu, P.; Zhao, J.; Chang, Y. NLRP3 and downstream cytokine expression elevated in the monocytes of patients with coronary artery disease. Arch. Med. Sci. 2014, 10, 791–800. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.-F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arter. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel insights into the nlrp3 Inflammasome in atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef]
- Bleda, S.; De Haro, J.; Varela, C.; Esparza, L.; Ferruelo, A.; Acín, F. NLRP1 inflammasome, and not NLRP3, is the key in the shift to proinflammatory state on endothelial cells in peripheral arterial disease. Int. J. Cardiol. 2014, 172, e282–e284. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Yaron, J.R.; Gangaraju, S.; Rao, M.; Kong, X.; Zhang, L.; Su, F.; Tian, Y.; Glenn, H.L.; Meldrum, D.R. K(+) regulates Ca(2+) to drive inflammasome signaling: Dynamic visualization of ion flux in live cells. Cell Death Dis. 2015, 6, e1954. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2017, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized Low-Density Lipoprotein. Methods Mol. Biol. 2009, 610, 403–417. [Google Scholar] [CrossRef]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Núñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Rajamäki, K.; Nordström, T.; Nurmi, K.; Åkerman, K.E.O.; Kovanen, P.T.; Öörni, K.; Eklund, K.K. Extracellular Acidosis Is a Novel Danger Signal Alerting Innate Immunity via the NLRP3 Inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.-C.; Marin, T.; Shentu, T.-P.; Wen, L.; Gongol, B.; et al. Sterol regulatory element binding protein 2 activation of nlrp3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegård, K.T.; Brekke, O.-L.; Lambris, J.D.; Damås, J.K.; et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef]
- Menu, P.; Pellegrin, M.; Aubert, J.-F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef]
- Sheedy, F.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, U.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Bhaskar, V.; Yin, J.; Mirza, A.M.; Phan, D.; Vanegas, S.; Issafras, H.; Michelson, K.; Hunter, J.J.; Kantak, S.S. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis 2011, 216, 313–320. [Google Scholar] [CrossRef]
- Lin, J.; Shou, X.; Mao, X.; Dong, J.; Mohabeer, N.; Kushwaha, K.K.; Wang, L.; Su, Y.; Fang, H.; Li, D. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: Implication in lesion instability? PLoS ONE 2013, 8, e62148. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Lin, J.; Dong, J.; Li, D. Pyroptosis: An inflammatory cell death implicates in atherosclerosis. Med Hypotheses 2013, 81, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Nagumo, Y.; Watanabe, T.; Imaizumi, T.; Shibuya, M.; Kataoka, T.; Kanoh, N.; Iwabuchi, Y.; Usui, T. Irciniastatin A induces JNK activation that is involved in caspase-8-dependent apoptosis via the mitochondrial pathway. Toxicol. Lett. 2010, 199, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Margaritis, M.; Lee, R.; Channon, K.M.; Antoniades, C. Statins as anti-inflammatory agents in atherogenesis: Molecular mechanisms and lessons from the recent clinical trials. Curr. Pharm. Des. 2012, 18, 1519–1530. [Google Scholar] [CrossRef]
- Peng, S.; Xu, L.-W.; Che, X.-Y.; Xiao, Q.-Q.; Pu, J.; Shao, Q.; He, B. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Boland, A.; Gangadharan, N.; Kavanagh, P.; Hemeryck, L.; Kieran, J.; Barry, M.; Walsh, P.T.; Lucitt, M. Simvastatin Suppresses Interleukin Iβ Release in Human Peripheral Blood Mononuclear Cells Stimulated With Cholesterol Crystals. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 509–517. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, L.; Zhong, X.; Yue, Y.; Hong, Y.; Li, Y.; Li, Y. Metformin reduced NLRP3 inflammasome activity in Ox-LDL stimulated macrophages through adenosine monophosphate activated protein kinase and protein phosphatase 2A. Eur. J. Pharmacol. 2019, 852, 99–106. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE-/- mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Jiang, Y.; Du, H.; Liu, X.; Fu, X.; Li, X.; Cao, Q. Artemisinin alleviates atherosclerotic lesion by reducing macrophage inflammation via regulation of AMPK/NF-κB/NLRP3 inflammasomes pathway. J. Drug Target. 2019, 28, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Ye, B.; Cao, J.; Cai, X.; Lin, L.; Huang, S.; Huang, W.; Huang, Z. Curcumin Represses NLRP3 Inflammasome Activation via TLR4/MyD88/NF-κB and P2X7R Signaling in PMA-Induced Macrophages. Front. Pharmacol. 2016, 7, 1524. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Mao, J.; Xu, S.; Zhao, L.; Long, L.; Chen, L.; Li, D.; Lu, S. Rosmarinic acid inhibits nicotine-induced C-reactive protein generation by inhibiting NLRP3 inflammasome activation in smooth muscle cells. J. Cell. Physiol. 2018, 234, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Zoccai, G.B.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef]
- Morton, A.C.; Rothman, A.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, I.P.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2014, 36, 377–384. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M. Closing the Loop on Inflammation and Atherothrombosis: Why Perform the Cirt and Cantos Trials? Trans. Am. Clin. Clim. Assoc. 2013, 124, 174–190. [Google Scholar]
- Choudhury, R.; Birks, J.S.; Mani, V.; Biasiolli, L.; Robson, M.D.; L’Allier, P.L.; Gingras, M.-A.; Alie, N.; McLaughlin, M.A.; Basson, C.T.; et al. Arterial Effects of Canakinumab in Patients With Atherosclerosis and Type 2 Diabetes or Glucose Intolerance. J. Am. Coll. Cardiol. 2016, 68, 1769–1780. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Melnichenko, A.A.; Wetzker, R.; Gerasimova, E.V.; Orekhov, A.N. NLPR3 Inflammasomes and Their Significance for Atherosclerosis. Biomedicines 2020, 8, 205. https://doi.org/10.3390/biomedicines8070205
Poznyak AV, Melnichenko AA, Wetzker R, Gerasimova EV, Orekhov AN. NLPR3 Inflammasomes and Their Significance for Atherosclerosis. Biomedicines. 2020; 8(7):205. https://doi.org/10.3390/biomedicines8070205
Chicago/Turabian StylePoznyak, Anastasia V., Alexandra A. Melnichenko, Reinhard Wetzker, Elena V. Gerasimova, and Alexander N. Orekhov. 2020. "NLPR3 Inflammasomes and Their Significance for Atherosclerosis" Biomedicines 8, no. 7: 205. https://doi.org/10.3390/biomedicines8070205
APA StylePoznyak, A. V., Melnichenko, A. A., Wetzker, R., Gerasimova, E. V., & Orekhov, A. N. (2020). NLPR3 Inflammasomes and Their Significance for Atherosclerosis. Biomedicines, 8(7), 205. https://doi.org/10.3390/biomedicines8070205