Endothelial Dysfunction in Diabetes


 ,
, {kind=link}
Abstract
1. Introduction
2. Insulin Resistance and Endothelial Dysfunction
2.1. Insulin Signaling in Endothelium
2.2. Insulin Resistance in the Endothelium
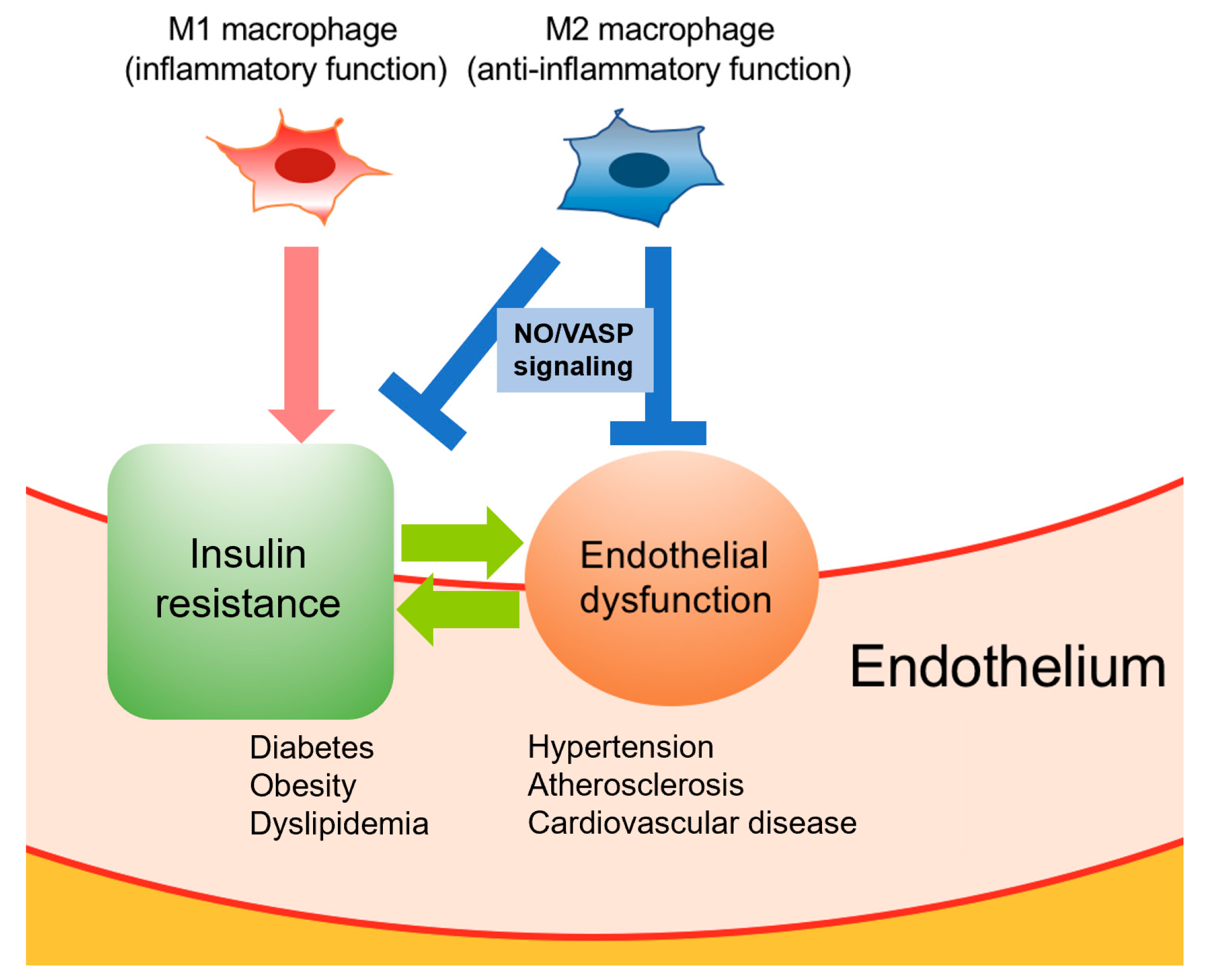
3. Crosstalk between Macrophage Polarization and Endothelial Cells
4. Targeting Endothelial Dysfunction
4.1. SGLT2 Inhibitors
4.2. GLP-1 Receptor Agonists
4.3. DPP-4 Inhibitors
4.4. Biguanides
4.5. Thiazolidinediones
4.6. Sulfonylureas
4.7. Medical Nutrition Therapy and Physical Activity
5. ROCK Inhibitors as Preclinical Agents
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas 9th Edition 2019. Available online: https://www.diabetesatlas.org/en/resources/ (accessed on 20 May 2020).
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. 2), S285–S290. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Kirpichnikov, D.; Sowers, J.R. Diabetes mellitus and diabetes-associated vascular disease. Trends Endocrinol. Metab. TEM 2001, 12, 225–230. [Google Scholar] [CrossRef]
- Siasos, G.; Tousoulis, D.; Antoniades, C.; Stefanadi, E.; Stefanadis, C. L-Arginine, the substrate for NO synthesis: An alternative treatment for premature atherosclerosis. Int. J. Cardiol. 2007, 116, 300–308. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Xu, J.; Zou, M.H. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 2009, 120, 1266–1286. [Google Scholar] [CrossRef]
- van Guilder, G.P.; Hoetzer, G.L.; Dengel, D.R.; Stauffer, B.L.; DeSouza, C.A. Impaired endothelium-dependent vasodilation in normotensive and normoglycemic obese adult humans. J. Cardiovasc. Pharmacol. 2006, 47, 310–313. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial cell metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Fujii, N.; Reinke, M.C.; Brunt, V.E.; Minson, C.T. Impaired acetylcholine-induced cutaneous vasodilation in young smokers: Roles of nitric oxide and prostanoids. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H667–H673. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Schmitz, B.; Brand, E. Salt controls endothelial and vascular phenotype. Pflugers Arch. 2015, 467, 499–512. [Google Scholar] [CrossRef]
- Bender, S.B.; Laughlin, M.H. Modulation of endothelial cell phenotype by physical activity: Impact on obesity-related endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1–H8. [Google Scholar] [CrossRef]
- Moreau, K.L.; Hildreth, K.L. Vascular aging across the menopause transition in healthy women. Adv. Vasc. Med. 2014, 2014, 204390. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Demosthenous, M.; Tousoulis, D.; Antonopoulos, A.S.; Vlachopoulos, C.; Toutouza, M.; Marinou, K.; Bakogiannis, C.; Mavragani, K.; Lazaros, G.; et al. Role of asymmetrical dimethylarginine in inflammation-induced endothelial dysfunction in human atherosclerosis. Hypertension 2011, 58, 93–98. [Google Scholar] [CrossRef]
- Kharbanda, R.K.; Walton, B.; Allen, M.; Klein, N.; Hingorani, A.D.; MacAllister, R.J.; Vallance, P. Prevention of inflammation-induced endothelial dysfunction: A novel vasculo-protective action of aspirin. Circulation 2002, 105, 2600–2604. [Google Scholar] [CrossRef] [PubMed]
- Channon, K.M.; Guzik, T.J. Mechanisms of superoxide production in human blood vessels: Relationship to endothelial dysfunction, clinical and genetic risk factors. J. Physiol. Pharmacol. 2002, 53, 515–524. [Google Scholar]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Targeting redox imbalance as an approach for diabetic kidney disease. Biomedicines 2020, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Wu, F.; Szczepaniak, W.S.; Shiva, S.; Liu, H.; Wang, Y.; Wang, L.; Wang, Y.; Kelley, E.E.; Chen, A.F.; Gladwin, M.T.; et al. Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L987–L997. [Google Scholar] [CrossRef]
- Zhang, Y.; Janssens, S.P.; Wingler, K.; Schmidt, H.H.H.W.; Moens, A.L. Modulating endothelial nitric oxide synthase: A new cardiovascular therapeutic strategy. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H634–H646. [Google Scholar] [CrossRef]
- Virdis, A.; Bacca, A.; Colucci, R.; Duranti, E.; Fornai, M.; Materazzi, G.; Ippolito, C.; Bernardini, N.; Blandizzi, C.; Bernini, G.; et al. Endothelial dysfunction in small arteries of essential hypertensive patients: Role of cyclooxygenase-2 in oxidative stress generation. Hypertension 2013, 62, 337–344. [Google Scholar] [CrossRef]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Barac, A.; Campia, U.; Panza, J.A. Methods for evaluating endothelial function in humans. Hypertension 2007, 49, 748–760. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, J.T. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Corretti, M.C.; Anderson, T.J.; Benjamin, E.J.; Celermajer, D.; Charbonneau, F.; Creager, M.A.; Deanfield, J.; Drexler, H.; Gerhard-Herman, M.; Herrington, D.; et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the International Brachial Artery Reactivity Task Force. J. Am. Coll. Cardiol. 2002, 39, 257–265. [Google Scholar] [CrossRef]
- Hamburg, N.M.; Palmisano, J.; Larson, M.G.; Sullivan, L.M.; Lehman, B.T.; Vasan, R.S.; Levy, D.; Mitchell, G.F.; Vita, J.A.; Benjamin, E.J. Relation of brachial and digital measures of vascular function in the community: The Framingham heart study. Hypertension 2011, 57, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Ghiadoni, L.; Versari, D.; Giannarelli, C.; Faita, F.; Taddei, S. Non-invasive diagnostic tools for investigating endothelial dysfunction. Curr. Pharm. Des. 2008, 14, 3715–3722. [Google Scholar] [CrossRef] [PubMed]
- Moerland, M.; Kales, A.J.; Schrier, L.; van Dongen, M.G.; Bradnock, D.; Burggraaf, J. Evaluation of the EndoPAT as a tool to assess endothelial function. Int. J. Vasc. Med. 2012, 2012, 904141. [Google Scholar] [CrossRef]
- Youngren, J.F. Regulation of insulin receptor function. Cell. Mol. Life Sci. CMLS 2007, 64, 873–891. [Google Scholar] [CrossRef]
- Withers, D.J.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.M.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.I.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Miao, B.; Skidan, I.; Yang, J.; Lugovskoy, A.; Reibarkh, M.; Long, K.; Brazell, T.; Durugkar, K.A.; Maki, J.; Ramana, C.V.; et al. Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc. Natl. Acad. Sci. USA 2010, 107, 20126–20131. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.A.; Alessi, D.R. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar] [PubMed]
- Park, K.; Li, Q.; Rask-Madsen, C.; Mima, A.; Mizutani, K.; Winnay, J.; Maeda, Y.; D’Aquino, K.; White, M.F.; Feener, E.P.; et al. Serine phosphorylation sites on IRS2 activated by angiotensin II and protein kinase C to induce selective insulin resistance in endothelial cells. Mol. Cell. Biol. 2013, 33, 3227–3241. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H.; Jaffrey, S.R. Vessels vivified by Akt acting on NO synthase. Nat. Cell Biol. 1999, 1, E95–E96. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Ferri, C.; Pittoni, V.; Piccoli, A.; Laurenti, O.; Cassone, M.R.; Bellini, C.; Properzi, G.; Valesini, G.; de Mattia, G.; Santucci, A. Insulin stimulates endothelin-1 secretion from human endothelial cells and modulates its circulating levels in vivo. J. Clin. Endocrinol. Metab. 1995, 80, 829–835. [Google Scholar]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Meza, C.A.; la Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Janus, A.; Szahidewicz-Krupska, E.; Mazur, G.; Doroszko, A. Insulin resistance and endothelial dysfunction constitute a common therapeutic target in cardiometabolic disorders. Mediat. Inflamm. 2016, 2016, 3634948. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.D.; Rahmutula, D.; Gardner, D.G. Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells. J. Biol. Chem. 2004, 279, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wheatley, C.M.; Richards, S.M.; Barrett, E.J.; Clark, M.G.; Rattigan, S. TNF-alpha acutely inhibits vascular effects of physiological but not high insulin or contraction. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E654–E660. [Google Scholar] [CrossRef] [PubMed]
- Kougias, P.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J. Vasc. Surg. 2005, 41, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Hennige, A.M.; Stefan, N.; Kapp, K.; Lehmann, R.; Weigert, C.; Beck, A.; Moeschel, K.; Mushack, J.; Schleicher, E.; Häring, H.U. Leptin down-regulates insulin action through phosphorylation of serine-318 in insulin receptor substrate 1. FASEB J. 2006, 20, 1206–1208. [Google Scholar] [CrossRef]
- Verma, S.; Li, S.H.; Wang, C.H.; Fedak, P.W.; Li, R.K.; Weisel, R.D.; Mickle, D.A. Resistin promotes endothelial cell activation: Further evidence of adipokine-endothelial interaction. Circulation 2003, 108, 736–740. [Google Scholar] [CrossRef]
- Chen, H.; Montagnani, M.; Funahashi, T.; Shimomura, I.; Quon, M.J. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J. Biol. Chem. 2003, 278, 45021–45026. [Google Scholar] [CrossRef]
- Tesauro, M.; Schinzari, F.; Iantorno, M.; Rizza, S.; Melina, D.; Lauro, D.; Cardillo, C. Ghrelin improves endothelial function in patients with metabolic syndrome. Circulation 2005, 112, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Li, P.P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-DeLeon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K.; et al. M2 macrophage polarization mediates anti-inflammatory effects of endothelial nitric oxide signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef]
- Trichet, L.; Sykes, C.; Plastino, J. Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J. Cell Biol. 2008, 181, 19–25. [Google Scholar] [CrossRef]
- Handa, H.; Tateya, S.; Rizzo, N.O.; Cheng, A.M.; Morgan-Stevenson, V.; Han, C.Y.; Clowes, A.W.; Daum, G.; O’Brien, K.D.; Schwartz, M.W.; et al. Reduced vascular nitric oxide-cGMP signaling contributes to adipose tissue inflammation during high-fat feeding. Arter. Thromb. Vasc. Biol. 2011, 31, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Tateya, S.; Rizzo, N.O.; Handa, P.; Cheng, A.M.; Morgan-Stevenson, V.; Daum, G.; Clowes, A.W.; Morton, G.J.; Schwartz, M.W.; Kim, F. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 2011, 60, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Hollmann, M.W.; Zuurbier, C.J.; Weber, N.C. Administration of SGLT2 inhibitor empagliflozin against TNF-α induced endothelial dysfunction in human venous and arterial endothelial cells. FASEB J. 2018, 32, 25. [Google Scholar] [CrossRef]
- Xu, L.; Ota, T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte 2018, 7, 121–128. [Google Scholar] [CrossRef]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Seino, Y.; Yabe, D. Glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1: Incretin actions beyond the pancreas. J. Diabetes Investig. 2013, 4, 108–130. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Drucker, D.J. Incretin action in the pancreas: Potential promise, possible perils, and pathological pitfalls. Diabetes 2013, 62, 3316–3323. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, A.; Oyama, J.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Mori, Y. Anti-atherogenic and anti-inflammatory properties of glucagon-like peptide-1, glucose-dependent insulinotropic polypepide, and dipeptidyl peptidase-4 inhibitors in experimental animals. J. Diabetes Investig. 2016, 1 (Suppl. 7), 80–86. [Google Scholar] [CrossRef] [PubMed]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor mediates cardiovascular protection by liraglutide in mice with experimental arterial hypertension. Arter. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef]
- Erdogdu, O.; Nathanson, D.; Sjöholm, A.; Nyström, T.; Zhang, Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol. Cell. Endocrinol. 2010, 325, 26–35. [Google Scholar] [CrossRef]
- Cai, X.; She, M.; Xu, M.; Chen, H.; Li, J.; Chen, X.; Zheng, D.; Liu, J.; Chen, S.; Zhu, J.; et al. GLP-1 treatment protects endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction. Int. J. Biol. Sci. 2018, 14, 1696–1708. [Google Scholar] [CrossRef]
- Lovshin, J.; Cherney, D. GLP-1R agonists and endothelial dysfunction: More than just glucose lowering? Diabetes 2015, 64, 2319. [Google Scholar] [CrossRef]
- Irace, C.; de Luca, S.; Shehaj, E.; Carallo, C.; Loprete, A.; Scavelli, F.; Gnasso, A. Exenatide improves endothelial function assessed by flow mediated dilation technique in subjects with type 2 diabetes: Results from an observational research. Diabetes Vasc. Dis. Res. 2013, 10, 72–77. [Google Scholar] [CrossRef]
- Kelly, A.S.; Bergenstal, R.M.; Gonzalez-Campoy, J.M.; Katz, H.; Bank, A.J. Effects of exenatide vs. metformin on endothelial function in obese patients with pre-diabetes: A randomized trial. Cardiovasc. Diabetol. 2012, 11, 64. [Google Scholar] [CrossRef]
- Vinué, Á.; Navarro, J.; Herrero-Cervera, A.; García-Cubas, M.; Andrés-Blasco, I.; Martínez-Hervás, S.; Real, J.T.; Ascaso, J.F.; González-Navarro, H. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia 2017, 60, 1801–1812. [Google Scholar]
- Bruen, R.; Curley, S.; Kajani, S.; Crean, D.; O’Reilly, M.E.; Lucitt, M.B.; Godson, C.G.; McGillicuddy, F.C.; Belton, O. Liraglutide dictates macrophage phenotype in apolipoprotein E null mice during early atherosclerosis. Cardiovasc. Diabetol. 2017, 16, 143. [Google Scholar] [CrossRef]
- Chowdhury, H.H.; Velebit, J.; Radić, N.; Frančič, V.; Kreft, M.; Zorec, R. Hypoxia alters the expression of dipeptidyl peptidase 4 and induces developmental remodeling of human preadipocytes. J. Diabetes Res. 2016, 2016, 7481470. [Google Scholar] [CrossRef]
- Varin, E.M.; Mulvihill, E.E.; Beaudry, J.L.; Pujadas, G.; Fuchs, S.; Tanti, J.-F.; Fazio, S.; Kaur, K.; Cao, X.; Baggio, L.L.; et al. Circulating levels of soluble dipeptidyl peptidase-4 are dissociated from inflammation and induced by enzymatic DPP4 inhibition. Cell Metab. 2019, 29, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.; Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 2012, 59, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Castillo, A.C.; Qian, J.; Ling, S.; Ye, H.; Perez-Polo, J.R.; Bajaj, M.; Ye, Y. Phosphodiesterase III inhibition increases cAMP levels and augments the infarct size limiting effect of a DPP-4 inhibitor in mice with type-2 diabetes mellitus. Cardiovasc. Drugs Ther. 2012, 26, 445–456. [Google Scholar] [CrossRef]
- Zhuge, F.; Ni, Y.; Nagashimada, M.; Nagata, N.; Xu, L.; Mukaida, N.; Kaneko, S.; Ota, T. DPP-4 Inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes 2016, 65, 2966–2979. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Akiyama, E.; Iwashita, S.; Kurokawa, H.; Ohba, K.; Maeda, H.; Fujisue, K.; Yamamoto, E.; Kaikita, K.; et al. Dipeptidyl peptidase-4 inhibitor, sitagliptin, improves endothelial dysfunction in association with its anti-inflammatory effects in patients with coronary artery disease and uncontrolled diabetes. Circ. J. 2013, 77, 1337–1344. [Google Scholar] [CrossRef]
- Leung, M.; Leung, D.Y.; Wong, V.W. Effects of dipeptidyl peptidase-4 inhibitors on cardiac and endothelial function in type 2 diabetes mellitus: A pilot study. Diabetes Vasc. Dis. Res. 2016, 13, 236–243. [Google Scholar] [CrossRef]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Iga, R.; Kobayashi, Y.; Kanda, E.; Uchino, H.; Hirose, T. Linagliptin improves endothelial function in patients with type 2 diabetes: A randomized study of linagliptin effectiveness on endothelial function. J. Diabetes Investig. 2017, 8, 330–340. [Google Scholar] [CrossRef]
- Kajikawa, M.; Maruhashi, T.; Hidaka, T.; Matsui, S.; Hashimoto, H.; Takaeko, Y.; Nakano, Y.; Kurisu, S.; Kihara, Y.; Yusoff, F.M.; et al. Effect of Saxagliptin on Endothelial Function in Patients with Type 2 Diabetes: A Prospective Multicenter Study. Sci. Rep. 2019, 9, 10206. [Google Scholar] [CrossRef]
- Nakamura, K.; Oe, H.; Kihara, H.; Shimada, K.; Fukuda, S.; Watanabe, K.; Takagi, T.; Yunoki, K.; Miyoshi, T.; Hirata, K.; et al. DPP-4 inhibitor and alpha-glucosidase inhibitor equally improve endothelial function in patients with type 2 diabetes: EDGE study. Cardiovasc. Diabetol. 2014, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.A.; Gudbjörnsdóttir, H.S.; Andersson, O.K.; Lönnroth, P.N. The effect of metformin on adipose tissue metabolism and peripheral blood flow in subjects with NIDDM. Diabetes Care 1996, 19, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Nafisa, A.; Gray, S.G.; Cao, Y.; Wang, T.; Xu, S.; Wattoo, F.H.; Barras, M.; Cohen, N.; Kamato, D.; Little, P.J. Endothelial function and dysfunction: Impact of metformin. Pharmacol. Ther. 2018, 192, 150–162. [Google Scholar] [CrossRef]
- de Jager, J.; Kooy, A.; Schalkwijk, C.; van der Kolk, J.; Lehert, P.; Bets, D.; Wulffelé, M.G.; Donker, A.J.; Stehouwer, C.D. Long-term effects of metformin on endothelial function in type 2 diabetes: A randomized controlled trial. J. Intern. Med. 2014, 275, 59–70. [Google Scholar] [CrossRef]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An old drug with new applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Mather, K.J.; Verma, S.; Anderson, T.J. Improved endothelial function with metformin in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2001, 37, 1344–1350. [Google Scholar] [CrossRef]
- Shih, M.H.; Xu, Y.Y.; Yang, Y.S.; Lin, G.L. A facile synthesis and antimicrobial activity evaluation of sydnonyl-substituted thiazolidine derivatives. Molecules 2015, 20, 6520–6532. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Knouff, C.; Auwerx, J. Peroxisome proliferator-activated receptor-gamma calls for activation in moderation: Lessons from genetics and pharmacology. Endocr. Rev. 2004, 25, 899–918. [Google Scholar] [CrossRef]
- del Valle, H.F.; Lascano, E.C.; Negroni, J.A.; Crottogini, A.J. Glibenclamide effects on reperfusion-induced malignant arrhythmias and left ventricular mechanical recovery from stunning in conscious sheep. Cardiovasc. Res. 2001, 50, 474–485. [Google Scholar] [CrossRef][Green Version]
- McAlister, F.A.; Eurich, D.T.; Majumdar, S.R.; Johnson, J.A. The risk of heart failure in patients with type 2 diabetes treated with oral agent monotherapy. Eur. J. Heart Fail. 2008, 10, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Pagano, P.J.; Griswold, M.C.; Ravel, D.; Cohen, R.A. Vascular action of the hypoglycaemic agent gliclazide in diabetic rabbits. Diabetologia 1998, 41, 9–15. [Google Scholar] [CrossRef]
- Katakami, N.; Yamasaki, Y.; Hayaishi-Okano, R.; Ohtoshi, K.; Kaneto, H.; Matsuhisa, M.; Kosugi, K.; Hori, M. Metformin or gliclazide, rather than glibenclamide, attenuate progression of carotid intima-media thickness in subjects with type 2 diabetes. Diabetologia 2004, 47, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Corgnali, M.; Piconi, L.; Ihnat, M.; Ceriello, A. Evaluation of gliclazide ability to attenuate the hyperglycaemic ‘memory’ induced by high glucose in isolated human endothelial cells. Diabetes/Metab. Res. Rev. 2008, 24, 301–309. [Google Scholar] [CrossRef]
- Jojima, T.; Suzuki, K.; Hirama, N.; Uchida, K.; Hattori, Y. Glimepiride upregulates eNOS activity and inhibits cytokine-induced NF-kappaB activation through a phosphoinoside 3-kinase-Akt-dependent pathway. Diabetes Obes. Metab. 2009, 11, 143–149. [Google Scholar] [CrossRef]
- American Diabetes Association. Standards of medical care in diabetes—2010. Diabetes Care 2010, 1 (Suppl. 33), 11–61. [Google Scholar]
- Hamdy, O.; Ledbury, S.; Mullooly, C.; Jarema, C.; Porter, S.; Ovalle, K.; Moussa, A.; Caselli, A.; Caballero, A.E.; Economides, P.A.; et al. Lifestyle modification improves endothelial function in obese subjects with the insulin resistance syndrome. Diabetes Care 2003, 26, 2119–2125. [Google Scholar] [CrossRef]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arter. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef]
- Hambrecht, R.; Adams, V.; Erbs, S.; Linke, A.; Kränkel, N.; Shu, Y.; Baither, Y.; Gielen, S.; Thiele, H.; Gummert, J.F.; et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation 2003, 107, 3152–3158. [Google Scholar] [CrossRef]
- Montero, D.; Walther, G.; Benamo, E.; Perez-Martin, A.; Vinet, A. Effects of exercise training on arterial function in type 2 diabetes mellitus: A systematic review and meta-analysis. Sports Med. 2013, 43, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, X.; Yin, H.; Sun, Z.; Zügel, M.; Steinacker, J.M.; Schumann, U. Exercise training and endothelial function in patients with type 2 diabetes: A meta-analysis. Cardiovasc. Diabetol. 2018, 17, 64. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Nagai, Y.; Takeda, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-Kinase blockade attenuates podocyte apoptosis by inhibiting the notch signaling pathway in diabetic nephropathy. Int. J. Mol. Sci. 2017, 18, 1795. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Matoba, K.; Kawanami, D.; Takeda, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. ROCK2 regulates TGF-β-induced expression of CTGF and profibrotic genes via NF-κB and cytoskeleton dynamics in mesangial cells. Am. J. Physiol. Renal. Physiol. 2019, 317, F839–F851. [Google Scholar] [CrossRef]
- Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 regulates monocyte migration and cell to cell adhesion in vascular endothelial Cells. Int. J. Mol. Sci. 2019, 20, 1331. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of RhoA/Rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar]
- Zhuang, R.; Wu, J.; Lin, F.; Han, L.; Liang, X.; Meng, Q.; Jiang, Y.; Wang, Z.; Yue, A.; Gu, Y.; et al. Fasudil preserves lung endothelial function and reduces pulmonary vascular remodeling in a rat model of end-stage pulmonary hypertension with left heart disease. Int. J. Mol. Med. 2018, 42, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Kanazawa, Y.; Ishizawa, S.; Yokota, T.; Utsunomiya, K. Thrombin induces MCP-1 expression through Rho-kinase and subsequent p38MAPK/NF-kappaB signaling pathway activation in vascular endothelial cells. Biochem. Biophys Res. Commun. 2011, 411, 798–803. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; Matsufuji, S.; et al. Rho-kinase regulation of TNF-α-induced nuclear translocation of NF-κB RelA/p65 and M-CSF expression via p38 MAPK in mesangial cells. Am. J. Physiol. Ren. Physiol. 2014, 307, F571–F580. [Google Scholar] [CrossRef]
- Silveira, A.A.A.; Dominical, V.M.; Vital, D.M.; Ferreira, W.A., Jr.; Costa, F.T.M.; Werneck, C.C.; Costa, F.F.; Conran, N. Attenuation of TNF-induced neutrophil adhesion by simvastatin is associated with the inhibition of Rho-GTPase activity, p50 activity and morphological changes. Int. Immunopharmacol. 2018, 58, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Dai, F.; Tang, L.; Bao, X.; Liu, Z.; Huang, C.; Zhang, T.; Yao, W. Distinct roles for ROCK1 and ROCK2 in the regulation of oxldl-mediated endothelial dysfunction. Cell Physiol. Biochem. 2018, 49, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, M.; Marampon, F.; Zani, B.M.; Prudente, S.; Perlas, E.; Caputo, V.; Cianetti, L.; Berno, V.; Narumiya, S.; Kang, S.W.; et al. ROCK2 and its alternatively spliced isoform ROCK2m positively control the maturation of the myogenic program. Mol. Cell. Biol. 2007, 27, 6163–6176. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Rajagopalan, L.E. Rho-kinase mediates lysophosphatidic acid-induced IL-8 and MCP-1 production via p38 and JNK pathways in human endothelial cells. FEBS Lett. 2010, 584, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukumoto, Y.; Tanaka, S.; Satoh, K.; Ikeda, S.; Shimokawa, H. Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Liao, J.K. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid. Redox Signal. 2014, 20, 1251–1267. [Google Scholar] [CrossRef]
- Zandi, S.; Nakao, S.; Chun, K.H.; Fiorina, P.; Sun, D.; Arita, R.; Zhao, M.; Kim, E.; Schueller, O.; Campbell, S.; et al. ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep. 2015, 10, 1173–1186. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial Dysfunction in Diabetes. Biomedicines 2020, 8, 182. https://doi.org/10.3390/biomedicines8070182
Takeda Y, Matoba K, Sekiguchi K, Nagai Y, Yokota T, Utsunomiya K, Nishimura R. Endothelial Dysfunction in Diabetes. Biomedicines. 2020; 8(7):182. https://doi.org/10.3390/biomedicines8070182
Chicago/Turabian StyleTakeda, Yusuke, Keiichiro Matoba, Kensuke Sekiguchi, Yosuke Nagai, Tamotsu Yokota, Kazunori Utsunomiya, and Rimei Nishimura. 2020. "Endothelial Dysfunction in Diabetes" Biomedicines 8, no. 7: 182. https://doi.org/10.3390/biomedicines8070182
APA StyleTakeda, Y., Matoba, K., Sekiguchi, K., Nagai, Y., Yokota, T., Utsunomiya, K., & Nishimura, R. (2020). Endothelial Dysfunction in Diabetes. Biomedicines, 8(7), 182. https://doi.org/10.3390/biomedicines8070182